
Abstract
Zanthoxylum nitidum (Roxb.) is a commonly used traditional Chinese medicine. However, the collection and protection of wild germplasm resources of Z. nitidum are still insufficient, and there is limited research on its genetic diversity and fingerprint. In the present study, 15 simple sequence repeat (SSR) markers were developed by genotyping based on multiplexed shotgun sequencing. The genetic diversity of 51 populations (142 individuals) of Z. nitidum was evaluated using these 15 SSRs. A total of 245 alleles (Na) were detected, with an average value of 16.333, and the average polymorphism information content was 0.756. The genetic distance among 51 populations was 0.164~1.000, with an average of 0.659. Analysis of molecular variance showed low genetic differentiation (40%) and high genetic differentiation (60%) between populations and individuals, respectively. The genetic differentiation coefficient (Fst) of the population was 0.338, indicating that 66.2% of the genetic variation occurred within the population, and the gene flow (Nm) was 0.636, demonstrating that the gene exchange between populations was low. Clustering analysis revealed that the genetic similarity coefficient was 0.30, dividing the 51 populations into 4 groups of 2, 17, 3, and 29 populations. There was no specific relationship between geographical location differences and genetic distance. The genetic diversity level of Z. nitidum is relatively high, and our results provide a theoretical basis for the rapid identification of Z. nitidum germplasm resources and variety selection.
Similar content being viewed by others
Introduction
Zanthoxylum nitidum (Roxb.) DC., a notable medicinal plant, belongs to the genus Zanthoxylum in the Rutaceae family, the root used as traditional medicine is named Liang-Mian-Zhen1. Z. nitidum was first recorded in "Shennong Materia Medica Classic" during the Qin and Han dynasties under the name "Manjiao"2. It mainly grows in Guangxi, Guangdong, and other locations in China3. The roots mainly contain alkaloids4,5, sesquiterpenoids6, coumarins7, lignans8, and other components that have various pharmacological activities, such as antiinflammatory9,10, antibacterial11,12, anticancer13 and analgesic14 activities. Z. nitidum is not only the main raw material of more than 60 famous traditional Chinese patent medicines and simple preparations, such as Sanjiu Weitai Granules, LiangMianZhen Analgesic Tablets, and Dieda Wanhua Oil, but its extracts are widely used in toothpaste, soap, shampoo and other daily personal products15,16.
Zanthoxylum nitidum can be subdivided into Z. nitidum var. nitidum and Z. nitidum var. tomentosum according to whether each section of the plant has short, rough hairs, particularly on both sides of the leaves. Furthermore, Z. nitidum var. nitidum is separated into three types based on the number of thorns on branches and leaf axes, as well as the size and thickness of leaflets3,17. Due to the high demand for Z. nitidum in the market, wild resources have been plundered recklessly for a long time, and have become increasingly scarce17,18. The fundamental measure to solve the resource crisis is to transform the source from wild to artificially planted. However, in the process of introducing and domesticating wild Z. nitidum germplasm, it is difficult to accurately distinguish resources with such similar traits using traditional morphological identification methods. Hence, there is a serious shortage of excellent germplasm basic materials for artificial cultivation, and the unclear phenotypic characteristics and genetic background of cultivated germplasm are unclear, which seriously limits the development of related industries for Z. nitidum.
Some of the advantages of simple sequence repeat (SSR) molecular markers are rich polymorphism, wide distribution, easy operation, and high sensitivity19. SSR molecular markers have been widely used in genetic diversity analysis, population structure analysis, and fingerprint construction of medicinal plants, such as for Astragali Radix20, Akebia trifoliata21, and Paris polyphylla22, providing new ideas for the protection of core germplasms, functional gene identification, variety identification, and molecular marker assisted breeding of medicinal plants. However, studies on the genetic diversity of Z. nitidum are limited, and one of the most important reasons is the lack of effective molecular markers for this species.
In this study, great efforts were made to collect wild germplasm resources of Z. nitidum from Guangxi and Guangdong. We detected an abundance of SSRs based on the multiplexed shotgun genotyping (MSG) data, and we developed SSR molecular markers to assess the level of genetic diversity for the collected germplasm resources. Our data will provide a scientific basis for species identification and selective breeding of Z. nitidum.
Materials and methods
Plant materials and DNA extraction
From September to October 2022, a total of 142 Z. nitidum var. nitidum and Z. nitidum var. tomentosum individuals from 51 populations in Guangdong and Guangxi were collected, including 16 germplasms from 7 artificially cultivated populations and 126 germplasms from 44 wild populations. The germplasm numbers and source information are shown in Table S1. Fresh young leaves were collected and stored at − 80 °C for DNA extraction. Total genomic DNA was extracted from the frozen leaves following the instructions of Magnetic Bead Method using a Plant Genomic Extraction Kit (NanoMagBio, Wuhan, China). The concentration and purity of DNA were determined by using an ultramicro spectrophotometer (NanoDrop ONE, Thermo Fisher, USA).
MSG library construction and sequencing
Three germplasms with significant phenotypic differences (GX0826-1, bl0915-1, 3-1) were selected for constructing the library. We prepared the sequencing library using the MSG method proposed by Andolfatto et al.23. We purified the libraries and selected DNA fragments in the 400 bp size range using AMPure XP beads (Beckman Coulter, Inc., USA), and then amplified them using PCR for 14 cycles. The Illumina NovaSeq platform (Illumina, Inc., San Diego, USA) was used to obtain raw sequence data. The original data were filtered using the sliding window analysis method of fastp (v0.20.0)24, and the sequences were integrated using FLASH (v1.2.11)25 to obtain high-quality data. The sequencing data have been deposited in China National GeneBank (CNGB) with project accession number CNP0004226 (https://db.cngb.org/search/project/CNP0004226/).
SSR primer pair design
The Microsatellite Identification Tool (MISA) (http://pgrc.ipk-gatersleben.de/misa/) was used to search for SSR loci in all high-quality sequences. Cd-hit software26 was used to cluster the sequences. The Perl program was used to analyze the clustering results and evaluate polymorphisms27. Finally, PCR primer pairs flanking the SSR repeats were designed using primer 3 (v2.3.6)28.
SSR analysis and screening
SSRs were classified based on the length of the SSR motifs in their sequences. Different repeats of the SSR motifs were used to analyze the characteristics of the SSRs. We randomly selected 192 pairs of primers, and preliminarily screened 10 germplasms with significant phenotypic differences (GX0826-1, bl0915-1, 3-1, te0920-1, jx0928-1, pg1001-1, tl1004-1, qb1007-1-1, nv-1, py1-1). PCR reaction was performed using a Veriti 384 PCR instrument(Veriti 384, AppliedBiosystem,USA). The PCR reaction system was as follows: template DNA 1 μL, forward and reverse primers are both 10 pmol/μL (Shanghai, Shanghai), 0.5 μL each, 5 μL of MIX enzyme (2 × Taq PCR Master Mix, GeneTech, USA) supplemented with 3 μL ddH2O (total 10 μL). The reaction conditions were as follows: predenaturation at 95 °C for 5 min, 25 cycles of denaturation at 95 °C for 30 s, gradient annealing at 62~52 °C for 30 s, extension at 72 °C for 30 s, followed by extension at 72 °C for 20 min. Polymorphism of each successfully amplified marker was evaluated by GeneMarker software.
Fluorescence capillary electrophoresis detection
Fluorescent primers were obtained from Wuhan Tianyi Huiyuan Biotechnology Co., Ltd. (Wuhan, China), and the fluorescent dyes used were FAM, HEX, and TAMRA. One microliter of fluorescent PCR product, 0.5 μL of GeneScan™500 LIZ, and 8.5 μL of Hi-Di™ formamide were added to the upper plate, centrifuged, denatured (95 °C for 5 min), and cooled. Finally, the samples were analyzed using an ABI3731XL sequence analyzer (AppliedBiosystem, USA).
Genetic diversity analysis
GenAlEx (v6.501) software was used to calculate genetic diversity indicators29, including the number of observed alleles (Na), effective alleles (Ne), observed heterozygosity (Ho), expected heterozygosity (He), fixation index (F), Shannon’s information index (I), genetic differentiation coefficient (Fst), gene flow (Nm) and analysis of molecular variance (AMOVA). Linkage Disequilibrium between the SSR loci was carried out using the SHEsis plus30. The genetic distance among populations was calculated using Powermarker (V3.25)31. The genetic structure of the germplasm was analyzed using STRUCTURE software (v2.3.4)32, and cluster analysis between populations was based on unweighted pair group with arithmetic average (UPGMA).
Plant collecting permit declaration
The plant materials used in this article did not involve disputes. We hereby declare that all of the plant materials (Z. nitidum var. nitidum and Z. nitidum var. tomentosum) were collected in compliance with institutional, national, and international guidelines and legislation. The plant material in our collection is preserved in the Germplasm Repository of Guangxi Botanical Garden of Medicinal Plants, Guangxi Province, China. The voucher ID for each sample is shown in Table S1. The formal identification of these plant materials was performed by Prof. Yudeng Peng (Guangxi Botanical Garden of Medicinal Plants, China).
Result analysis
SSR quantity analysis
MSG data of the three samples (GX0826-1, bl0915-1, and 3-1) were 8.63 Gb, 6.66 Gb, and 9.35 Gb, respectively. After filtering, high-quality data were generated, which were 1.30 Gb, 0.93 Gb, and 1.20 Gb, respectively. Furthermore, 2,903,693 pairs of read pairs were merged from a total of 12,014,757 pairs. Using MISA software, 261,267 SSR loci were found, distributed among 227,023 unigenes. The frequency of SSR occurrence (the proportion of sequences containing SSR loci to all sequences) was 7.82%. The average distance (total sequence length divided by the total number of SSRs) was 2.73 kb. Among them, there were 30,512 unigenes containing more than one SSR locus, and 29,314 SSRs were present in composite form (Table 1).
SSR polymorphisms
SSRs with mono, di-, tri-, tetra-, penta-, and hexanucleotide repeat units were identified based on MSG data. Among the SSRs, mononucleotide (207,994, 79.61%) was the most abundant repeat type, followed by dinucleotide loci (32,332, 12.38%), trinucleotide loci (17,637, 6.75%), tetranucleotide loci (2265, 0.87%), hexanucleotide loci (525, 0.20%), and pentanucleotide loci (514, 0.20%). Moreover, the three types of repetitive motifs with the highest number were A/T (207,789, 79.53%), AT/AT (19,881, 7.61%), and AAT/ATT (8245, 3.16%) (Table 2).
SSR marker development
On the basis of the identified SSR loci, 768 primer pairs were designed (Table S2). Among the 192 randomly selected SSR markers (Table S3) screened using 10 selected populations, 39 SSR markers with polymorphisms were preliminarily screened, with a polymorphism rate of 20.31%. Then, 15 randomly selected SSR markers were used for genetic diversity analysis of 51 Z. nitidum populations (Table 3).
SSR marker detection
A total of 245 alleles (Na) from 15 developed SSR markers were amplified from the 51 populations (142 individuals). The total effective alleles (Ne) were 85.828, with an average of 5.722, and the proportion of effective alleles was 35.03%. Shannon’s information index (I) ranged from 1.313 to 2.994, with an average value of 1.984. The polymorphism information content (PIC) ranged from 0.531 to 0.922, with an average of 0.756. All loci were not conforming to the Hardy–Weinberg equilibrium (Table 4). The linkage disequilibrium distribution pattern based on R2 values between SSR loci is shown in Fig. 1. R2 ranged between 0.01 and 0.39, indicating a relatively low degree of linkage. These results show that the 15 loci those were chosen have a high degree of polymorphism and can be utilized for further research on Z. nitidum's genetic diversity.

Linkage disequilibrium distribution pattern based on R2 values between SSR loci. The color of the box ranges from white to red, representing the level of linkage disequilibrium from low to high.
Population genetic diversity
Statistical analysis showed that the maximum genetic distance among 51 populations was 1.000, the minimum was 0.164, and the average was 0.659 (Table S4). The results obtained for population-level indices of genetic diversity in each population are shown in Table 5. The average values of Na, Ne, I, Ho, He and F at the population level were 2.220, 1.964, 0.619, 0.478, 0.380, and − 0.268, respectively. Taken together, these results indicate that POP15 and POP1 exhibit the highest and lowest levels of genetic diversity, respectively.
Genetic differentiation
According to AMOVA, 40% of the total genetic variation originated from variability among the populations, whereas 60% was attributed to within-individual differences (Table 6). F-statistical analysis revealed that the genetic differentiation coefficient (Fst) of the population was 0.338, and the gene flow (Nm) was 0.636 (Table S5).
Phylogeny tree
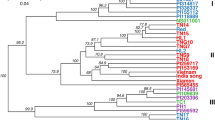
According to Nei's genetic similarity coefficient, the UPGMA method was used to construct a phylogenetic tree. The genetic similarity coefficient variation of 51 populations ranged from 0 to 0.44, and when the similarity coefficient was 0.30, they could be clustered into 4 clusters (Fig. 2). Cluster I contained 2 populations, both of which have been identified as Z. nitidum var. tomentosum. Cluster II contained 17 populations, all of which have been identified as type 1 of Z. nitidum var. nitidum. Cluster III included 3 populations from Guangdong, all of which have been identified as type 3 of Z. nitidum var. nitidum. Cluster IV contained 29 populations, including all 7 cultivated populations, all of which have been identified as type 2 of Z. nitidum var. nitidum.

Phylogeny tree of 51 populations.
Discussion
SSRs, as important molecular markers, have been widely used in genetic diversity evaluation, genetic map, construction, and finger mapping in medicinal plants, such as for the medicinal plants Aristolochia delavayi33, Andrographis paniculata34, and Paris polyphylla35. However, genetic research on Z. nitidum has been hampered due to limited genetic information. MSGs were used for high-throughput discovery of SSRs23. It was confirmed that MSG is similar in essence to restriction site-associated DNA (RAD) sequencing36 and whole-genome resequencing (WGD)37, and it is more effective and flexible for detecting SSRs. In this study, 261,267 SSR loci by MSG were discovered.
The number of Na, Ne, Ho, He, F, I, and PIC of primers are important indicators for measuring polymorphism of SSR markers. When PIC > 0.5, it indicates that the primer has a high degree of polymorphism38. In this study, we designed 768 SSRs based on MSG data, and screened out specific markers of Z. nitidum form them. To evaluate the effectiveness of the designed SSR primers, we randomly selected 192 SSRs and conducted two rounds of screening based on 10 germplasms with significant phenotypic differences. Twenty-six SSRs were found to be effective. Fifteen SSRs were selected from 26 SSRs for genetic diversity analysis on 142 individuals. The PIC of these 15 SSRs ranged from 0.531 to 0.922, with an average of 0.756. These 15 SSRs have high polymorphism and can effectively reveal the genetic diversity of Z. nitidum. This is the first time SSR markers have been used for genetic diversity analysis of Z. nitidum populations; at the same time, our results confirm that SSR markers have the advantages of a large number of loci and high identification efficiency.
Among the indicators of population genetic diversity, expected heterozygosity (He)39 and Shannon’s information index (I)40 are important. The larger their values are, the richer the genetic diversity of the population is. This study found that the He of these 51 populations ranged from 0.133 to 0.600, with an average of 0.380; the I ranged from 0.185 to 1.043, with an average of 0.619. These data indicate that the 51 populations have rich genetic diversity. In general, Fst > 0.25 indicates a high level of genetic differentiation among populations41; Nm < 1 indicates that the genetic differentiation among populations is caused by migration or genetic drift42. In this study, the Fst of 51 populations was 0.338, and the Nm was 0.636, which indicated that the genetic differentiation among populations of Z. nitidum is at a high level. Additionally, the genetic differentiation between populations was caused by migration or genetic drift, which is consistent with the conclusions reached by previous researchers using ISSR molecular markers43, and similar to the results of genetic differentiation studies of other Zanthoxylum plants44,45.
Conducting variety identification by combining molecular marker technology with phenotypic analysis is reliable and convenient46,47. In our study, we constructed a UPGMA clustering tree using the 15 SSRs based on Nei's genetic similarity coefficient. Fifty-one populations could be clearly clustered into 4 clusters. Among them, the first cluster was identified as Z. nitidum var. tomentosum, indicating distant genetic relationship between Z. nitidum var. tomentosum and the rest of the germplasm with relatively clear separation. The second cluster was identified as Z. nitidum var. nitidum (type 1), indicating a distant genetic relationship between type 1 and the other two types. The third cluster was identified as Z. nitidum var. nitidum (type 3), all of which originated from Guangdong, showing obvious regional characteristics. The cluster was identified as Z. nitidum var. nitidum (type 2), with the closest genetic relationship to type 3, and included 7 artificially cultivated populations, indicating a decrease in genetic diversity after transitioning from wild to domestic. The conclusion of the genetic relationship between the four types of Z. nitidum through cluster analysis is consistent with traditional morphological identification methods and proves the effectiveness of the SSR molecular markers selected in this study for identifying Z. nitidum germplasm.
Conclusion
In this study, MSG data were used to develop SSRs, and 261,267 SSRs were detected. Fifteen SSRs were powerful molecular markers for genetic diversity analysis and variety identification in Z. nitidum. Our data will be useful for germplasm identification, genetic improvement, and variety selection.
Data availability
The data that support the findings of this study are available in the supplementary material of this article.
References
Commission, S. P. Pharmacopoeia of the People’s Republic of China (Pharmacopoeia of the People’s Republic of China, 2020).
Sun, X. & Sun, F. Shennong Materia Medica Classic (People’s Health Publishing House, 1963).
Huang, C. Flora of China Vol. 43 (Science Press, 1997).
Hu, J. et al. Benzophenanthridine alkaloids from Zanthoxylum nitidum (Roxb.) DC, and their analgesic and anti-inflammatory activities. Chem. Biodivers. 3, 990–995 (2006).
Yang, G. & Chen, D. Alkaloids from the roots of Zanthoxylum nitidum and their antiviral and antifungal effects. Chem. Biodivers. 5, 1718–1722 (2008).
Qin, F. et al. Four new sesquiterpenoids from Zanthoxylum nitidum. Chem. Biodivers. 19, e202200449 (2022).
Zuo, G.-Y., Wang, C.-J., Han, J., Li, Y.-Q. & Wang, G.-C. Synergism of coumarins from the Chinese drug Zanthoxylum nitidum with antibacterial agents against methicillin-resistant Staphylococcus aureus (MRSA). Phytomedicine 23, 1814–1820 (2016).
Su, J.-H. et al. Coumarins and lignans from the roots of Zanthoxylum nitidum. Biochem. Syst. Ecol. 101, 104399 (2022).
Qin, F. et al. Anti-inflammatory activity of isobutylamides from Zanthoxylum nitidum var. tomentosum. Fitoterapia 142, 104486 (2020).
Bhattacharya, S., Haldar, P. K. & Zaman, M. K. Anti-inflammatory and in vitro antioxidant property of Zanthoxylum nitidum root. Curr. Trends Biotechnol. Pharm. 4, 774–783 (2010).
Bhattacharya, S., Zaman, M. & Haldar, P. Antibacterial activity of stem bark and root of Indian Zanthoxylum nitidum. Asian J. Pharm. Clin. Res. 2, 30–34 (2009).
Zhao, L.-N. et al. (±)-Zanthonitidine A, a pair of enantiomeric furoquinoline alkaloids from Zanthoxylum nitidum with antibacterial activity. Nat. Prod. Bioprospect. 8, 361–367 (2018).
Qin, F. et al. Undescribed isoquinolines from Zanthoxylum nitidum and their antiproliferative effects against human cancer cell lines. Phytochemistry 205, 113476 (2023).
Qin, F. et al. Analgesic effect of Zanthoxylum nitidum extract in inflammatory pain models through targeting of ERK and NF-κB signaling. Front. Pharmacol. 10, 359 (2019).
Chen, Y., Li, M., Qin, F. & Wang, H. Research progress on chemical components and pharmacological effects of Zanthoxylum nitidum (Roxb.) DC. in recent five years. J. Guangxi Norm. Univ. (Nat. Sci. Ed.) 41, 24–37 (2023).
He, J. et al. Research progress on pharmacological effects of Liangmianzhen (Zanthoxyli Radix) based on efficacy and clinical application analysis. J. Liaoning Univ. TCM 25, 93–97 (2023).
Yu, L., Huang, B., Tan, X. & Guo, B. Survey of wild germplasm of Zanthoxylum nitidum in Guangxi. Guangxi Zhiwu/Guihaia 29, 231–284 (2009).
Peng, Z., Wu, M., Xie, Z., Yang, X. & Lai, M. Investigation of wild resource of Zanthoxylum nitidum. Pharm. Today 28, 500–504 (2018).
Kalia, R. K., Rai, M. K., Kalia, S., Singh, R. & Dhawan, A. Microsatellite markers: An overview of the recent progress in plants. Euphytica 177, 309–334 (2011).
Jiang, M. et al. Genetic diversity of the Chinese medicinal plant Astragali Radix based on transcriptome-derived SSR markers. Electron. J. Biotechnol. 62, 13–20 (2023).
Zhang, Z. et al. Genome survey sequencing and genetic diversity of cultivated Akebia trifoliata assessed via phenotypes and SSR markers. Mol. Biol. Rep. 48, 241–250 (2021).
Gao, X. et al. Development of EST-SSR markers related to polyphyllin biosynthesis reveals genetic diversity and population structure in Paris polyphylla. Diversity 14, 589 (2022).
Andolfatto, P. et al. Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res. 21, 610–617 (2011).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Magoč, T. & Salzberg, S. L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Stajich, J. E. et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 12, 1611–1618 (2002).
Untergasser, A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115–e115 (2012).
Peakall, R. & Smouse, P. E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295 (2006).
Shen, J. et al. SHEsisPlus, a toolset for genetic studies on polyploid species. Sci. Rep. 6, 24095 (2016).
Liu, K. & Muse, S. V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 21, 2128–2129 (2005).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Yu, Y.-L. et al. Genetic diversity and structure of the endemic and endangered species Aristolochia delavayi growing along the Jinsha River. Plant Divers. 43, 225–233 (2021).
Kumar, R. et al. Development of novel genomic simple sequence repeat (g-SSR) markers and their validation for genetic diversity analyses in Kalmegh [Andrographis paniculata (Burm. F.) Nees]. Plants 9, 1734 (2020).
Oliya, B. K. & Manandhar, L. Pant B: Genetic diversity and population structure analysis of Paris polyphylla Sm revealed by SSR marker (2022)
Baird, N. A. et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PloS One 3, e3376 (2008).
Huang, X. et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 19, 1068–1076 (2009).
Botstein, D., White, R. L., Skolnick, M. & Davis, R. W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Human Genet. 32, 314 (1980).
Reddy, S. B. & Rosenberg, N. A. Refining the relationship between homozygosity and the frequency of the most frequent allele. J. Math. Biol. 64, 87–108 (2012).
Spellerberg, I. F. & Fedor, P. J. A tribute to Claude Shannon (1916–2001) and a plea for more rigorous use of species richness, species diversity and the ‘Shannon–Wiener’Index. Glob. Ecol. Biogeogr. 12, 177–179 (2003).
Wright, S. Evolution and the Genetics of Populations, Volume 4: Variability Within and Among Natural Populations (University of Chicago Press, 1984).
Govindaraju, D. R. Variation in gene flow levels among predominantly self-pollinated plants. J. Evolut. Biol. 2, 173–181 (1989).
Xiang, Q., Huang, X., Li, H. & Jiang, S. ISSR analysis on genetic diversity of medically important Zanthoxylum nitidum (Roxb). DC. in Guangxi. Guangxi Sci. 21, 541–549 (2014).
Feng, S. et al. Genetic diversity and evolutionary relationships of chinese pepper based on nrDNA markers. Forests 11, 543 (2020).
Feng, S. et al. Genetic diversity and relationships of wild and cultivated Zanthoxylum germplasms based on sequence-related amplified polymorphism (SRAP) markers. Genet. Resour. Crop Evolut. 62, 1193–1204 (2015).
Luo, Y. et al. Phenotypic and molecular marker analysis uncovers the genetic diversity of the grass Stenotaphrum secundatum. BMC Genet. 21, 1–12 (2020).
Abdelsalam, N. R. et al. Morphological, pomological, and specific molecular marker resources for genetic diversity analyses in fig (Ficus carica L.). HortScience 54, 1299–1309 (2019).
Funding
This research was funded by the Scientific Research and Technology Development Program of Guangxi (GUIKE AB22035068).
Author information
Authors and Affiliations
Contributions
Conceptualization: Y.Z. and J.J.; methodology: Y.Z. and Y.H.; formal analysis: Y.Z.; resources: Y.Z., T.M., Y.L. and Y.P.; writing-original draft preparation: Y.Z. and T.M.; writing-review and editing: Y.Z. and J.J. All authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, Y., Ma, T., Lin, Y. et al. SSR molecular marker developments and genetic diversity analysis of Zanthoxylum nitidum (Roxb.) DC. Sci Rep 13, 20767 (2023). https://doi.org/10.1038/s41598-023-48022-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-48022-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
