
Abstract
WHO 2016 classified glioblastomas into IDH-mutant and IDH-wildtype with the former having a better prognosis but there was no study on IDH-mutant primary glioblastomas only, as previous series included secondary glioblastomas. We recruited a series of 67 IDH-mutant primary glioblastomas/astrocytoma IV without a prior low-grade astrocytoma and examined them using DNA-methylation profiling, targeted sequencing, RNA sequencing and TERT promoter sequencing, and correlated the molecular findings with clinical parameters. The median OS of 39.4 months of 64 cases and PFS of 25.9 months of 57 cases were better than the survival data of IDH-wildtype glioblastomas and IDH-mutant secondary glioblastomas retrieved from datasets. The molecular features often seen in glioblastomas, such as EGFR amplification, combined +7/−10, and TERT promoter mutations were only observed in 6/53 (11.3%), 4/53 (7.5%), and 2/67 (3.0%) cases, respectively, and gene fusions were found only in two cases. The main mechanism for telomere maintenance appeared to be alternative lengthening of telomeres as ATRX mutation was found in 34/53 (64.2%) cases. In t-SNE analyses of DNA-methylation profiles, with an exceptional of one case, a majority of our cases clustered to IDH-mutant high-grade astrocytoma subclass (40/53; 75.5%) and the rest to IDH-mutant astrocytoma subclass (12/53; 22.6%). The latter was also enriched with G-CIMP high cases (12/12; 100%). G-CIMP-high status and MGMT promoter methylation were independent good prognosticators for OS (p = 0.022 and p = 0.002, respectively) and TP53 mutation was an independent poor prognosticator (p = 0.013) when correlated with other clinical parameters. Homozygous deletion of CDKN2A/B was not correlated with OS (p = 0.197) and PFS (p = 0.278). PDGFRA amplification or mutation was found in 16/59 (27.1%) of cases and was correlated with G-CIMP-low status (p = 0.010). Aside from the three well-known pathways of pathogenesis in glioblastomas, chromatin modifying and mismatch repair pathways were common aberrations (88.7% and 20.8%, respectively), the former due to high frequency of ATRX involvement. We conclude that IDH-mutant primary glioblastomas have better prognosis than secondary glioblastomas and have major molecular differences from other commoner glioblastomas. G-CIMP subgroups, MGMT promoter methylation, and TP53 mutation are useful prognostic adjuncts.
Similar content being viewed by others


Introduction
The WHO 2016 Classification of Tumours of the Central Nervous System classified glioblastomas by the IDH status. [1] IDH-wildtype glioblastomas make up the majority of the tumours and have a poorer prognosis than IDH-mutant glioblastomas. Secondary glioblastomas, which arise from a previous low-grade astrocytoma, are regarded as mostly IDH-mutant. Increasingly, IDH-mutant glioblastomas and IDH-mutant low-grade astrocytomas have undergone scrutiny as only a small number of IDH-mutant glioblastomas were evaluated for molecular pathology prior to the WHO 2016 classification [2]. Since then, there have been three sizeable series of IDH-mutant glioblastomas including ours [3,4,5]. Two studies [3, 4] have characterized the cohort more extensively and shown that IDH-mutant glioblastomas are a more heterogenous group for risk than hitherto thought, with some having a poor prognosis, especially those with CDKN2A/B homozygous deletion. A few studies have also identified homozygous deletion of CDKN2A/B, alone or in combination with other biomarkers, as a poor prognosticator in IDH-mutant histologically low-grade diffuse astrocytomas [3, 6,7,8,9,10]. These studies prompted the cIMPACT-NOW group to propose a replacement of the term IDH-mutant glioblastoma with IDH-mutant astrocytoma, WHO Grade IV, and with CDKN2A/B homozygous deletion being a diagnostic hallmark irrespective of whether other features of malignancy in astrocytomas, such as necrosis or endothelial proliferation, are present or not [11].
In a large series of 188 secondary glioblastomas, Hu et al. showed that only 67% of them were IDH-mutant and their prognosis was seemingly not dissimilar to that of IDH-wildtype glioblastomas [12]. By the same token, Miller et al. showed that once IDH-mutant low-grade gliomas progressed, presumably with many becoming high-grade gliomas, clinical progression was rapid [13]. Hence, it seems that IDH-mutant glioblastomas or IDH-mutant astrocytoma, Grade IV, can be separated clinically into primary and secondary IDH-mutant glioblastomas. Hu et al. extensively evaluated the mutational landscape of secondary glioblastomas and identified MET-exon14 skipping (METex14) as a novel therapeutic target [12]. In this paper, we collected a cohort of 67 IDH-mutant primary glioblastomas and evaluated its molecular landscape by DNA-methylation profiling, targeted sequencing, and RNA sequencing, and in addition performed Sanger sequencing for TERT promoter. We established G-CIMP status and copy number alterations (CNA) for genes regarded as critical in the pathogenesis of glioblastomas. Finally, we correlated the molecular landscape with the clinical variables. This is the largest single series of IDH-mutant glioblastoma to our knowledge.
Materials and methods
Tumour samples
Tumour samples were retrieved from Prince of Wales Hospital (Hong Kong, China), Hua Shan Hospital (Shanghai, China) and the First Affiliated Hospital of Zhengzhou University (Zhengzhou, China) from 2008 onwards and cases of primary IDH-mutant glioblastomas were selected. They were from adult patients (>17 years old) with a histological diagnosis of glioblastomas and with a clinical history of <3 months [3] and there was no previous history of a low grade astrocytoma. The cohort included 27 cases of a previous study [4] but the secondary glioblastomas of that series were excluded. Previous diagnostic workups of these cases included Sanger sequencing for IDH1 and IDH2 and they were shown to be IDH-mutant. Ethics approvals were obtained from The Joint Chinese University of Hong Kong–New Territories East Cluster Clinical Research Ethics Committee, and the Ethics Committees of Huashan Hospital, Shanghai and The First Affiliated Hospital of Zhengzhou University, Zhengzhou. The study was performed in accordance with the Declaration of Helsinki. Hematoxylin-eosin stained sections were reviewed and the diagnosis of glioblastoma was confirmed by three pathologists (HKN, HC, WWW). The histological diagnoses were as per WHO 2016 Classification [1]. Data on patient demographics and therapeutic treatment were retrieved from institutional paper and electronic records. Survival data were obtained from follow-up clinic visits and direct contact with patients or close relatives via phone.
IDH1, IDH2, and TERT promoter mutation analysis
Only samples with IDH1 or IDH2 mutation were included in this study. IDH1/2 and TERT promoter mutation was evaluated by Sanger sequencing. In brief, crude cell lysate was prepared from formalin-fixed paraffin-embedded (FFPE) sections accordingly [14]. DNA from the lysate was amplified using forward primer (IDH1: 5′-CGGTCTTCAGAGAAGCCATT-3′; IDH2 5′-AGCCCATCATCTGCAAAAAC-3′; and, TERT 5′-GTCCTGCCCCTTCACCTT-3′), reverse primer (IDH1: 5′-CACATTATTGCCAACATGAC-3′; IDH2 5′-CTAGGCGAGGAGCTCCAGT-3′; and, TERT 5′-CAGCGCTGCCTGAAACTC-3′), KAPA Robust HotStart ReadyMix (Sigma)/KAPA HiFi HotStart ReadyMix (Sigma). Amplification was conducted on a thermal cycler according to the manufacturer’s recommendation. PCR products were visualized on electrophoresis gel, cleaned with a spin column-based PCR product purification kit (iNtRON Biotechnology) and sequenced with BigDye Terminator Cycle Sequencing kit v1.1 (Life Technologies).
Illumina infinium methylationEPIC BeadChip array
FFPE sections were sent to Shanghai, China (Sinotech Genomics Co., Ltd), where the DNA was extracted, bisulfite modified and subjected to DNA-methylation profiling by EPIC Illumina Infinium Human (850k) Array following the manufacture’s recommendation (Illumina). Unprocessed IDAT files can be downloaded from http://www.acp.cuhk.edu.hk/hkng. Background correction, global dye-bias normalization, and calculation of DNA-methylation level were parts of EPIC array preprocessing, and were performed according to the previous publications [15, 16]. G-CIMP status were determined using a random forest (RF) machine learning algorithm as described in previous publications [15, 16]. The most variable probes were used to generate t-SNE plot according to Capper et al. study [17]. The t-SNE (t-distributed stochastic neighbour embedding) plot was generated with Rtsne R package. The raw IDAT files were uploaded to DKFZ ‘Classifier’ tool (https://www.molecularneuropathology.org) for supervised analysis using the RF methylation class prediction algorithm [17]. MGMT promoter methylation status was computed by DKFZ ‘Classifier’ tool.
Determination of copy number variations with EPIC 850k array
To determine copy number variations, probe-level signal intensities were retrieved from the IDAT files and then subjected to background correction and dye-bias normalization. Probes targeting the sex chromosomes, containing single-nucleotide polymorphisms, or mapping to multiple locations in the human genome were excluded to avoid inaccurate assessment. Two sets of 50 control samples derived from male and female donors were used for normalization [17]. Copy number variation analysis were performed using the ‘conumee’ R package in Bioconductor (http://www.bioconductor.org/packages/release/bioc/html/conumee.html). A log2 ratio +0.35 was used as a cutoff for amplification and a log2 ratio −0.415 was used for the cutoff of homozygous losses [9].
GISTIC v2.0 analysis was conducted to identify significantly recurrent copy number amplification and deletions at arm-level and focal-level, defined as affected regions spanning <50% of a chromosome arm [18, 19].
Targeted sequencing
DNA was obtained from FFPE sections using GeneRead DNA FFPE kit (Qiagen). The quality and quantity of extracted DNA were assessed by QIAseq DNA QuantiMIZE Assay (Qiagen). Samples that passed quality control were subjected to library preparation with a custom QIAseq Targeted DNA Panel, covering the coding exons of 74 genes altered in gliomas and other CNS tumours. The DNA libraries were then further assessed for quality and quantity. Sequencing of libraries passing quality control was performed on MiSeq v3 (Illumina).
Paired-end reads were aligned to the hg19 (GRCh37) build of the human reference genome with BWA-MEM algorithm on GeneGlobe platform (Qiagen). Variants were called using smCounter2 [20] and annotated using wANNOVAR [21]. Variants with the following criteria were excluded: not passing quality filters, with variant allele fractions of <5%, with variant allele counts of <5, or with minor allele frequencies of >1% in overall human population or East Asians or documented in public databases (1000 Genomes, ExAc, gnomAD exome and genome databases).
RNA sequencing
RNA was extracted from FFPE sections using RNeasy FFPE kit (Qiagen). RNA passing quality control examination was subjected to cDNA synthesis and molecular barcode-indexed ligation library preparation. Target capture was done using TruSight RNA Pan-Cancer Panel (Illumina), which covered 1385 genes commonly involved in cancer. Libraries were sequenced on MiSeq instrument (Illumina).
Paired-end reads were aligned to human genome assembly GRCh37 (hg19) and fusion genes were called using the STAR aligner and STAR fusion caller [22, 23]. In-frame fusion genes with at least five junction reads were further confirmed by conventional RT-PCR.
Statistical analysis
Statistical analysis was performed on IBM SPSS software v22. Overall survival (OS) was defined as the period of time between operation and death or the last follow-up. Progression-free survival (PFS) was defined as the time between operation and recurrence revealed by neuroradiological finding. Chi-square or Fisher’s test was applied to determine correlation between molecular alterations and clinical parameters and between different molecular alterations. Survival curves were evaluated by the Kaplan–Meier (KM) method, and log-rank test was done to compare survival distribution between groups. Multivariable analysis was performed by Cox proportional hazards model. P < 0.05 (two-sided) was considered statistically significant.
Results
Samples and clinical data
A summary of clinical features of this study cohort is shown in Table 1. A total of 67 IDH-mutant primary glioblastoma samples were collected in this study. MRI studies showed 37/42 cases (88.1%) with enhancement on T1 but the imaging records of 25 cases were no longer available for review. For histological review, all cases showed variable degrees of hypercellularity and cellular anaplasia. All except 3 cases showed necrotic foci and 43 cases showed microvascular proliferation, including the 3 cases without necrosis. Male-to-female ratio was 1.6:1. The mean and median age at diagnosis were 39.3 years old and 38.0 years old, respectively. Most patients received gross total resection (56/67; 83.6%). Most patients (n = 50; 74.6%) were given both chemotherapy and radiotherapy. Radiotherapy alone was given to 2 (3.0%) patients. Follow-up data for overall survival (OS) and progression-free survival (PFS) were available for 64 and 57 cases, respectively. The mean and median follow-up periods were 58.9 months and 60.1 months, respectively. Median OS was 39.4 months, with a 2-year survival of 64.1% and a 5-year survival of 30.8%. Median PFS was 25.9 months. Univariate Cox regression was performed according to the clinical features, including gender, age at diagnosis, tumour location, operation, chemotherapy and radiotherapy (Supplementary Table 1). Only being female was significantly associated with a better PFS (p = 0.011). Age had no correlation with survival (OS: p = 0.297; PFS: p = 0.769).
IDH-mutant primary glioblastomas had a better prognosis than IDH-wildtype glioblastomas and IDH-mutant secondary glioblastomas
Our cohort of IDH-mutant primary glioblastomas showed a median OS of 39.4 months and median PFS of 25.9 months. This was a longer median OS than IDH-wildtype glioblastomas (13.6 months, p < 0.001, log-rank test; Fig. 1A) as we could retrieve from established databases [24, 25] and IDH-mutant secondary glioblastomas (18.8 months, p = 0.032, log-rank test; Fig. 1C) as we could retrieve from Hu et al. [12]. Our cohort also had a longer median PFS than IDH-wildtype glioblastomas (8.7 months, p < 0.001, log-rank test; Fig. 1B) as in established databases [24, 25] and IDH-mutant secondary glioblastomas (11.3 months, p = 0.002, log-rank test; Fig. 1D) as in Hu et al. [12].

Classification of G-CIMP groups based on genome-wide DNA-methylation profiling
We determined genome-wide DNA-methylation profiling in 53 IDH-mutant primary glioblastomas with sufficient tissue material for Illumina MethylationEPIC (850k) arrays. We applied RF with a two-step process and assigned the samples into one of the two IDH-mutant methylation-based glioma subgroups (G-CIMP-high and G-CIMP-low) according to the previous publication [15]. The majority of the samples belonged to G-CIMP-high group (36/53; 67.9%), and G-CIMP-low was present in 17/53 (32.1%) of cases. As described below, G-CIMP-low tumours were associated with PDGFRA amplification (p = 0.005). The correlations between G-CIMP-low and PDGFRA mutation, and between G-CIMP-low and TP53 mutation were not quite significant (p = 0.060 and p = 0.056, respectively) (also see the section Targeted Sequencing below).
DNA methylomes clustered with two main DNA-methylation classes
We then conducted an unsupervised clustering of our samples using the reference cohorts and classes of the German Cancer Research Centre (DKFZ) as per Capper et al. [17], and also using the method of t-SNE dimensionality reduction as per that paper. As shown in Fig. 2, with the exception of one case, all samples were clustered or in close proximity to either the methylation class called IDH glioma, subclass astrocytoma (A_IDH) or the methylation class called IDH glioma, subclass high-grade astrocytoma (A_IDH_HG). With the DBSCAN algorithm described previously [26], the majority of our cohort were annotated to IDH glioma, subclass high-grade astrocytoma (40/53; 75.5%), and 12/53 cases (22.6%) were annotated to IDH glioma, subclass astrocytoma. There was one outlier clustered in proximity to the DKFZ classifier glioblastoma, IDH wildtype, subclass mesenchymal but Sanger sequencing and targeted sequencing both confirmed an IDH1-R132H mutation for this case. A review of the histology confirmed regular features of glioblastomas and the H&E of this case is provided in Supplementary Fig. 1. In view of the t-SNE findings, we evaluated histologically all the cases designated to the methylation class IDH glioma, subclass astrocytoma (A_IDH) and confirmed that the cases satisfied WHO 2016 histological criteria for glioblastomas. Representative photomicrographs from these cases showing necrotic foci are included in Supplementary Fig. 2. In spite of the cohorts being IDH-mutant, no case was clustered with 1p19q codeleted oligodendroglioma class.

The reference cohort of the DKFZ CNS tumour classifier includes 82 tumour and 9 non-tumour classes and they are shown as circles of different colours. Except one case, all IDH-mutant primary glioblastomas in this study (shown in red triangle or green diamond) clustered close to the methylation class called IDH glioma, subclass astrocytoma (A_IDH) or IDH glioma, subclass high-grade astrocytoma (A_IDH_HG). Mutations of IDH in our samples were tested and confirmed by independent PCR and Sanger sequencing.
Korshunov et al. studied 97 IDH-mutant glioblastomas, which included both primary and secondary tumours, by 450k methylation array. They selected the 10 000 most variably methylated CpG probes for unsupervised hierarchical clustering [3]. The results showed 84/97 cases (86.6%) clustered to IDH-mutant high-grade glioma and 11/97 cases (11.3%) to IDH-mutant astrocytoma class. They also had the odd outliers similar to us.
We then correlated the DNA-methylation classes to G-CIMP status. We found a strong correlation between G-CIMP status and DNA-methylation class in IDH-mutant glioblastomas (p = 0.017; Supplementary Table 2). G-CIMP-low tumours (17/17) were clustered or near the IDH glioma, subclass high-grade astrocytoma (A_IDH_HG). All tumours clustered to IDH glioma, subclass astrocytoma (A_IDH) were G-CIMP-high tumours. However, DNA-methylation classes were not associated with OS (p = 0.104) and PFS (p = 0.181) in our cohort. Methylation classes were also not associated with CDKN2A/B deletion, PDGFRA amplification, and TP53 mutation (see below). MGMT promoter methylation status can be derived directly from the methylation profiling [17, 27]. Overall, 46/53 (86.8%) cases were found to be MGMT promoter methylated. MGMT promoter methylation was not associated with G-CIMP status or methylation classes.
Copy number variation analysis showed significant differences from IDH-wildtype glioblastomas
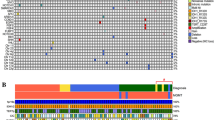
A summary of the molecular findings in this cohort, together with copy number variations (CNVs) is depicted in Fig. 3.

The top black bar chart represents the OS of each sample in months. The coloured bar chart on the right represents the frequency of tumours with the genetic and genomic aberrations found in this study. The bottom black bar chart indicates the number of mutations in each tumour identified by targeted sequencing. The central heatmap illustrates the molecular alterations. Each column represents one sample.
CNVs were found to be extremely useful derivatives of DNA-methylation profiles in gliomas [9, 28]. We thus determined CNVs from EPIC 850k array data according to previous research [17]. We employed GISTIC analysis to determine statistically significant recurrent amplifications and losses at arm-level and focal-level. At the arm-level, we found that the IDH-mutant primary glioblastomas showed significant amplification at chromosomes 8q, 10p, and 12p and significant loss at chromosomes 3p, 4q, 9p, 10p, 10q, 11p, 13q, 14q, and 19q (Fig. 4A; q < 0.05). At focal-level, we found recurrent amplification regions on chromosomes 4q12, 8q24.21, 11q13.3, 11q14.1, 12p13.32, 12q14.1 and 13q33.3. Genes located in these regions include PDGFRA, MYC, CCND1, CCND2, and CDK4 (Fig. 4B). We also identified eight recurrent loss regions and they were on chromosomes 4q35.1, 5q34, 6p21.32, 8p23.3, 9p21.3, 10q26.3, 11p15.4, and 13q14.2. Genes located in these regions included the well-known CDKN2A and CDKN2B. Chromosome 7 gain and chromosome 10 loss are molecular hallmark of many glioblastomas, and cIMPACT-NOW 3 recommended that IDH-wildtype diffuse astrocytic glioma with their combined whole chromosome loss would follow an aggressive clinical course closely resembling that of an IDH-wildtype glioblastoma despite their grade II or III histology [29]. Thus, we in particular determined the frequency of +7/−10 in our cohort and found that only 4/53 cases (7.5%) had +7/−10.

A Significant arm-level CNVs in IDH-mutant primary glioblastomas. Frequencies of CNVs are shown on the x-axes, while the human chromosomes 1–22 are illustrated along the y-axis. Arms with significant alterations (q < 0.05) are marked with an asterisk. Data on 13p, 14, 15p and 22p are not shown and were not significant. B Significant focal-level CNVs in IDH-mutant primary glioblastomas. Statistical significance of CNVs is indicated by false discovery rate Q values on the x-axes.
At gene-level, we investigated genes with established relevance in gliomas [9]. We employed the cutoff established in Shirahata et al. study to determine amplification and deletion. The results revealed that EGFR amplification which is frequently found in the regular glioblastomas was infrequent in our cohort (6/53; 11.3%). Instead, in our cohort, we detected amplifications of CDK4 (15/53; 28.3%), CCND2 (12/53; 22.6%), ERBB2 (11/53; 20.8%), MYC (10/53; 18.9%), MET (9/53, 17.0%) PDGFRA (8/53; 15.1%), and PIK3CA (2/53; 3.8%). PTEN deletion was identified in 6/53 cases (11.3%), and homozygous deletion of CDKN2A/B was identified in 23/53 cases (43.4%) (Table 2). In our cohort, we did not observe a difference in EGFR promoter methylation between EGFR amplified and non-amplified cases (p = 0.388).
When compared with IDH-wildtype glioblastomas in TCGA databases [24, 25], our cohort had much fewer EGFR amplifications (11.3% vs. 43.2%). Our cohort however showed more amplifications of CCND2 (22.6% vs. 2.1%), ERBB2 (20.8% vs. 0%), MYC (18.9% vs. 1.2%), and MET (17.0% vs. 4.0%). Similarly, some of the other genetic alterations mentioned by the cIMPACT-NOW update 5 to possibly be important in the pathogenesis of IDH-mutant astrocytoma, Grade IV, were not very common in our IDH-mutant primary glioblastomas: deletion of RB1 (9/53; 17.0%), amplification of MYCN (4/53; 7.5%), and deletion of PIK3R1 (3/53; 5.7%). No case displayed 1p19q codeletion.
In G-CIMP-low tumours (n = 17), we identified significant PDGFRA amplification (p = 0.005). G-CIMP status was not associated with other CNVs.
We also computed the frequency of CNVs in our cohort by established method [30, 31]. We found that G-CIMP high group had a significantly lower frequency of CNVs compared to the G-CIMP low group (p = 0.013; 4.43% +/− 4.51% vs. 8.37% +/− 6.46%).
Gene fusion was very rare in IDH-mutant primary glioblastomas
RNA sequencing was performed on 41 IDH-mutant primary glioblastomas in our cohort with sufficient materials. Surprisingly, only 4 intrachromosomal fusions were detected in 2/41 (4.9%) cases. The fusion genes were PTPRZ1-MET, UBTD2-CSF1R, SARNP-MYL2 and LRP1-TRHDE. One tumour carried three fusion genes. The PTPRZ1-MET fusion gene was identified in 1/41 (2.0%) cases and the same fusion was also found by Hu et al. in secondary glioblastomas [12]. All four fusion genes were confirmed by RT-PCR and Sanger sequencing (Fig. 3; Supplementary Fig. 3). The fact that gene fusion was a rare event in our cohort is different from what was found in glioblastomas in general where gene fusions are abundant and fusion genes are potential targets for therapy [32,33,34].
Targeted sequencing showed differences from IDH-wildtype glioblastomas and IDH-mutant secondary glioblastomas
Targeted next-generation sequencing was performed on 53 samples in our cohort using an in-house panel which contains 74 genes relevant to the pathogenesis of brain tumours (Supplementary Table 3). Overall, there are 7.98 ± 4.57 mutations/sample. All mutations are listed in Supplementary Table 4. Sanger sequencing was used to identify TERT promoter mutations in 67 samples. The most significant finding was the extremely low occurrence of TERT promoter mutation (2/67; 3.0%). Both TERT promoter mutations found were C228T. Other mutations that were, interestingly, rarely found in our cohort were EGFR (2/53; 3.8%) MET (4/53; 7.5%), FGFR3 (5/53; 9.4%), and PTEN (2/53; 3.8%). Another significant finding was the high incidence of ATRX (34/53; 64.2%) and TP53 (30/53, 56.6%) mutations (Table 3). Other recurrent mutations in our cohort included KMT2D (18/53; 34.0%), FAT1 (14/53; 26.4%), POLE (12/53; 22.6%), KMT2C (11/53; 20.8%), PTCH1 (11/53; 20.8%), PIK3CA (11/53; 20.8%), and PDGFRA (10/53; 18.9%). DNA mismatch repair (MMR) genes were mutated in a small proportion of cases (11 cases) including MSH6 (8/53; 15.1%), MSH2 (2/53; 3.8%), PMS2 (2/53, 3.8/%), and MLH1 (1/53; 1.9%). All except two cases showed only single MMR mutation. For these two cases, one tumour had mutations of the MSH2 and PMS2 while another had mutations of MSH2 and MSH6. Germline DNA was not available for testing. No prior chemotherapy was given as these were primary tumours with first presentations.
When compared with IDH-wildtype primary glioblastomas in the literature [35], our cohort of IDH-mutant primary glioblastomas showed very few TERT promoter mutations (3.0% vs. 80.4%). When compared with IDH-wildtype glioblastomas in TCGA databases [24, 25], our cohort also showed more mutations in ATRX (64.2% vs. 0%), TP53 (56.6% vs. 7.1%), KMT2D (34.0% vs. 0.4%), FAT1 (26.4% vs. 0.4%), POLE (22.6% vs. 0.1%), PDGFRA (18.9% vs. 1.8%), and MSH6 (15.1% vs. 0.4%). When compared with IDH-mutant secondary glioblastomas in literature [12], our samples had more mutations in PDGFRA (18.9% vs. 5.2%) but fewer TP53 mutations (56.6% vs. 80%). Supplementary Table 5 summarizes the prevalence of genetic and genomic alterations among IDH-mutant primary glioblastomas, IDH-wildtype glioblastomas, and IDH-mutant secondary glioblastomas. We identified cases as suggestive of hypermutation when the number of mutations was greater than the mean+2 S.D [36]. Three cases in this cohort displayed a potential hypermutation state (3/53; 5.7%), and the frequency is lower than that in IDH-mutant secondary glioblastomas (5.7% vs. 18.6%) [12]. Two of the three potentially hypermutated cases incidentally also showed MSH6 mutation as described above. Given the facts that the size of our target panel was relatively small covering 74 genes and we lacked tissue materials, we could not further confirm hypermutation status. The two other genes mentioned by cIMPACT-NOW update 5 [11] as having possible pathogenetic significance in IDH-mutant astrocytoma, Grade IV, were as follows: PIK3CA was mutated in 11/53 (20.8%) cases, and PIK3R1 was mutated in 4/53 (7.5%) cases.
Canonical pathways altered in IDH-mutant primary glioblastomas
Three canonical pathways are well known to be aberrated in glioblastomas, namely RTK/RAS/PI3K/AKT pathway, TP53 pathway, and RB pathway. When we combined copy number variation analysis with targeted sequencing results, RTK/RAS/PI3K/AKT pathway was altered in 67.2% samples, with PDGFRA aberrations being the most common (27%) (Fig. 5). TP53 pathway was altered in 59.7% samples, with TP53 aberrations being the most common (53%). RB pathway was altered in 65.7% samples, with CDKN2B deletion being the most common (42%). The proportions of all three were lower than those observed in IDH-wildtype glioblastomas as retrieved from TCGA: RTK/RAS/PI3K/AKT (90%), TP53 (86%) and RB (79%) pathways [24]. Two other pathways significantly involved in the pathogenesis of IDH-mutant primary glioblastomas included chromatin modifiers (88.7%) and DNA mismatch repair genes (20.8%). The former’s high level of involvement was due to the frequent mutations of ATRX as mentioned above. The alteration of signalling pathways in this cohort of IDH-mutant primary glioblastomas is depicted in Fig. 5.

Frequency of pathway alterations in IDH-mutant primary glioblastomas, including chromatin modifiers, DNA mismatch repair genes, RTK/RAS/PI3K/AKT pathway, TP53 pathway, and RB pathway.
Clinical correlations of molecular parameters
G-CIMP-high tumours showed a significantly longer OS than G-CIMP-low ones (median: 44.5 months vs. 24.7 months, p = 0.015, log-rank test; Fig. 6A). G-CIMP-high tumours also displayed a trend towards longer PFS compared to the G-CIMP-low tumours (Fig. 6B). MGMT promoter methylation was correlated with a better OS (p = 0.001) and a better PFS (p = 0.042) (Fig. 6C, D).

A OS and B PFS of G-CIMP-high and G-CIMP-low IDH-mutant primary glioblastomas. C OS and D PFS of MGMT promoter methylation in IDH-mutant primary glioblastomas. E OS and F PFS of TP53 mutation in IDH-mutant primary glioblastomas.
TP53 mutation was correlated with a worse OS (p = 0.020; Fig. 6E) but not PFS (p = 0.331; Fig. 6F). It is the only major pathogenetic gene of which mutation or copy number alteration was correlated with survival. Also, PIK3R1 amplification showed a trend toward poor overall survival (p = 0.079).
In multivariate analysis, G-CIMP-high subgroup (p = 0.022; Supplementary Table 6A) and MGMT promoter methylation (p = 0.002; Supplementary Table 6B) were both independent prognostic factors for better OS. TP53 mutation was an independent poor prognosticator for OS (p = 0.013) (Supplementary Table 6C).
Correlation with OS and PFS of other major genetic aberrations, including copy number alterations and mutations, is depicted in Tables 2 and 3. Only genetic events with significant hits are included. Details can also be referred to Fig. 3. None of the following genes or chromosomal arms which were discussed in previous publications as of potential pathogenetic significance in IDH-mutant gliomas was correlated with survival: ATRX, CDK4, MET, MYCN, PIK3R1, or 14q [10, 11, 36]. Frequency of CNVs was also not correlated with survival (OS: p = 0.645; PFS: p = 0.598).
Among IDH-mutant glioblastomas including both primary and secondary glioblastomas, two studies including ours concluded that CDKN2A was a marker for poor prognosis [3, 4]. Both studies included secondary glioblastomas in the cohorts. However, in our cohort where only IDH-mutant primary glioblastomas were studied, homozygous deletion of CDKN2A interestingly was not correlated with OS (p = 0.290) and PFS (p = 0.138) (Table 2). CDKN2B homozygous deletion showed a trend of poor OS (p = 0.077) and PFS (p = 0.097) (Table 2). Deletion of CDKN2A/B was not correlated with OS (p = 0.197) or PFS (p = 0.278) (Supplementary Fig. 4A, B).
Discussion
This study showed that IDH-mutant primary glioblastomas had a better survival than the more common IDH-wildtype glioblastomas as retrieved from the TGCA datasets [24, 25]. While this is as expected, as a matter of fact, the number of IDH-mutant glioblastomas with survival data as examined by Yan et al. in their seminal paper was only 14 [2]. Since then there have been only been three sizeable series of IDH-mutant glioblastomas [3,4,5]. Two of these series [3, 4] contained both primary and secondary IDH-mutant glioblastomas and the situation is uncertain for the other [5]. And the former two conducted more extensive characterization than the latter. Our present cohort, while overlapping with one of the previous series [4], contained only primary tumours with the secondary tumours removed. Additional new cases of IDH-mutant primary glioblastomas have been added to the series in our present study. Secondary glioblastomas were already extensively investigated by Hu et al. for molecular landscape [12] and they found MET-exon 14 skipping (METex14) as a potential therapeutic target for secondary glioblastomas. Interestingly, our cohort showed for the first time that primary glioblastomas, when they were of the IDH-mutant genotype, had a better prognosis than IDH-mutant secondary glioblastomas as documented by Hu et al. This corroborates the clinical findings that IDH-mutant diffuse astrocytomas, once they progress presumably with many becoming high-grade gliomas, will become aggressive [13]. This may be due to the fact that most Grades 2–3 astrocytomas would have been treated with chemoradiotherapy before progressing to glioblastomas and studies have demonstrated that IDH-mutant cells can undergo clonally expansion to obtain growth advantage and post-treatment mutant gliomas responded to PD-1 blockade poorly [37, 38]. Our study is the largest single series of IDH-mutant glioblastoma to our knowledge.
In this study, although the prognosis of IDH-mutant primary glioblastomas was relatively good compared with the regular glioblastomas, with a median OS of 39.4 months and a 5-year survival of only 30.8%, it was still not a low-grade tumour. The prognosis we showed in this cohort was still much worse than the prognosis of low-grade IDH-mutant astrocytomas as recorded in the literature [9, 10, 30]. However, it is possible that some IDH-mutant primary glioblastomas may be molecularly akin to IDH-mutant astrocytoma and similar clustering was also observed by Korshunov et al. [3]. It is hard to be certain whether intra-tumoral heterogeneity and regional variation of molecular changes among tumour clones have affected the results of ours and Korshunov’s [3]. But our genome-wide DNA-methylation analysis also showed that many G-CIMP-high cases were clustered to the IDH-mutant astrocytoma methylation class. Furthermore, G-CIMP-high tumours were associated with a favourable outcome and G-CIMP subgroups and MGMT promoter methylation were independent favourable prognosticators for overall survival.
Surprisingly, fusion genes were identified in only two cases in this study. This is in contrast to the common fusion events involving genes like EGFR, FGFR3, MET, and NTRK1/2/3 in glioblastomas [24, 32, 33]. IDH-mutant primary glioblastomas showed extensive differences from IDH-wildtype glioblastomas. The most significant findings were that the key molecular markers for the diagnosis of the common glioblastoma, namely EGFR amplification or mutation, 10q loss or PTEN mutation, and TERT promoter mutation, were rare in IDH-mutant primary glioblastomas [24, 25, 35]. Like us, Korshunov et al. also found only a very small number of cases with TERT promoter mutations in their series [3]. Telomere maintenance seemed to be done via the alternative lengthening of telomeres (ALT) mechanism with a high percentage of ATRX mutation (64.2%) [39,40,41]. ATRX mutations are known to be associated with IDH-mutant gliomas [42, 43]. Very significantly, EGFR amplification, combined 7+/10− and TERT promoter mutation [11, 44, 45] were only found in 6/53 (11.3%), 4/53 (7.5%), and 2/67 (3.0%) cases, respectively, in our cohort.
The three well-known pathways involved in glioblastoma pathogenesis, namely RTK/RAS/PI3K/AKT, TP53, and RB pathways, were involved to a lesser extent in our cohort when compared with TCGA [25] while mismatch repair genes and chromatin modifying pathways were significantly involved. The latter was involved in a majority of cases because of the high incidence of ATRX mutation, suggesting that IDH-mutant primary glioblastomas make use of the ALT (alternative lengthening for telomeres) mechanism for telomere maintenance instead of TERT promoter mutation [38, 40]. For the mismatch repair genes, their mutations did not seem to lead onto hypermutations as can be seen in other gliomas with mutations of these genes [38, 46].
cIMPACT-NOW update 5 proposed CDKN2A/B deletion to be a diagnostic marker for IDH-mutant astrocytoma IV/IDH-mutant glioblastoma [11] and two studies examining IDH-mutant glioblastomas, inclusive of both primary and secondary tumours, concluded CDKN2A deletion as a prognosticator for this group of tumours [3, 4]. Our findings were consistent with the cIMPACT-NOW’s proposal to use CDKN2A/B deletion as a diagnostic criteria for an IDH-mutant astrocytoma being Grade IV, as this was found in 43.4% of this series. G-CIMP subgroup and MGMT promoter methylation were independent good prognosticators and TP53 mutation was an independent poor prognosticator. MGMT promoter methylation has been an established prognosticator for glioblastoma [47, 48] and Ceccarelli et al. have shown the utility of G-CIMP status in the prognostication of IDH-mutant gliomas though their series consisted mostly of lower grade IDH-mutant gliomas [15]. Our findings showed that in IDH-mutant primary glioblastoma itself, prognostication should be based on G-CIMP subgroup, MGMT promoter methylation, and TP53 mutation instead of relying on CDKN2A homozygous deletion alone. Clinical management of IDH-mutant primary glioblastomas which are G-CIMP-low or MGMT promoter unmethylated or TP53 mutated should take into consideration of the poorer prognosis of these tumours in spite of their mutant IDH genotype. Figure 7 depicts the prognostic significance of molecular features identified in this study.

IDH-mutant glioma can be classified as glioblastoma or Grade IV astrocytoma with the following criteria. The prognosis of IDH-mutant glioblastomas can be stratified by clinical history and molecular testing of MGMT promoter methylation, G-CIMP subgroup, and TP53 mutation.
Data availability
Unprocessed IDAT files can be downloaded from http://www.acp.cuhk.edu.hk/hkng.
Change history
15 April 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41379-021-00802-0
References
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131:803–20.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N. Engl J Med. 2009;360:765–73.
Korshunov A, Casalini B, Chavez L, Hielscher T, Sill M, Ryzhova M, et al. Integrated molecular characterization of IDH-mutant glioblastomas. Neuropathol Appl Neurobiol. 2019;45:108–18.
Li KK-W, Shi Z-F, Malta TM, Chan AK-Y, Cheng, S, Kwan JSH et al. Identification of subsets of IDH-mutant glioblastomas with distinct epigenetic and copy number alterations and stratified clinical risks. Neuro-Oncology Adv. 2019;1:1–11.
Pekmezci M, Rice T, Molinaro AM, Walsh KM, Decker PA, Hansen H, et al. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: additional prognostic roles of ATRX and TERT. Acta Neuropathol. 2017;133:1001–16.
Aoki K, Nakamura H, Suzuki H, Matsuo K, Kataoka K, Shimamura T, et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol. 2018;20:66–77.
Reis GF, Pekmezci M, Hansen HM, Rice T, Marshall RE, Molinaro AM, et al. CDKN2A loss is associated with shortened overall survival in lower-grade (World Health Organization Grades II-III) astrocytomas. J Neuropathol Exp Neurol. 2015;74:442–52.
Richardson TE, Sathe AA, Kanchwala M, Jia G, Habib AA, Xiao G, et al. Genetic and epigenetic features of rapidly progressing IDH-mutant astrocytomas. J Neuropathol Exp Neurol. 2018;77:542–8.
Shirahata M, Ono T, Stichel D, Schrimpf D, Reuss DE, Sahm F, et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 2018;136:153–66.
Yang RR, Shi ZF, Zhang ZY, Chan AK, Aibaidula A, Wang WW, et al. IDH mutant lower grade (WHO Grades II/III) astrocytomas can be stratified for risk by CDKN2A, CDK4 and PDGFRA copy number alterations. Brain Pathol. 2020;30:541–53.
Brat DJ, Aldape K, Colman H, Figrarella-Branger D, Fuller GN, Giannini C, et al. cIMPACT-NOW update 5: recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. 2020;139:603–8.
Hu H, Mu Q, Bao Z, Chen Y, Liu Y, Chen J, et al. Mutational landscape of secondary glioblastoma guides MET-targeted trial in brain tumor. Cell. 2018;175:1665–78.e1618.
Miller JJ, Loebel F, Juratli TA, Tummala SS, Williams EA, Batchelor TT, et al. Accelerated progression of IDH mutant glioma after first recurrence. Neuro Oncol. 2019;21:669–77.
Li YX, Shi Z, Aibaidula A, Chen H, Tang Q, Li KK, et al. Not all 1p/19q non-codeleted oligodendroglial tumors are astrocytic. Oncotarget. 2016;7:64615–30.
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164:550–63.
de Souza CF, Sabedot TS, Malta TM, Stetson L, Morozova O, Sokolov A, et al. A distinct DNA methylation shift in a subset of glioma CpG island methylator phenotypes during tumor recurrence. Cell Rep. 2018;23:637–51.
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555:469–74.
Franch-Expósito S, Bassaganyas L, Vila-Casadesús M, Hernández-Illán E, Esteban-Fabró R, Díaz-Gay M, et al. CNApp, a tool for the quantification of copy number alterations and integrative analysis revealing clinical implications. Elife 2020;9:e50267.
Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41.
Xu C, Gu X, Padmanabhan R, Wu Z, Peng Q, DiCarlo J, et al. smCounter2: an accurate low-frequency variant caller for targeted sequencing data with unique molecular identifiers. Bioinformatics. 2019;35:1299–309.
Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10:1556–66.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Haas BJ, Dobin A, Li B, Stransky N, Pochet N, Regev A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019;20:213.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77.
Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8.
Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547:311–7.
Bady P, Sciuscio D, Diserens AC, Bloch J, van den Bent MJ, Marosi C, et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol. 2012;124:547–60.
Cimino PJ, Holland EC. Targeted copy number analysis outperforms histologic grading in predicting patient survival for WHO grades II/III IDH-mutant astrocytomas. Neuro Oncol. 2019;21:819–21.
Brat DJ, Aldape K, Colman H, Holland EC, Louis DN, Jenkins RB, et al. cIMPACT-NOW update 3: recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018;136:805–10.
Mirchia K, Sathe AA, Walker JM, Fudym Y, Galbraith K, Viapiano MS, et al. Total copy number variation as a prognostic factor in adult astrocytoma subtypes. Acta Neuropathol Commun. 2019;7:92.
Mirchia K, Snuderl M, Galbraith K, Hatanpaa KJ, Walker JM, Richardson TE. Establishing a prognostic threshold for total copy number variation within adult IDH-mutant grade II/III astrocytomas. Acta Neuropathol Commun. 2019;7:121.
Shah N, Lankerovich M, Lee H, Yoon JG, Schroeder B, Foltz G. Exploration of the gene fusion landscape of glioblastoma using transcriptome sequencing and copy number data. BMC Genomics. 2013;14:818.
Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–5.
Xu T, Wang H, Huang X, Li W, Huang Q, Yan Y, et al. Gene fusion in malignant glioma: an emerging target for next-generation personalized treatment. Transl Oncol. 2018;11:609–18.
Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr., et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013;110:6021–6.
Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, et al. Comprehensive analysis of hypermutation in human cancer. Cell. 2017;171:1042–.e1010.
Mazor T, Chesnelong C, Pankov A, Jalbert LE, Hong C, Hayes J, et al. Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc Natl Acad Sci USA. 2017;114:10743–8.
Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature. 2020;580:517–23.
Abedalthagafi M, Phillips JJ, Kim GE, Mueller S, Haas-Kogen DA, Marshall RE, et al. The alternative lengthening of telomere phenotype is significantly associated with loss of ATRX expression in high-grade pediatric and adult astrocytomas: a multi-institutional study of 214 astrocytomas. Mod Pathol. 2013;26:1425–32.
Lee J, Solomon DA, Tihan T. The role of histone modifications and telomere alterations in the pathogenesis of diffuse gliomas in adults and children. J Neurooncol. 2017;132:1–11.
Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T, et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012;8:e1002772.
Ebrahimi A, Skardelly M, Bonzheim I, Ott I, Mühleisen H, Eckert F, et al. ATRX immunostaining predicts IDH and H3F3A status in gliomas. Acta Neuropathol Commun. 2016;4:60.
Wiestler B, Capper D, Holland-Letz T, Korshunov A, von Deimling A, Pfister SM, et al. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. 2013;126:443–51.
Louis DN, Wesseling P, Aldape K, Brat DJ, Capper D, Cree IA, et al. cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020;30:844–56.
Stichel D, Ebrahimi A, Reuss D, Schrimpf D, Ono T, Shirahata M, et al. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol. 2018;136:793–803.
McCord M, Steffens A, Javier R, Kam KL, McCortney K, Horbinski C. The efficacy of DNA mismatch repair enzyme immunohistochemistry as a screening test for hypermutated gliomas. Acta Neuropathol Commun. 2020;8:15.
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66.
Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J, et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol. 2009;27:5743–50.
Acknowledgements
This study was supported by National Natural Science Foundation of China (No. 81702471, 81702465, U1804172 and U1904148); Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX01); Health and Medical Research Fund (HMRF), the Food and Health Bureau of Hong Kong (reference number: 07180736); and the Children’s Cancer Foundation, Hong Kong.
Author information
Authors and Affiliations
Contributions
The authors confirm contribution to the paper as follows: Study conception and design: KKWL, HKN. Acquisition of data: QHWW, KKWL, WWW, DTMC, YM, ZYZ, ZFS, HKN. Analysis and interpretation of data: QHWW, KKWL, WWW, TMM, HN, YG, CJ, AKYC, JSHM, QJQH, GCHW, WCL, XZL, HC, HKN. Drafting of manuscript: QHWW, KKWL, HKN.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics
Ethics approvals were obtained from The Joint Chinese University of Hong Kong—New Territories East Cluster Clinical Research Ethics Committee, and the Ethics Committees of Huashan Hospital, Shanghai and The First Affiliated Hospital of Zhengzhou University, Zhengzhou. The study was performed in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: The three co-first authors (i.e. authors who contributed equally) should be: Queenie Hoi-Wing Wong, Kay Ka-Wai Li and Wei-Wei Wang. The three co-corresponding authors should be: Ying Mao, Zhen-Yu Zhang and Zhi-Feng Shi.
Supplementary information
Rights and permissions
About this article

Cite this article
Wong, Q.HW., Li, K.KW., Wang, WW. et al. Molecular landscape of IDH-mutant primary astrocytoma Grade IV/glioblastomas. Mod Pathol 34, 1245–1260 (2021). https://doi.org/10.1038/s41379-021-00778-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00778-x
This article is cited by
-
Improved prognostic stratification of patients with isocitrate dehydrogenase-mutant astrocytoma
Acta Neuropathologica (2024)
-
Exploration of the radiosensitivity-related prognostic risk signature in patients with glioma: evidence from microarray data
Journal of Translational Medicine (2023)
-
Radiomic features from dynamic susceptibility contrast perfusion-weighted imaging improve the three-class prediction of molecular subtypes in patients with adult diffuse gliomas
European Radiology (2023)
-
Assessment of Ki-67 expression levels in IDH-wildtype glioblastoma using logistic regression modelling of VASARI features
Neurosurgical Review (2023)
-
Molecular landscape of pediatric type IDH wildtype, H3 wildtype hemispheric glioblastomas
Laboratory Investigation (2022)
