
Abstract
Ailanthus altissima var. erythrocarpa is an A. altissima variety with high economic, ecological and ornamental value, but there have been no reports on the development of SSR primers for it. According to the SSR primer information provided by the transcriptome of A. altissima var. erythrocarpa, 120 individuals with different redness levels were used to screen polymorphic primers. Transcriptomic analysis revealed 10,681 SSR loci, of which mononucleotide repeats were dominant (58.3%), followed by dinucleotide and trinucleotide repeats (16.6%, 15.1%) and pentanucleotide repeats (0.2%). Among 140 pairs of randomly selected primers, nineteen pairs of core primers with high polymorphism were obtained. The average number of alleles (Na), average number of effective alleles (Ne), average Shannon’s diversity index (I), average observed heterozygosity (Ho), average expected heterozygosity (He), fixation index (F) and polymorphic information content (PIC) were 11.623, 4.098, 1.626, 0.516, 0.696, 0.232 and 0.671, respectively. Nineteen EST-SSR markers were used to study the genetic diversity and population structure of A. altissima var. erythrocarpa. The phylogenetic tree, PCoA, and structure analysis all divided the tested resources into two categories, clearly showing the genetic variation between individuals. The population showed high genetic diversity, mainly derived from intraspecific variation. Among nineteen pairs of primers, 4 pairs (p33, p15, p46, p92) could effectively distinguish and be used for fingerprinting of the tested materials. This study is of great significance for genetic diversity analysis and molecular-assisted breeding of A. altissima var. erythrocarpa.
Similar content being viewed by others


Introduction
Ailanthus altissima (Mill) Swingle, belonging to Simaroubaceae and Ailanthus, is widely distributed on all continents except Antarctica and is a pioneer tree used in greening and barren mountain afforestation. It has high economic, medicinal, ecological and ornamental value1,2,3,4,5,6,7. Wild related species often become important resources for innovative plant breeding due to their higher resistance and richer genetic characteristics than those of this species. Ailanthus altissima var. erythrocarpa is a variety of A. altissima (Mill) Swingle; it is usually recognized as a single population or multiple very small populations in the northern region of China. A. altissima var. erythrocarpa has significantly better physical and chemical properties8 and medicinal and ornamental value than A. altissima (Mill) Swingle due to its higher contents of flavonoids and other substances. The fruiting period of A. altissima var. erythrocarpa is from June to September. Because the fruit is red and persists for months, it has high ornamental value and a high profile8. There are significant differences in the appearance time, duration and chroma of fruit color (data not yet published). Genetic composition and the environment can lead to stability or variability in plant morphology9, which necessitates scientific identification and evaluation of germplasm of A. altissima var. erythrocarpa.
Expressed Sequence Tag-SSRs (EST-SSRs) are derived from mRNA transcription sequences and are applicable to genetic linkage map construction, genetic diversity analysis, population genetic structure research, and studies of species evolution and variation in plants with or without reference genomes10,11,12. Compared with g-SSRs, EST-SSRs are more efficient, require less time and have a lower cost. They also have stronger transferability among species13,14,15,16,17,18,19 and have been widely used in plant variety identification, genetic structure analysis and fingerprint development20,21,22,23,24. To date, due to the lack of genomic and genetic information, there have been few studies on the genetic diversity of A. altissima (Mill) Swingle in China and elsewhere. The research that has been performed concerned the development of nuclear microsatellite markers for Ailanthus on Mediterranean islands and in eastern Austria; population structure and genetic diversity analyses of North American A. altissima (Mill) Swingle; patterns of regional expansion and genetic differentiation among populations of Japanese highland A. altissima (Mill) Swingle; the origin, expansion and genetic structure of Chinese squash introduced from Austria; the development of 10 cpSSR markers and 13 EST-SSR markers; and genetic diversity analysis of 14 population groups in different regions of China18,25,26,27,28,29,30,31,32. These findings have promoted the application of SSR labeling in A. altissima (Mill) Swingle. Nevertheless, there are few reports on the genetic diversity of A. altissima var. erythrocarpa and no reports on the development of polymorphic SSR primers or fingerprints.
The number of available expressed sequence tags in A. altissima (Mill) Swingle is limited, and EST-SSRs of A. altissima var. erythrocarpa have not been reported. Therefore, transcriptome sequencing was used to provide SSR marker information for A. altissima var. erythrocarpa. We developed a total of nineteen pairs of EST-SSR markers and conducted genetic diversity and population structure analyses on the basis of 120 wild individuals of A. altissima var. erythrocarpa from 7 regions in China, constructing fingerprint maps. This study will provide technical support for resource collection and protection, germplasm bank establishment and management, germplasm identification and intellectual property protection in the future and will lay a theoretical foundation for breeding and germplasm innovation of A. altissima var. erythrocarpa and A. altissima (Mill) Swingle.
Results
Number and distribution of EST-SSR loci in A. altissima var. erythrocarpa
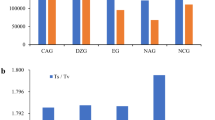
By using the Illumina HiSeq™ high-throughput sequencing platform, transcriptome sequencing of A. altissima var. erythrocarpa was performed, and an SSR search of the spliced and assembled unigenes revealed 9828 accurate SSRs and 853 compound SSRs, totaling 10,681 SSR loci. The EST-SSR types of A. altissima var. erythrocarpa were abundant, and their distribution showed (Fig. 1a) trinucleotide repeats, dinucleotide repeats and mononucleotide repeats in the largest numbers, among which 6228 mononucleotides accounted for the highest proportion (58.31%); 1777 dinucleotides accounted for 16.64%; 1621 trinucleotides accounted for 15.18%; and tetra-hexanucleotide repeats were rare, accounting for 0.99%, 0.24% and 0.66%, respectively. Other complex nucleotide repeats accounted for 7.99% of the total SSRs, indicating that mono-trinucleotides were the main repeat motifs of EST-SSRs in A. altissima var. erythrocarpa.

Distribution of different types of EST-SSRs in A. altissima var. erythrocarpa. Mono mononucleotide repeat motif, Di dinucleotide repeat motif, Tri Trinucleotide repeat motif, Tetra tetranucleotide repeat motif, Penta pentanucleotide repeat motif, Hexa hexanucleotide repeat motif, Com compound nucleotide repeat motif.
According to EST-SSR repeat motif type analysis, there were 166 exact SSR motifs, and the numbers of mononucleotide to hexanucleotide repeats were 2, 6, 30, 39, 23 and 66, respectively (Table 1). Mononucleotides were mainly the A/T repeat type (6214), accounting for 58.18% of the total number of mononucleotides. Dinucleotides were mainly the AT/AT (33.54%) and AG/CT (32.58%) repeat motifs, accounting for 5.58% and 5.42% of the total SSRs, respectively. The specific percentages of the most common trinucleotides, AGA/TCT, AAG/CTT and CTT/AAG, were 1.40%, 1.35% and 1.33%, respectively. The AAAT/ATTT and AAAG/CTTT tetranucleotides accounted for 0.18% and 0.08%, respectively. The penta- to hexanucleotide repeat motifs appeared in many forms, but their frequency was relatively low. Among all repeat types (Fig. 1b), the number of repeats of SSR loci motifs was 5 or more, and the frequency of nucleotide repeats decreased gradually with an increase in the number of repeats. The types with more than 10 repetitions accounted for 63.16% of the total, the largest percentage. This was followed by those with 5 and 6, accounting for 9.90% and 9.14% of the total, respectively. The lowest percentage was observed for those with 9 repetitions, accounting for only 2.32%. The number of mononucleotide repeats was 10 or more, that of dinucleotides was 6 or more, and the highest repetition rate was 6 times. The trinucleotide frequency was highest for 5 or more repetitions. The number of tetra- to hexanucleotides was mainly 5, which indicated that the polymorphism potential of A. altissima var. erythrocarpa was high.
Screening and validation of EST-SSR primers for A. altissima var. erythrocarpa
Based on transcriptome sequencing data analysis, SSR primers were designed, their information was obtained, and 140 pairs of primers were randomly selected for effectiveness verification. Initial screening was performed by PCR amplification and agarose gel electrophoresis, in which 106 pairs of primers were able to amplify bands with the expected fragment size, eventually resulting in amplification in 120 A. altissima var. erythrocarpa materials. DNA samples were tested by polyacrylamide gel electrophoresis and capillary electrophoresis. The analysis results for nineteen polymorphic primers (Table 2) showed clear and rich polymorphisms. The effective amplification rate was 75.71%, and the primer polymorphism rate was 13.57%.
Primer polymorphism analysis
Based on capillary electrophoresis, correlation analysis of 7 natural populations of A. altissima var. erythrocarpa was carried out with microsatellite sites showing polymorphism (Table 3 and Fig. 2). There were 221 alleles identified among the nineteen EST-SSR markers. All 120 individuals could be uniquely genotyped using these 221 alleles, demonstrating the high discrimination capacity of the nineteen EST-SSR markers. The markers showed considerable variation, with the number of alleles (Na) ranging from 7 to 20 (mean 11.632), number of effective alleles (Ne) from 1.550 to 11.942 (mean 4.098), observed heterozygosity (Ho) from 0.191 to 0.931 (mean 0.516), expected heterozygosity (He) from 0.355 to 0.916 (mean 0.696), unbiased expected heterozygosity (uHe) from 0.356 to 0.920 (mean 0.699), Shannon’s information index (I) from 0.797 to 2.675 (mean 1.626), and polymorphism information content (PIC) from 0.340 to 0.912 (mean 0.671). The marker with the most alleles was p33 (20), and the markers with the fewest alleles were p30, p40 and p95 (7). The PIC values of the nineteen EST-SSR markers ranged from 0.340 to 0.912, with an average of 0.671. All markers exhibited high polymorphism, with the exception of p30 and p49 (PIC > 0.5).

Allelic distribution patterns of nineteen polymorphic loci in 7 natural populations.
As shown in Table 4, the average inbreeding coefficients Fis and Fit were − 0.018 and 0.199, respectively, indicating that the number of homozygous individuals was greater than that of heterozygous individuals in the seven natural populations of A. altissima var. erythrocarpa. Among the seven natural populations, the Fst differentiation coefficient of the nineteen EST-SSRs showed a variation range of 0.084–0.456, with an average value of 0.237, which is between 0.15 and 0.25. The variation range of gene flow at polymorphic sites was 0.298–2.737, with an average value of 1.056.
Development of DNA fingerprints for A. altissima var. erythrocarpa
According to the genotype statistics of nineteen pairs of SSR primers, the primers with higher PIC values and more genotypes were selected to distinguish 120 A. altissima var. erythrocarpa accessions. According to the PIC values of the SSR primers, the test materials were added successively from high to low and identified gradually until they were completely separated. Table 5 shows that SSR primers p33 and p15 could distinguish 92.5% of the materials (111). SSR primers p33, p15 and p46 could distinguish 99.17% of the materials (119). Four pairs of SSR primers, p33, p15, p46 and p92, could be used to distinguish the 120 test materials. To facilitate promotion and application, the fingerprint information and names of 120 individuals of A. altissima var. erythrocarpa were imported into QR code software to generate fingerprint QR codes, wherein the primers p33, p15, p46 and p92 were represented by A, B, C and D, respectively (Supplementary Fig. 1). Please see the appendix for all fingerprints. In this study, nineteen pairs of core primers were screened to completely separate 120 test samples of A. altissima var. erythrocarpa, and the obtained DNA fingerprints were used to evaluate and identify the genotypes of this variety at the molecular level.
Genetic diversity analysis of A. altissima var. erythrocarpa
There were significant differences in genetic diversity among A. altissima var. erythrocarpa from 7 different populations (Table 6 and Fig. 3). Compared with those of A. altissima var. erythrocarpa in the other six populations, the values of Ne, I, He, uHe and F in the HB population were the highest, and the values of Ho were the lowest. There were differences in indicators of genetic diversity between populations: HB (Ne = 4.439) > NX (Ne = 3.330) > SHX (Ne = 3.224) > HN (Ne = 3.202) > BJ (Ne = 2.829) > TJ and SHD (Ne = 1.421). In contrast, the level of genetic diversity in SHD and TJ was low, which may be due to the small number of individuals. The fixation index was less than 0 in populations TJ, BJ and SHD, indicating that the number of heterozygotes was greater than that of homozygotes, while the number of homozygotes was greater than that of heterozygotes in the other four populations. The average paired FST coefficient among the 7 populations of A. altissima var. erythrocarpa was 0.163. The differentiation between TJ and SHD was the highest (0.600), which may be related to too few samples. The differentiation between BJ and SHD was the second highest (0.342), and the differentiation coefficient between NX and SHX (0.016) was the lowest, but their Nm was the highest (15.401), indicating that the genetic differentiation between the NX and SHX populations was small and that gene exchange is frequent (Fig. 4 and Supplementary Table 2).

Allelic patterns across populations of A. altissima var. erythrocarpa.

Fst and Nm values detected for population pairs of A. altissima var. erythrocarpa. Fst: above diagonal; Nm: below diagonal.
Population Structure of A. altissima var. erythrocarpa
The range of genetic distances between the seven natural populations of A. altissima var. erythrocarpa was 0.042–1.386, and the variation range of genetic similarity was 0.250–0.938 (Table 7). Among the populations, NX and SHX had the highest genetic similarity, and the genetic distance was relatively short, indicating that they were more genetically similar than other pairs. TJ and SHD had the highest genetic similarity and the greatest genetic distance, making them the most distant relatives.
According to the genetic similarity coefficients among the test materials of A. altissima var. erythrocarpa, using MEGA7 software, the UPGMA algorithm was used to constructed a dendrogram for 120 A. altissima var. erythrocarpa materials (Fig. 5a), which could be divided into two groups (A and B) through cluster analysis. Group A was mainly composed of 24 materials from the Handan and Pingshan areas of Hebei Province and Shandong Province, accounting for 20% of the total test materials. There were 96 materials from the 7 natural populations of A. altissima var. erythrocarpa in Group B, accounting for 80% of the total materials tested. In Group B, the test materials from Henan Province were scattered in each subgroup, while those in Hebei Province were more clustered, which may be related to the genetic background of the samples and the environment at the sample collection sites. Using GenAlEx 6.502 software to carry out principal coordinate analysis (Fig. 5b), the variation contribution rates of the first and second principal components were 23.22% and 5.4%, respectively, and the classification results were consistent with those of the cluster analysis. This further proves that the primers developed in this study are effective and will not mistakenly indicate obvious inter-population variation due to genetic drift. STRUCTURE 2.3.4 The software results were processed by Structure Harvester and found that (Fig. 4d) the value of LnP(D) continued to increase, there was no inflection point, and the Delta K values could be used to determine the optimal K value (Supplementary Table 3, Fig. 5c,d). STRUCTURE analysis showed that the seven natural populations of A. altissima var. erythrocarpa could be genetically divided into two different subpopulations using the ΔK method (Fig. 5c), and the clustering pattern was the same as that in the dendrogram. When K was equal to 3–5, some individuals in subgroup 1 were divided into several subgroups, indicating that these individuals were hybrids and that the other individuals had a pure pedigree (Fig. 5e). Individuals HX01, WX04, and ZHL04 in subgroup 1 consistently originated from two subgroups. The individuals in subgroup 2 originated from a single ancestral group, except that P15 and P16 were divided among three subgroups (K = 3–5).

Structure analysis of 120 A. altissima var. erythrocarpa individuals. (a) Dendrogram based on the genetic distance of individuals in different populations (UPGMA); (b) principal coordinate analysis plot; (c) delta K distribution based on the rate of change in L(K) between consecutive K values. (d) Estimated average likelihood L(K) distribution(mean ± SD) from 2 to 10 possible clusters (K). (e) histogram of the structure assignment when K = 2–5.
Discussion
In recent years, SSR molecular markers have been widely used in studies of species genetic diversity due to their advantages, such as high species specificity and cross-related species transfer, and primer development is their primary use. To date, only JosphatK. Saina et al.18,33 identified 219 SSR loci based on the chloroplast genome of A. altissima (Mill.) Swingle in 2018. In 2021, 13 EST-SSRs were developed, and 10 polymorphic chloroplast microsatellite (cpSSR) markers were constructed based on the transcriptome of A. altissima (Mill.) Swingle and 219 cpSSR markers. The distribution characteristics of cpSSRs and EST-SSRs were similar, but cpSSRs did not show hexanucleotide and compound nucleotide repetitions. In this study, 10,681 potential microsatellite loci were obtained based on transcriptome sequencing results of tissue-cultured seedlings from young leaves of A. altissima var. erythrocarpa, which was far lower than that reported by JosphatK. Saina et al.18 (33,084 potential SSR loci), and the EST-SSR distribution characteristics were similar. The SSR markers developed in this study had a high effective amplification rate, which is helpful for analyzing the genetic background and performing auxiliary breeding of A. altissima var. erythrocarpa.
Most plants show mononucleotides, dinucleotides and trinucleotides as dominant repeat motifs, such as Stephanandra incisa23, Bougainvillea cultivars21, Cocos nucifera L.34, A. altissima (Mill.) Swingle18, Pinus bungeana11, Ulmus pumila L.35, and A. altissima var. erythrocarpa. The results of the transcriptome analysis showed the same pattern, with these three types accounting for 90.12% of the total SSRs. AG/CT is the most frequent dinucleotide repeat motif in most plants, and the homopurine–homopyrimidine extension is often found in the 5′ untranslated region, which plays an important role in gene expression and nucleic acid metabolism regulation in plants. AAG/CTT is the main nucleotide repeat type in dicotyledons18,23,24,33,36,37,38,39,40,41. In this study, the dinucleotide AT/AT repeat type had the highest frequency, which was consistent with the results for Pinus bungeana11,42, Bougainvillea cultivars21. The majority of the trinucleotide repeats were AGA/TCT, followed by AAG/CTT. Among the 140 pairs of primers screened and synthesized, 106 yielded amplification bands, and the effective amplification rate reached 75.71%, which was similar to that in A. altissima (Mill.) Swingle (80%)18 and Cinnamomum chago (70.59%)43. The polymorphism rate of the primers was 13.57%, which was significantly lower than that reported by Saina et al.18 (65%), which may be caused by the use of more randomly selected primers, small genetic differences between samples and different environmental selection pressure. Therefore, EST-SSRs are still the most economical and effective tool for developing a large number of primers at the same time using transcriptome data. The chloroplast genome of A. altissima is a circular molecule with a size of 160,815 base pairs and a tetrad structure. The chloroplast genome size of A. altissima was similar to that of Arabidopsis thaliana (L.) Heynh. (120–217 kb) and Populus trichocarpa (155–159 kb), so it was relatively easy to develop cpSSR markers for A. altissima. Compared with cpSSRs, EST-SSRs exist in the gene regions of the nuclear genome and are highly polymorphic18,44. In this study, EST-SSRs were combined with molecular markers of A. altissima var. erythrocarpa to study the point mutations in A. altissima var. erythrocarpa, providing important resources for subsequent germplasm resource protection and evolutionary research in populations. Phenotypic traits combined with molecular markers could be used to analyze the correlations of traits, providing guidance for the utilization of germplasm resources and breeding of new varieties.
PIC can measure the polymorphism of primers: PIC > 0.5 indicates highly polymorphic, 0.25 < PIC < 0.5 indicates moderately polymorphic, and PIC < 0.25 indicates weakly polymorphic45. In this study, 19 pairs of clearly polymorphic primers with the expected size were screened, and the average PIC value was 0.671, lower than the average PIC value of A. altissima (0.819)18. PIC > 0.5 indicated that the identified SSR loci had high genetic resolution, making them conducive to population genetic analysis and revealing that the studied A. altissima var. erythrocarpa accessions had high genetic diversity. This may be related to the complex living environment of wild A. altissima var. erythrocarpa. The materials of wild A. altissima var. erythrocarpa used in this experiment are widely distributed in Ningxia, Shanxi, Shandong, Henan, Tianjin, Beijing, Hebei and other places. The sampling points were mostly on hillsides, on roadsides and in other similar places. The diversity of the geographical environment and the characteristics of climate variability jointly determine the diversity of the living environment of A. altissima var. erythrocarpa. Therefore, this study further reveals the genetic diversity and structure of this species.
The average expected heterozygosity is less affected by sampling than other metrics. The higher the value is, the lower the genetic consistency and the higher the level of genetic diversity. The average expected heterozygosity of A. altissima var. erythrocarpa from 7 natural populations was 0.509, similar to the results of Saina et al.18, which may be related to the wide distribution and complex living environment of wild A. altissima var. erythrocarpa. The mean He was slightly lower than that reported in previous studies18,28,30, which may be related to the fact that A. altissima var. erythrocarpa is a variety of A. altissima and originated in the same place. In the study of Saina et al.18, the mean Ne of A. altissima (Mill.) Swingle is 3.757. In this study, the mean Ne of A. altissima var. erythrocarpa is 2.838. This may be due to the fact that the population size (14) and sample size (165) of A. altissima (Mill.) Swingle are larger than that of this study (7 and 120), and the distribution range (99–120° N and 29–37° E) is wider than that of A. altissima var. erythrocarpa (105–117° N and 33–41° E). Among the seven natural populations, HB (Ne = 4.439, I = 1.625) had the highest genetic diversity, TJ (Ne = 1.421, I = 0.292) and SHD (Ne = 1.421, I = 0.292) had the lowest genetic diversity. This may be because each group has different site conditions (city streets, mountains, rocks), and its growing environment is different; Samples from city streets, there is human interference. These all influence the measurement of genetic diversity. The FST comparison between nineteen pairs of EST-SSRs and 7 natural populations (Fst mean = 0.163, Nm mean = 3.783) showed a high degree of genetic differentiation (Table 4 and Fig. 4) and frequent gene exchange, indicating that there was strong gene flow (Nm > 1) among the seven natural populations, which inhibited genetic differentiation caused by genetic drift, with low differentiation level and high intra-population differentiation level. This is supported by PCoA (Fig. 5b) and dendrogram based on the UPGMA algorithm (Fig. 5a).
According to genetic distance and genetic similarity analysis, NX and SHX had the closest genetic relationship. To effectively analyze A. altissima var. erythrocarpa germplasm resources, information about natural habitat, genetic evolution, genetic diversity, and the degree of species mixing is required46. For principal coordinate analysis, structure analysis, and cluster analysis based on genetic distance as determined by UPGMA, the seven natural populations of red-fruited Ailanthus could be divided into two subgroups; one was subgroup A dominated by materials from Handan and Pingshan County, Hebei Province, and the other was subgroup B (Fig. 5a,b,e) dominated by materials other than those from SHD, which may be because the material for transcriptome sequencing was taken from Handan, and subgroup A was highly represented among the samples. Structure-based clustering is a reliable tool for estimating the degree of species mixing, and the kinship of a small number of samples in subpopulation A and subpopulation B was complex (Fig. 5e), indicating that during the evolution of the species, frequent gene exchange and recombination between individuals in the region led to a richer genetic structure. Some germplasm resources are introduced to different places via wind and animal and human activities. NX, SHX, HN, TJ, and BJ were clustered in subgroup A, which may not be geographically isolated, and subgroup A and subgroup B included samples of the HB natural population. When K = 3–5, individuals HX01, WX04, and ZHL04 in subgroup 1 have complex lineages and are always related to both subpopulations. Individuals in subgroup 2, except for P15 and P16, which can be divided into three subgroups, all originated from a single ancestral group, these results can explain that there is no geographical separation between the 7 natural groups, and the kinship and complexity of each individual can be clearly known. The resources of the same area were almost always clustered together (Fig. 5a,b,e). This finding is similar to the classification results of Astragali Radix47, indicating high genetic similarity and frequent gene exchange.
DNA fingerprinting technology analyzes the genetic material itself, avoiding the influences of environmental factors, sample morphology, material source and other factors, and identifies the germplasm genotypes as indicators at the molecular level. It has been widely used in grain, oil, vegetable and fruit crops, flowers, shrubs, arbors and other plants16,20,21,48. DNA fingerprinting has high simplicity and stability, variability, and multiple loci and can distinguish members of the same family, such as different species, varieties, and forms and even the same strain and subtle variation between individuals. It can be used in germplasm genetic relationship inference, genetic breeding, population genetic structure determination, ecological and evolutionary studies, classification and other valuable genetic marker methods48,49,50. However, there have been no reports on the fingerprint information of A. altissima var. erythrocarpa. In this study, four of nineteen EST-SSR core primers (p33, p15, p46 and p92) were screened to distinguish 120 tested A. altissima var. erythrocarpa materials and construct a unique molecular ID card. The establishment of fingerprinting for A. altissima var. erythrocarpa is of great significance for its subsequent breeding, as the resource can be used to identify varieties and excellent genes as well as their variation and transmission in offspring, and lays a foundation for the establishment and improvement of fingerprinting databases of A. altissima var. erythrocarpa variety resources, genetic resource management and intellectual property protection. However, in this study, there were few test materials, the sample collection area was only in northern China, and the fingerprint background of the "core germplasms” of A. altissima var. erythrocarpa lacked in-depth research. As a result, further verification of the reliability of the results is required.
Conclusion
In this study, nineteen pairs of EST-SSRs revealed high genetic diversity and significant genetic structure in A. altissima var. erythrocarpa. Using four pairs of SSR core primers, a total of 120 wild individuals from 7 regions of A. altissima var. erythrocarpa were 100% distinguished and assigned unique identification IDs. The experimental resources of A. altissima var. erythrocarpa have rich genetic variations, mainly derived from intraspecific variations, and do not exhibit geographical variation. The research results presented in this article provide a public resource of SSR primers in A. altissima var. erythrocarpa, in turn providing a theoretical basis for effective management, protection, and utilization of its germplasms. The excellent varieties studied according to the SSR primers of A. altissima var. erythrocarpa can be introduced by biotechnological means and can also be preserved ex situ or in situ, which would be conducive to subsequent research. The findings also provide genetic resources for an in-depth understanding of population genetics and evolutionary history, joint genomics research, the selection of dominant parents for the cultivation of new varieties, and intellectual property protection in A. altissima var. erythrocarpa.
Materials and methods
Plant materials
A total of 120 wild individuals of Ailanthus altissima var. erythrocarpa selected based on four indicators were collected from 7 provinces in China (Ningxia, Shanxi, Henan, Tianjin, Beijing, Hebei, and Shandong). The geographical locations of the sample spanned 106.00–117.72 east longitude, 33.76–41.12 north latitude, and 34.7–1858.2 m altitude, and the local soil was mostly red soil. When sampling, the latitude, longitude and altitude of the sample were recorded with a GPS device. The HN, TJ, BJ and SHD sampling sites were distributed discontinuously, with 6–10 plants per hectare. The number of individuals was small., the distribution was scattered, and the distance between the trees collected was not less than 50 m to prevent half-sib relationship between the samples. (Supplementary Table 1, Figs. 6 and 7).

Screening index system for 120 A. altissima var. erythrocarpa individuals. Redness: degree of redness of the samara; type: full red and gradually red; fruiting period: early red (June-July) and late red (July August); measurement: large fruit and small fruit.

Geographic distribution of 120 A. altissima var. erythrocarpa wild individual samples from 7 provinces in China. NX: Ningxia Province, China; SHX: Shanxi Province, China; HN: Henan Province, China; TJ: Tianjin Province, China; BJ: Beijing Province, China; HB: Hebei Province, China; and SHD: Shandong Province, China. The map was generated using ArcMap 10.2 software.
DNA extraction
Fresh and tender Ailanthus altissima var. erythrocarpa leaves were selected and brought back into the laboratory in an ice box. Genomic DNA of Ailanthus altissima var. erythrocarpa was extracted by the improved CTAB method and an Omega kit. DNA concentration and quality were determined by a microspectrophotometer (model: Nano300). After quality determination, the DNA was diluted to 30 ng/μl in each tube. A total of 5 tubes (150 μl/tube) were used as working liquid. The original liquid and working liquid were stored in a freezer at − 20 °C for later use.
Development of SSR primers
Due to the lack of a reference genome sequence, leaves of tissue-cultured plantlets were sent to Wuhan Matwell Biotechnology Co., Ltd., for transcriptome sequencing and primer design. SSR primer design data were provided by Professor Dukejiu of Hebei Agricultural University. A total of 140 pairs of primers were randomly selected from 10,681 pairs of primers by Excel, Sequon Bioengineering (Shanghai) Co., Ltd., and Tsingke Biotechnology Co., Ltd., were commissioned to synthesize primers.
SSR primer screening and PCR amplification
First, the mixtures of 8 samples were used as templates to screen out primers with clear and bright bands that matched the size of the product. Second, primers with clear bands and good polymorphism were selected for amplification in 6 samples. Finally, the core primers were screened from 120 samples, which were verified with the primers.
-
(1)
Nondenaturing polyacrylamide gel electrophoresis. The optimized PCR system was carried out in a volume of 20 µl: DNA template 1.5 µl (30 ng/μl), 2 × Taq PCR Master Mix 10 μl, upstream and downstream primers 1 μl (10 µmol/µl), and ddH2O 6.5 µl. The thermal cycling parameters used in the PCR procedure were 95 °C predenaturation for 5 min, followed by 33 cycles of 95 °C for 1 min, 55 °C annealing for 30 s, and 72 °C extension for 45 s, with a final elongation step at 72 °C for 8 min. The samples were then stored at 4 °C. The initial screening conditions for polymorphic primers were as follows: 2% agarose gel electrophoresis, voltage 110 V, 30 min, real-time photography; screening and verification with 8% nondenatured polyacrylamide gel electrophoresis, 200 V, 100 mA, 20 W, 1 h and 300 V, 100 mA, 20 W, 2.5 h; silver staining (AgNO3); and photo storage for subsequent analysis.
-
(2)
Fluorescent capillary electrophoresis The PCR systems were similar to those used for nondenaturing polyacrylamide gel electrophoresis, with the only modification being the addition of 1 μl (10 µmol/μl) fluorescent primer (FAM/TAMRA/HEX/POX) to each plate system. The procedure was as follows: 95 °C predenaturation (5 min), followed by 25 cycles of 95 °C denaturation (1 min), 58 °C annealing (30 s), and 72 °C extension (45 s) and then 10 cycles of 95 °C denaturation (1 min), 58 °C annealing (30 s), and 72 °C extension (45 s), with a final elongation step at 72 °C for 8 min. The products amplified by PCR were sent to Taihe Biotechnology Co., Ltd., for testing.
Data analysis
The fluorescence capillary electrophoresis detection results were viewed using GeneMarker V2.4.0 software and collated in Excel. GenAlEx 6.502 software was used to calculate the average number of alleles (Na), average effective number of alleles (Ne), Shannon's information index (I), average observed heterozygosity (Ho), average expected heterozygosity (He), and fixation index (F). Hardy–Weinberg equilibrium tests were performed18,51. The polymorphism information content (PIC) was calculated using Cervus 307 software18,51. In GenAlExv6.5, F and Nm (Nm = (0.156/SHUA)2 statistical calculations (Fis, Fit and Fst) and principal coordinate analysis (PCoA) were performed in combination with Microsoft Excel. The paired heat map of Fst and Nm was constructed in R 4.2.2. Used Populations v1.2.32 to calculate genetic distances, based on genetic similarity coefficients, a phylogenetic tree was drawn using MEGA752. Mixed analysis based on Bayesian models was used for population structure analysis in Structure v2.3.453. Then, the number of Markov chain Monte Carlo (MCMC) iterations after burn-in was set to 100,000, with a run length of 100,000 and a genetic uniform cluster number (K value) between 1 and 10. Each analysis consisted of 10 repeated runs. The optimal K value was determined using the best-K method in StructureHarvester54. Then used CLUMPP Windows.1.1.2b software55 to found the real K value, replaced the K value in STRUCTURE, and generated a genetic structure map.
Statement
Permission to collect plant samples was obtained for this study. Voucher specimens were identified by Alfred Rehder and deposited in the Chinese Field Herbarium (CFH). This study complied with local and national guidelines.
Data availability
The dataset is available from the NCBI BioProject (PRJNA1006443) and NCBI Short Read Archive (SRA) with accession number SRR25900069.
References
Gürbüz, R. & Kahramanolu, I. Possibility of using leaf extracts of tree-of-heaven (Ailanthus altissima (Mill.) Swingle) for the postharvest quality preservation of fresh apricot fruits. Physiol. Mol. Plant Pathol. 113, 101594. https://doi.org/10.1016/j.pmpp.2020.101594 (2020).
Shu, M. et al. Physiological response of main greening tree species to dust pollution in northwest liaoning province. Acta Bot. Bor Occid. Sin. 40, 1740–1750 (2020).
Liu, L. J., Leng, P. S., Hu, Z. H., Zhang, C. L. & Yang, Y. Influences of growth media mixed from municipal raw sludge and construction waste on growth and heavy metal transferring of Ailanthus altissima. Chin. J. Appl. Environ. Biol. 24, 1390–1397. https://doi.org/10.19675/j.cnki.1006-687x.2018.02031 (2018).
Tan, Q. W., Ni, J. C., Shi, J. T., Zhu, J. X. & Chen, Q. J. Two novel quassinoid glycosides with antiviral activity from the samara of Ailanthus altissima. Molecules 25, 5679. https://doi.org/10.3390/molecules25235679 (2020).
Yan, Z. Y. et al. Terpenylated coumarins from the root bark of Ailanthus altissima (Mill.) Swingle. Phytochemistry 175, 112361. https://doi.org/10.1016/j.phytochem.2020.112361 (2020).
Zhu, X. Q., Zhang, P. A., Chen, J. Y. & Zhao, T. B. Ornamental types of Ailanthus Desf. in Henan. J. Henan For. Sci. Technol. 20, 10–12 (2000).
Kowarik, I. & Säumel, I. Biological flora of centralEurope: Ailanthus altissima (Mill.) swingle. Perspect. Plant Ecol. Evol. Syst. 8, 207–237. https://doi.org/10.1016/j.ppees.2007.03.002 (2007).
Xu, H. Study on the Characteristics of Morphology and Physiological of Red Fruit Ailanthus altissima (Liaocheng University, 2015).
Coleman, J. S., McConnaughay, K. D. & Ackerly, D. D. Interpreting phenotypic variation in plants. Trends Ecol. Evol. 9, 187–191. https://doi.org/10.1016/0169-5347(94)90087-6 (1994).
Chen, M. et al. Development of EST-SSR markers based on transcriptome sequencing for germplasm evaluation of 65 lilies (Lilium). Mol. Biol. Rep. 50(4), 3259–3269. https://doi.org/10.1007/s11033-022-08083-9 (2023).
Yang, X. et al. Development and application of EST-SSR molecular markers in Pinus bungeana. J. Beijing For. Univ. 43, 1–11 (2021).
Wang, L. et al. Development and validation of EST-SSR markers of Magnolia wufengensis using de novo transcriptome sequencing. Trees 33, 1213–1223. https://doi.org/10.1007/s00468-019-01870-1 (2019).
Wang, X. et al. Development of EST-SSR markers and their application in an analysis of the genetic diversity of the endangered species Magnolia sinostellata. Mol. Genet. Genom. 294, 135–147. https://doi.org/10.1007/s00438-018-1493-7 (2019).
Zhang, J., Liu, T. & Rui, F. Development of EST-SSR markers derived from transcriptome of Saccharina japonica and their application in genetic diversity analysis. J. Appl. Phycol. 30, 2101–2109. https://doi.org/10.1007/s10811-017-1354-5 (2017).
Vašek, J. et al. New EST-SSR markers for individual genotyping of opium poppy cultivars (Papaver somniferum L.). Plants 9, 10. https://doi.org/10.3390/plants9010010 (2019).
Gao, P., Ma, H. Y., Luan, F. S. & Song, H. B. DNA fingerprinting of Chinese melon provides evidentiary support of seed quality appraisal. PLoS ONE 7, e52431. https://doi.org/10.1371/journal.pone.0052431 (2012).
Kumar, N. et al. SSR-based DNA fingerprinting and diversity assessment among Indian germplasm of Euryale ferox: An aquatic underutilized and neglected food crop. Appl. Biochem. Biotechnol. 185, 34–41. https://doi.org/10.1007/s12010-017-2643-9 (2018).
Saina, J. K., Li, Z. Z., Mekbib, Y., Gichira, A. W. & Liao, Y. Y. Transcriptome sequencing and microsatellite marker discovery in Ailanthus altissima (Mill.) Swingle (Simaroubaceae). Mol. Biol. Rep. 48, 2007–2023. https://doi.org/10.1007/s11033-020-05402-w (2021).
Kim, J. M. et al. Analysis of genetic diversity and relationships of Perilla frutescens using novel EST-SSR markers derived from transcriptome between wild-type and mutant Perilla. Mol. Biol. Rep. 48, 6387–6400. https://doi.org/10.1007/s11033-021-06639-9 (2021).
Li, Q. et al. Construction of fingerprint and genetic diversity analysis of 61 high-oil soybean varieties (lines). J. Henan Agric. Sci. 50, 53–62. https://doi.org/10.15933/j.cnki.1004-3268.2021.09.007 (2021).
Pavan Kumar, P., Janakiram, T. & Bhat, K. V. Microsatellite based DNA fingerprinting and assessment of genetic diversity in bougainvillea cultivars. Gene 30(753), 144794. https://doi.org/10.1016/j.gene.2020.144794 (2020).
Ma, S. et al. Fingerprint identification of white clover cultivars based on SSR molecular markers. Mol. Biol. Rep. 47, 8513–8521. https://doi.org/10.1007/s11033-020-05893-7 (2020).
Zhang, C. et al. De novo transcriptomic analysis and identification of EST-SSR markers in Stephanandra incisa. Sci. Rep. 11, 1059. https://doi.org/10.1038/s41598-020-80329-7 (2021).
Wu, Q. et al. Full-length transcriptome sequencing analysis and development of EST-SSR markers for the endangered species Populus wulianensis. Sci. Rep. 10, 16249. https://doi.org/10.1038/s41598-020-73289-5 (2020).
Dallas, J. F., Leitch, M. J. & Hulme, P. E. Microsatellites for tree of heaven (Ailanthus altissima). Mol. Ecol. Res. 5, 340–342. https://doi.org/10.1111/j.1471-8286.2005.00920.x (2005).
Neophytou, C. et al. Analysis of microsatellite loci in tree of heaven (Ailanthus altissima (Mill.) Swingle) using SSR-GBS. Tree Genet. Genomes 14, 82. https://doi.org/10.1007/s11295-018-1295-4 (2018).
Aldrich, P. R. et al. Genetic structure of the invasive tree Ailanthus altissima in eastern United States cities. J. Bot. https://doi.org/10.1155/2010/795735 (2010).
Brusa, A. & Holzapfel, C. Population structure of Ailanthus altissima (Simaroubaceae): The role of land-use history and management1. J Torrey Bot. Soc. 145, 55–69. https://doi.org/10.3159/TORREY-D-17-00012.1 (2018).
Chuman, M., Kurokochi, H., Saito, Y. & Ide, Y. Expansion of an invasive species, Ailanthus altissima, at a regional scale in Japan. J. Ecol. Environ. 38, 47–56 (2015).
Kurokochi, H., Saito, Y., Chuman, M. & Ide, Y. Low chloroplast diversity despite phylogenetically divergent haplotypes in Japanese populations of Ailanthus altissima (Simaroubaceae). Botany 91, 148–154. https://doi.org/10.1139/cjb-2012-0159 (2012).
Kurokochi, H., Saito, Y. & Ide, Y. Genetic structure of the introduced heaven tree (Ailanthus altissima) in Japan: Evidence for two distinct origins with limited admixture. Botany 93, 133–139 (2014).
Neophytou, C., Pötzelsberger, E., Curto, M., Meimberg, H. & Hasenauer, H. Population bottlenecks have shaped the genetic variation of Ailanthus altissima (Mill) Swingle in an area of early introduction. Forestry https://doi.org/10.1093/forestry/cpz019 (2019).
Saina, J. K., Li, Z. Z., Gichira, A. W. & Liao, Y. Y. The complete chloroplast genome sequence of tree of heaven (Ailanthus altissima (Mill) (Sapindales: Simaroubaceae), an important pantropical tree. Int. J. Mol. Sci. 19, 929. https://doi.org/10.1007/s11033-020-05402-w (2021).
Preethi, P. et al. Development of EST-SSR markers for genetic diversity analysis in coconut (Cocos nucifera L.). Mol. Biol. Rep. 47, 9385–9397. https://doi.org/10.1007/s11033-020-05981-8 (2020).
Zuo, L. H., Zhang, S. H., Liang, H. Y., Wang, J. M. & Yang, M. S. Development of the elm transcriptome with EST-SSR primers and genetic diversity analysis. J. Plant Genet. Resour. 19, 57–166. https://doi.org/10.13430/j.cnki.jpgr.2018.01.018 (2018).
Sharma, H. et al. Development of polymorphic EST-SSR markers and their applicability in genetic diversity evaluation in Rhododendron arboreum. Mol. Biol. Rep. 47, 2447–2457. https://doi.org/10.1007/s11033-020-05300-1 (2020).
Zhou, Q., Zhou, P. Y., Zou, W. T. & Li, Y. G. EST-SSR marker development based on transcriptome sequencing and genetic analyses of Phoebe bournei (Lauraceae). Mol. Biol. Rep. 48, 2201–2208. https://doi.org/10.1007/s11033-021-06228-w (2021).
Yin, Y. et al. SSR information in transcriptome and development of molecular markers in Lycium ruthenicum. J. Zhejiang A&F Univ. 36, 422–428 (2019).
Xiao, Y. S., Chen, X. Q., Li, H. C., Wang, K. L. & Song, L. Y. Development and identification of SSR molecular markers based on transcriptome sequencing of Wedelia trilobata. Mol. Plant Breed. 19, 2293–2299. https://doi.org/10.13271/j.mpb.019.002293 (2021).
Morgante, M., Hanafey, M. & Powell, W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 30, 194–200. https://doi.org/10.1038/ng822 (2002).
Martienssen, R. A. & Colot, V. DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 293, 1070–1074. https://doi.org/10.1126/science.293.5532.1070 (2001).
Duan, D., Jia, Y., Yang, J. & Li, Z. H. Comparative transcriptome analysis of male and female conelets and development of microsatellite markers in Pinus bungeana, an endemic Conifer in China. Genes (Basel) 8, 393. https://doi.org/10.3390/genes8120393 (2017).
Zhang, X., Zhang, Y., Wang, Y. H. & Shen, S. K. Transcriptome analysis of Cinnamomum chago: A revelation of candidate genes for abiotic stress response and terpenoid and fatty acid biosyntheses. Front. Gene 9, 505. https://doi.org/10.3389/fgene.2018.00505 (2018).
Thiel, T., Michalek, W., Varshney, R. K. & Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Gene 106, 411–422. https://doi.org/10.1007/s00122-002-1031-0 (2003).
Botstein, D., White, R. L., Skolnick, M. & Davis, R. W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 32(3), 314. https://doi.org/10.1016/0165-1161(81)90274-0 (1980).
Raja, W. H. et al. Morpho-molecular characterization and genetic diversity analysis across wild apple (Malus baccata) accessions using simple sequence repeat markers. S. Afr. J. Bot. 145, 378–385. https://doi.org/10.1016/j.sajb.2021.08.020 (2021).
Jiang, M. et al. Genetic diversity of the Chinese medicinal plant Astragali Radix based on transcriptome-derived SSR markers. Electron. J. Biotechnol. 62, 13–20. https://doi.org/10.1016/j.ejbt.2022.12.001 (2023).
Sarimana, U. et al. Analysis of genetic diversity and discrimination of Oil Palm DxP populations based on the origins of pisifera elite parents. Breed. Sci. 71, 134–143. https://doi.org/10.1270/jsbbs.20043 (2021).
Jiang, F. DNA fingerprint technology and its application in Chinese medicine. Strait Pharm. J. 17, 183–185. https://doi.org/10.3969/j.issn.1006-3765.2005.04.111 (2005).
Chen, J., Zheng, G. C., Jing, Y. H., Ma, Z. Q. & Zheng, Y. The analysis of DNA fingerprints in the four series maize inbred from different source. J. Inner Mong. Minzu Univ. (Nat. Sci.) https://doi.org/10.14045/j.cnki.15-1220.2007.03.015 (2007).
Shi, Y. T., Lai, B. W., Wang, X. J., Qin, H. Z. & Chai, S. F. Development EST-SSR markers based on transcriptome sequences in Keteleeria calcarea. Mol. Plant Breed. 43, 1–18 (2022).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Liu, J. et al. Genetic diversity analysis of Sapindus in China and extraction of a core germplasm collection using EST-SSR markers. Front. Plant Sci. 13, 857993. https://doi.org/10.3389/fpls.2022.857993 (2022).
Earl, D. A. & vonHoldt, B. M. Structure harvester: A website and program for visualizing structure output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. https://doi.org/10.1007/s12686-011-9548-7 (2012).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23(14), 1801–1806. https://doi.org/10.1093/bioinformatics/btm233 (2007).
Acknowledgements
This work was supported by the Hebei Provincial Innovation Ability Promotion Program (20567648H), Shijiazhuang Science and Technology Research and Development Program (201520322A), and Hebei Provincial High-level Talents Funding Project (A202001039), and Shijiazhuang Science and Technology Plan Project (231520132A). We would like to thank Professor Du Qingzhang for his review and suggestions for this article, as well as Mr. Dai Jianfeng for his assistance in the experiment.
Author information
Authors and Affiliations
Contributions
Z.X. and K.D. designed the experiments and proofread the article; M.Z. collected the plant materials, performed the experiments, conducted the data analysis, and wrote the manuscript; C.Z. collected part of the plant materials; J.L. extracted the DNA; X.W., C.L. and X.L. collected the plant materials.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, M., Zheng, C., Li, J. et al. Genetic diversity, population structure, and DNA fingerprinting of Ailanthus altissima var. erythrocarpa based on EST-SSR markers. Sci Rep 13, 19315 (2023). https://doi.org/10.1038/s41598-023-46798-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-46798-2
This article is cited by
-
Genetic diversity and population structure of Azadirachta indica using simple sequence repeat (SSR) markers for genetic resource conservation and utilization
Genetic Resources and Crop Evolution (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
