
Abstract
Multitarget anticancer drugs are more superior than single target drugs regarding patient compliance, drug adverse effects, drug-drug interactions, drug resistance as well as pharmaceutical industry economics. Dysregulation of both VEGFR-2 and c-Met tyrosine kinases (TKs) could result in development and progression of different human cancers. Herein, we reported a novel series of 3-phenylquinazolin-2,4(1H,3H)-diones with thiourea moiety as dual VEGFR-2/c-Met TKs. Compared to sorafenib, cabozantinib went behind VEGFR-2 inhibition to target c-Met TK. The dual VEGFR-2/c-Met inhibitory activity of cabozantinib is due to a longer HB domain than that of sorafenib. Based on pharmacophore of cabozantinib analogues, we designed new dual VEGFR-2/c-Met TKs. We synthesized the target compounds via a new single pot three-component reaction. The cytotoxic activity of synthesized compounds was conducted against HCT-116 colorectal cancer cell line. Compounds 3c and 3e exhibited the highest cytotoxic activity against HCT-116 cell line (IC50 1.184 and 3.403 µM, respectively). The in vitro enzyme inhibitory activity was carried out against both VEGFR-2 and c-Met TKs. Compound 3e has the highest inhibitory activity against both VEGFR-2/c-Met (IC50 = 83 and 48 nM, respectively). Docking studies showed that α-oxo moiety in quinazoline ring formed hydrogen bond HB with Met1160 residue in the adenine region of c-Met TK.
Similar content being viewed by others
Introduction
The most serious health challenge of humanity is cancer1. It was predicted that about 30 million people will be diagnosed as new cancer patients by 20401. As a result, identifying novel drug targets and developing more selective chemotherapeutic agents are important goals of current drug research2. Although recently developed single target drugs are more selective and safer than awkward conventional anticancer therapeutics, they can probably develop drug resistance3,4. One way to solve this issue is a combination therapy but this can increase undesired side effects and toxicity5. In contrast, several studies proved that multitarget small molecules can overcome this resistance with acceptable safety profile3,4. Compared to combination therapy, multitarget drugs enhance patient compliance, void detrimental off-target effects, decrease drug-drug interactions, and save excessive manufacturing costs5.
There are over five hundred protein kinases in our bodies representing the most abundant family amidst all function proteins. Protein kinases harmonically control vital cellular processes like cell division, proliferation, metabolism, migration, and apoptosis6. Protein kinases deregulation is responsible for a number of fatal illnesses, including cancer2. Accordingly, the inhibition of these deregulated protein kinases is a viable strategy to combat cancer6.
Binding of vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF) to their receptors VEGFR-2 and c-Met, respectively results in conformational changes and dimerization of the receptors followed by phosphorylation of multiple tyrosine residues in the intracellular domain2. Consequently, a cascade of intracellular signaling pathways were sparked off2.
Dysregulated VEGFR-2 and c-Met tyrosine kinases (TKs) work together to promote angiogenesis that result in development and progression of different human cancers7. Both tumor growth and metastasis are depend mainly on angiogenesis which is controlled by VEGFR-2 and c-Met signaling8. Consequently, multitarget molecules that can inhibit both VEGFR-2 and c-Met simultaneously may be more effective than single target molecules since they can shut down many signaling pathways implicated in tumor angiogenesis, proliferation, and metastasis7,9. Additionally, drug resistance is more common with single target drugs than multitarget ones9.
Rationale and molecular design
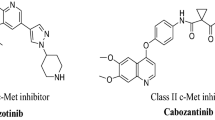
There are four pharmacophoric elements required for type II dual VEGFR-2/c-Met inhibitors namely nitrogenous heterocycle, linker, hydrogen bonding (HB) domain and hydrophobic moiety9. The first is a planar nitrogenous heterocyclic system (in blue) mainly six-membered monocyclic rings like pyridine10,11,12,13, pyrimidine7,10,14, and their fused benzo rings like quinoline10,12, quinazoline9,10,12,15, as well as fused hetero- rings like thienopyridine12,16, thienopyrimidine2, pyrrolopyridine17,18, pyrrolopyrimidine18, triazolopyrazine3, pyrrolotriazine12,19. Quinazoline ring is incorporated in several FDA approved 4-anilinoquinazoline EGFR inhibitors like lapatinib, erlotinib, gefitinib, dacomitinib, vandetanib, afatinib and icotinib as well as an VEGFR-2 inhibitor, cediranib (Fig. 1)12,20.

Clinically used quinazoline containing TK inhibitors.
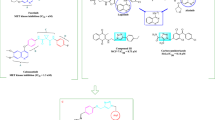
The heterocyclic ring occupies the hinge region of both TKs and forms HB via its nitrogen with the highly conserved Cys919 and Met1160 residues in VEGFR-2 and c-Met proteins, respectively. It is worth mentioning that several dual VEGFR-2/c-Met inhibitors contain HB group at α-position of the nitrogenous heterocycle that augment binding of inhibitor to the adenine region of the protein through forming additional HBs (Fig. 2). This α-HB group may be amino7,21, oxo9,22, amido19, carbamoyl11, triazolyl10,11, ureido10,19, or even the pyrrole nitrogen of pyrrolopyridine system23. In addition to HB of pyrimidine nitrogen with Cys919 and Met1160 in the adenine pocket of VEGFR-2 and c-Met TK, respectively, α-ureido group of compound II formed extra two HB with Cys919 in the hinge region of VEGFR-2 TK. Moreover, compound II formed HB with Met1160 in the hinge region of c-Met TK10. Carboxamido group of altiratinib III formed extra HB with Met1160 in the hinge region of c-Met TK24. α-Amino group in compound V act as hydrogen bond donor (HBD) forming HB with Met1160 in the hinge region of c-Met TK. Contrary, the N-methylated analogue of compound V showed no activity against both VEGFR-2 and c-Met TK7. The authors purposed that there is a relationship between HB of the α-amino with the adenine region of TKs and the inhibitory activity of the compound V7. α-Amino group of VII BMS-777607 act as HBD forming HB with Met1160 in the hinge region of c-Met TK25. Amino group of compound IX forms extra HB with Cys919 and Met1160 in the hinge region of VEGFR-2 and c-Met TKs, respectively14. Triazole ring of compound X formed extra HB with Cys919 in the hinge region of VEGFR-2 TK. In addition, It also form new HB with Tyr1159 in adenine pocket of c-Met TK11. Tang et al., synthesized several series of pyrrolo[2,3-b]pyridine c-Met inhibitors with considerable VEGFR-2 inhibitory activity23,26. Nitrogen of pyrrole ring in XI act as HBD forming HB with Met1160 in the hinge region of c-Met TK26.

Some reported dual VEGFR-2/c-Met inhibitors with α-HB group (in green).
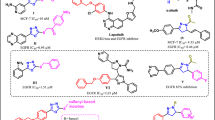
The second pharmacophoric element is a hydrophobic linker (in black) which connects nitrogenous heterocycle and HB domain. This liker has little importance in protein binding, but it puts other pharmacophoric elements in their correct position and orientations27,28. The third is a HB domain (in red) which forms HB with both Asp1046 and Glu885 of the VEGFR-2 TK as well as both Asp1222 and Lys1110 of the c-Met TK. In the type II TKIs, the main difference between VEGFR-2 inhibitors and dual VEGFR-2/c-Met inhibitors is the relatively longer HB domain of dual inhibitors7,12. VEGFR-2 inhibitors contain 2–3 atom HB domain like carboxamide (e.g., sunitinib and nintedanib) or urea (e.g., sorafenib, regorafenib and lenvatinib)28. On the other hand, the dual VEGFR-2/c-Met inhibition is originated from the presence of an HB domain with 4 or more atoms7,12. HB domain of dual VEGFR-2/c-Met is usually a dicarboxamide structure which can be a widespread cyclopropane-1,1-dicarboxamide like clinically used cabozantinib and foretinib as well as hundreds of other investigated compounds2,4,12,14,15,19,24. The dicarboxamide structure can be also involved in heteroaromatic anilides like N-phenyl-2-oxoimidazolidine-1-carboxamide29, N-phenyl-3-oxo-2,3-dihydro-1H-pyrazole-4-carboxamide (e.g., compound IV)4,11, N-phenyl-2-oxo-1,2-dihydropyridine-3-carboxamide (e.g., VII BMS-777607 and merestinib)3,25, N-phenyl-4-oxo-1,4-dihydropyridine-3-carboxamide (e.g., VIII BMS-794833) (Fig. 3)18. Finally, N-acyl(thio)urea can surrogate a dicarboxamide structure and exhibited a dual VEGFR-2/c-Met inhibitory activity (Fig. 4)16,30,31,32,33,34. The forth is a hydrophobic moiety (in purple) that occupies allosteric pocket of the TKs3,4,6,35.

HB domain requirements for dual VEGFR-2/c-Met TKs inhibition over VEGFR-2 single inhibition.

Some reported N-acyl(thio)urea containing c-Met and dual VEGFR-2/c-Met inhibitors.
Consequently, cabozantinib was selected as a lead compound, building up the pharmacophore for dual VEGFR-2/c-Met inhibition through incorporation of N-acylthiourea moiety as HB domain that mimics dicarboxamide structure of cabozantinib Fig. 53. Quinazolin-2,4(1H,3H)-dione ring was chosen to occupy adenine pocket of the TKs. α-Oxo moiety was involved in several reported TKIs like dovitinib, nintedanib, orantinib, and sunitinib36. Therefore, α-oxo moiety of target compounds was designed to augment HB and increase the affinity for both the VEGFR-2 and c-Met TKs. Finally, we different phenyl and heteroaryl rings with variable substituents were designed to occupy allosteric pocket.

Design of new quinazoline dual VEGFR-2/c-Met inhibitors.
Result and discussion
Chemistry
The synthetic pathway of 3-phenylquinazolin-2,4(1H,3H)-dione derivatives 2a,b, 3a-h and 4a,b is depicted in Scheme 137,38. The first step involves the synthesis of intermediate benzoyl isothiocyanate by the reaction of 4-(2,4-dioxo-1,4-dihydro-2H-quinazolin-3-yl)-benzoyl chloride 1 and ammonium thiocyanate under reflux. In the second step, the latter intermediate got readily transformed to the final products by nucleophilic attack of the amino groups of various used reagents such as urea, thiourea, aromatic and hetero amines via a single pot three-component reaction.

Synthetic routes of 3-phenylquinazolin-2,4(1H,3H)-dione derivatives 2a,b, 3a-h and 4a,b. Reagent and conditions: NH4SCN, (thio)urea or Ar-NH2, TEA, dioxane, reflux.
The benzoyl isothiocyanate has been employed in synthesis of some new six membered heterocyclic skeletons like thioxo-triazin-2-one and triazine-2,4-dithione, by nucleophilic addition with urea and thiourea, respectively. Herein, an equimolar reaction of benzoyl isothiocyanate with urea and/or thiourea under reflux for 6–8 h, yields an intermediate which underwent cyclodehydration to furnish compounds 2a,b, respectively. The success of cyclization by water elimination was supported by spectral data of the resulting triazines 2a,b. Their IR spectra showed absorption bands at (3406, 3226), (1716, 1655) and 1237 cm−1 for NH’s, C=O’s, and C=S, respectively. The 1H-NMR spectrum of triazine 2a exhibited their presence of two tautomeric forms from the downfield signals at 7.98, and 8.10 ppm for the NH protons. In the same manner, 1H-NMR of triazine 2b represented 8.06 and 8.08 for NH protons.
The formation of the afforded N-benzoyl-N′-phenylthiourea derivatives 3a-h was carried out by the nucleophilic reaction of different aromatic amines with benzoyl isothiocyanate in dioxane and few drops of TEA for 6–10 h. Their chemical structures were supported by IR and 1H-NMR spectra. For example, the IR bands of compound 3a illustrated the presence of NH, C=O’s and C=S at 3243, 1718, 1663 and 1268, respectively. In addition, 1H-NMR spectrum of compound 3a exhibited three singlet signals for NH protons at 12.63, 11.72, and 11.66 ppm, respectively, along with aromatic protons at 7.26–8.11 ppm.
Similarly, the intermediate benzoyl isothiocyanate reacts with some heterocyclic amines namely 5-amino-1,3,4-thiadiazole-2-thiol and 2-aminobenzothiazole under reflux for 10–12 h to furnish thiadiazole and benzothiazole 4a,b, respectively.
Biology
In vitro antiproliferative activity against HCT-116
HCT-116 colorectal cancer cell line is characterized by overexpression of both VEGFR-2 and c-Met TKs. Consequently, we selected it to study the cytotoxic activity of the target derivatives9. We tested the effect of several concentrations of the designed derivatives on HCT-116 cells by using MTT assay. Table 1 represents the IC50 of all synthesized derivatives as well as that of the reference drug, cabozantinib.
The designed compounds showed noticeable antiproliferative activities. 5 out of 12 derivatives namely, 2b, 3a, 3c, 3d, and 3e exhibited noteworthy cytotoxic activity (IC50 = 1.184–9.379 µM) that was more superior than that of the positive reference, cabozantinib. Additionally, compounds 4a and 4b (IC50 = 19.90 and 20.27 µM, respectively) displayed comparable cytotoxic activities to cabozantinib. The rest of target compounds, 2a, 3b, 3g, and 3h exhibited acceptable cytotoxic activity (IC50 = 27.030–53.39 µM). Compound 3f showed the least cytotoxic activity (IC50 = 113.500 µM). Compound 3c (with p-carboxyl moiety) represented higher cytotoxic activity than compound 3b (with m-carboxyl moiety) that suggests the regioisomerism effect on cytotoxic activity with p-substitution is superior to m-substitution. Apart from 3f, compounds with electron withdrawing group on phenyl ring represented higher cytotoxic activity as compared to unsubstituted phenyl.
In vitro toxicity against normal cells
The cytotoxic activity of selected two compounds namely, 3c and 3e against WI38 normal cell line was evaluated to study the safety of the designed compounds to the normal cell (Table 2). Although, both tested compounds exhibited higher cytotoxicity against normal cells than cabozantinib, they are better in terms of selectivity index. Compound 3c showed 20 times selective cytotoxicity against the colorectal cancer cells over the normal cells. Moreover, compound 3e showed more than 3 times selective cytotoxicity against the colorectal cancer cells over the normal cells. Compounds 3c and 3e as well as cabozantinib exhibited considerable safety as the values of SI of all of them are more than 239.
In vitro activity against c-Met and VEGFR-2 tyrosine kinases
Based on our rationale design of dual VEGFR-2/c-Met inhibition, we selected the most active cytotoxic derivatives to the HCT-116 cancer cells for scrutinization of their inhibition activity against both VEGFR-2 and c-Met TKs. Table 3 represents the inhibition activity of tested derivatives as well as cabozantinib. All tested derivatives showed acceptable inhibitory activity against the target TKs. Compound 3d exhibited comparable activity to cabozantinib against VEGFR-2 enzyme with (IC50 = 51 nM). In addition, compound 3e showed noticeable inhibitory activity against VEGFR-2 enzyme with (IC50 = 83 nM). Regarding the c-Met TK, both compounds 3c and 3e represented inhibition activity against c-Met TK with IC50 values 74 and 48 nM, respectively.
Apoptosis assay
To go deeper in the mechanism of designed derivatives, the cell cycle arrest study of compounds 3c and 3e was conducted by using Annexin V-FITC/PI staining (Table 4 and Fig. 6). HCT-116 cells were treated by cabozantinib and the two tested compounds at their IC50 concentrations. Compared with the control group, HCT-116 cell cycle was blocked in G0/G1 phase after treatment with 3c (55.41%). On another hand, compound 3e (26.51%) had a higher ability to enhance the population of HCT-116 cells in G2/M process than cabozantinib (24.72%).

Flow cytometric analysis of cell cycle phases after treatment with 3c, 3e and cabozantinib.
Moreover, we studied the apoptosis mechanism of HCT-116 that was induced by tested derivatives 3c and 3e as well as cabozantinib (Table 5 and Fig. 7). There was an increase in the number of both early and late apoptotic cells of both tested compounds 3c and 3e. The number of late apoptotic cells of 3e (35.39%) was higher than that of reference drug, cabozantinib (32.13%). Also, the number of late apoptotic cells of 3c (19.28%) was approximately equal to that of cabozantinib (19.61%). Totally, both designed compounds 3c and 3e were able to induce remarkable apoptosis. Moreover, compound 3e succeeded in apoptosis induction more efficiently than cabozantinib (Fig. 8).

Effect of 3c, 3e and cabozantinib on HCT-116 cells in annexin V-FITC staining test.

Representative cytograms of apoptotic HCT-116 cells induced by 3c and 3e compared to cabozantinib for 24 h.
In silico studies
Molecular modeling studies
Molecular docking of the target compounds was carried out in the active site of c- VEGFR-2 (PDB: 4ASD) and c-Met (PDB: 3LQ8) TKs. Docking styles of compounds 3c and 3e in the active site of VEGFR-2 TK are shown in Figs. 9 and 10, respectively. The thiourea group of compound 3c showed HB with a highly conserved residue Asp1046 in the HB region and α-oxo group of quinazoline formed HB with backbone amide of Phe918 in the hinge region. Moreover, compound 3c exhibited hydrophobic interactions with both Leu840 and HB with Lys868. 4-Oxo moiety of compound 3e declared HB with the conserved residue Cys919 in the hinge region. Compound 3e showed another two HB with Asp1046 and Lys868 in addition to hydrophobic interaction with Leu84027,28.

Docking style of compound 3c with VEGFR-2 TK (PDB: 4ASD).

Docking style of compound 3e with VEGFR-2 TK (PDB: 4ASD).
On the other hand, Figs. 11 and 12 represent docking representations of compounds 3c and 3e it the active site of c-Met TK. α-Oxo moiety of quinazoline ring of compound 3c showed HB with highly conserved residue Met1160 in the hinge region as well as thiourea group formed HB with the another highly conserved residue Lys1110 in the HB region. Further, compound 3c declared hydrophobic interaction with Ile1084. N-acylthiourea group of compound 3e exhibited dual HB with both Lys 1110 and Asp1222 in addition to hydrophobic interactions with both Ile1084 and Met1211 in the hinge region.

Docking style of compound 3c with c-Met TK (PDB: 3LQ8).

Docking style of compound 3e with c-Met TK (PDB: 3LQ8).
In silico prediction of physicochemical and pharmacokinetic properties
To reach advanced clinical phases, drug candidates have to possess an acceptable pharmacokinetic profile. Therefore, the physicochemical and pharmacokinetic properties of target derivatives were predicted using SwissADME (Tables 6, 7 and Fig. 13). All target compounds showed good druglikeness with no Lipinski violations except compound 3h. Regarding Abbott bioavailability score, most target derivatives have accepted oral absorption. All target compounds as well as cabozantinib were predicted not to cross BBB. Target compounds were predicted to have little effect on CYP450 enzymes like CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4 which minimizes the suspected drug-drug interactions.

Rader for 3c, 3e and cabozantinib.
Conclusion
In brief, based on the structure of a dual VEGFR-2/c-Met inhibitor, cabozantinib, we designed and synthesized a new series of novel 3-phenylquinazolin-2,4(1H,3H)-diones with (thio)urea scaffold. The cytotoxic activity of the target compounds was conducted against HCT-116 colorectal cancer cell line. Compounds contain electron-withdrawing groups on phenyl ring at position-4, declared the highest cytotoxic activity. The inhibitory activity was performed against both VEGFR-2 and c-Met TKs. α-Oxo moiety in quinazoline ring form HB with Met1160 residue in the adenine region of c-Met TK. Compound 3e has the highest inhibitory activity against both VEGFR-2/c-Met (IC50 = 83 and 48 nM, respectively).
Experimental
Chemistry
All reactions were observed on silica gel GF254 plate with thin layer chromatography (TLC). Melting points (uncorrected) were recorded on an electrothermal melting apparatus. FT-IR spectra were recorded on a Shimadzu 8101 PC spectrometer. 1H- and 13C-NMR spectra were determined on a Varian Mercury 400 MHz spectrophotometer in DMSO-d6. Chemical shifts are reported in parts per million with tetramethylsilane as an internal standard and are given in δ units. Electron impact mass spectra were obtained at 70 eV using a GCMS-QP 1000 EX spectrometer. Elemental analyses were carried out at the microanalytical center at Cairo University.
Synthesis of 4-(2,4-dioxo-1,4-dihydro-2H-quinazolin-3-yl)-benzoyl chloride 1
4-(2,4-dioxo-1,4-dihydro-2H-quinazolin-3-yl)-benzoic acid (0.013 mol, 1 gm) was dissolved in thionyl chloride, then the reaction mixture was refluxed for 5 h. After completion of the reaction, the excess of thionyl chloride was evaporated and the residue was allowed to stand at room temperature, then recrystallized from benzene to yield the product as yellow crystal. Yield: 85%. M.P: 270 °C. FT-IR (KBr, υ, cm−1) = 3195 (NH), 1763, 1725 (C = O’s), 751 (C–Cl). 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 11.65 (s, 1H, NH), 7.22–7.96 (m, 4H, Ar–H (quinazoline)), 7.49–8.07 (dd, 4H, Ar–H). 13C-NMR (DMSO): δ 114.74, 115.77, 123.09, 127.52, 128.04, 129.99, 130.30, 131.95, 135.81, 140.30, 150.43, 162.57, 167.35. MS (EI) (RT: 3.56–3.58 min): m/z = 300.22 (obs.), 300.03 (expected) [M+], 302.22 (obs.), 302.27 (expected) [M+ + 2]. Anal. Calcd for C15H9ClN2O3: C, 59.91; H, 3.02; Cl, 11.79; N, 9.32%. Found C, 60.16; H, 3.26; Cl, 11.84; N, 9.18%.
General procedures for the synthesis of 3-[4-(6-mercapto-4-oxo/thioxo-1,4-dihydro-[1,3,5]-triazin-2-yl)-phenyl]-1H-quinazolin-2,4-dione 2a,b
4-(2,4-dioxo-1,4-dihydro-2H-quinazolin-3-yl)-benzoyl chloride 1 (0.003 mol, 0.32 g) was allowed to react with ammonium thiocyanate (0.003 mol, 0.32 g) in dioxane (20 ml) under reflux for 2 h. After completion of the reaction, the formed precipitate of ammonium chloride was removed, then urea and/or thiourea was added to the filtrate in presence of few drops of TEA under reflux for 6 h. The solid formed was filtered off, dried, and recrystallized from ethanol to afford compounds 2a, b, respectively.
3-(4-(4-oxo-6-thioxo-1,4,5,6-tetrahydro-1,3,5-triazin-2-yl)phenyl)quinazolin-2,4(1H,3H)-dione 2a
Brown crystals. Yield 60%. MP > 300 °C. FT-IR (KBr, υ, cm−1) = 3406, 3226, 3060 (NH’s), 1717, 1654 (C = O’s), 1610 (C=N), 1280 (C=S). 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 11.64 (s, 1H, NH), 8.10 (s, 1H, NH), 7.98 (s, 1H, NH), 7.25–7.75 (m, 4H, Ar–H (quinazoline), 7.42–7.96 (dd, 4H, Ar–H). 13C-NMR (DMSO): δ 114.76, 115.75, 123.06, 128.05, 128.47, 129.52, 129.71, 129.78, 134.63, 135.78, 138.78, 140.31, 150.50, 162.60, 167.94. MS (EI) (RT: 2.90–3.00): m/z = 365.00 (obs.), 365.37 (expected) [M+], 367.00 (obs.), 367.37 (expected) [M+ + 2]. Anal. Calcd for C17H11N5O3S: C, 55.88; H, 3.03; N, 19.17; S, 8.78%. Found C, 55.99; H, 3.14; N, 19.10; S, 8.88%.
3-(4-(4,6-dithioxo-1,4,5,6-tetrahydro-1,3,5-triazin-2-yl)phenyl)quinazolin-2,4(1H,3H)-dione 2b
Pale brown crystals. Yield 60%. MP > 300 °C. FT-IR (KBr, υ, cm−1) = 3406, 3235, 3063 (NH's), 1716, 1655 (C=O’s), 1607 (C=N), 1275, 1237 (C=S’s). 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 11.64 (s, 1H, NH), 8.08 (s, 1H, NH), 8.06 (s, 1H, NH), 7.23–8.04 (m, 4H, Ar–H (quinazoline)), 7.42–7.98 (dd, 4H, Ar–H). 13C-NMR (DMSO): δ 114.76, 115.75, 123.06, 128.05, 128.48, 129.72, 132.45, 134.64, 135.68, 138.48, 140.30, 150.22, 162.17, 167.96, 182.55. MS (EI) (RT: 1.60–1.63): m/z = 381.00 (obs.), 381.44 (expected) [M+], 383.00 (obs.), 383.44 (expected) [M+ + 2]. Anal. Calcd for C17H11N5O2S2: C, 53.53; H, 2.91; N, 18.36; S, 16.81%. Found C, 53.66; H, 3.09; N, 18.25; S, 16.97%.
General procedures for the synthesis of arylquinazolin-2,4-diones 3a-h
Multi-component reaction of 4-(2,4-dioxo-1,4-dihydro-2H-quinazolin-3-yl)-benzoyl chloride 1 with ammonium thiocyanate and aromatic amines namely aniline, m-aminobenzoic acid, p-aminobenzoic acid, ethyl 4-aminobenzoate, p-chloroaniline, p-nitroaniline, p-aminophenol, and 4-amino-N-pyrimidin-2-yl-benzenesulfonamide in dioxane (20 ml) and few drops of TEA was heated under reflux for 10–12 h (monitored by TLC) to afforded compounds 3a–h, respectively.
4-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-(phenylcarbamothioyl)benzamide 3a
White crystals. Yield 75%. MP 210 °C. FT-IR (KBr, υ, cm−1) = 3243 (NH), 1716, 1663 (C=O’s), 1268 (C=S). 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 12.63 (s, 1H, NH), 11.72 (s, 1H, NH), 11.66 (s, 1H, NH), 7.26–8.05 (m, 9H, Ar–H), 7.72–8.11 (dd, 4H, Ar–H). 13C-NMR (DMSO): δ 114.78, 115.79, 123.10,124.89, 126.85, 128.06, 128.81, 129.18 129.71, 129.80, 132.45, 135.84, 140.33, 140.48, 150.43, 162.59, 168.27, 179.58. MS (EI) (RT: 4.47–4.50): m/z = 416.34 (obs.), 416.34.46 (expected) [M+]. Anal. Calcd for C22H16N4O3S: C, 63.45; H, 3.87; N, 13.45; S, 7.70%. Found C, 63.56; H, 4.01; N, 13.35; S, 7.83%.
3-(3-(4-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)benzoyl)thioureido)benzoic acid 3b
Pale brown crystals. Yield 75%. MP: > 300 °C. FT-IR (KBr, υ, cm−1) = 3405 (COOH), 3222, 3061 (NH's), 1716, 1656 (C=O’s), 1279 (C=S). 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 12.70 (s, 1H, COOH), 11.65 (s, 1H, NH), 11.61 (s, 1H, NH), 11.40 (s, 1H, NH), 7.23–8.12 (m, 8H, Ar–H), 7.42–7.98 (dd, 4H, Ar–H). 13C-NMR (DMSO): δ 114.75, 115.75, 123.06, 123.15, 128.05, 128.48, 129.52, 129.72, 129.78, 129.86, 134.63, 135.77, 138.79, 140.30, 150.51, 162.61, 167.97, 179.94, 182.54. MS (EI) (RT: 2.73–2.76): m/z = 460.48 (obs.), 460.47 (expected) [M+]. Anal. Calcd for C23H16N4O5S: C, 59.99; H, 3.50; N, 12.17; S, 6.96%. Found C, 60.12; H, 3.60; N, 12.09; S, 7.11%.
4-(3-(4-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)benzoyl)thioureido)benzoic acid 3c
Orange crystal crystals. Yield 70%. MP 258 °C. FT-IR (KBr, υ, cm−1) = 3355 (COOH), 3044 (NH), 1721, 1681 (C=O’s), 1264 (C=S). 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 12.80 (s, 1H, COOH), 12.68 (s, 1H, NH), 11.82 (s, 1H, NH), 11.65 (s, 1H, NH), 7.25–8.16 (m, 8H, Ar–H), 7.52–8.02 (dd, 4H, Ar–H). 13C-NMR (DMSO): δ 113.01, 114.77, 115.79, 117.30, 123.10, 124.04, 128.05, 128.57, 129.85, 130.42, 131.69, 132.39, 140.54, 142.43, 150.43, 162.59, 167.20, 168.18, 179.74. MS (EI) (RT: 4.09–4.40): m/z = 460.48 (obs.), 460.47(expected) [M+]. Anal. Calcd for C23H16N4O5S: C, 59.99; H, 3.50; N, 12.17; S, 6.96%. Found C, 60.12; H, 3.60; N, 12.09; S, 7.11%.
Ethyl4-(3-(4-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)benzoyl)thioureido)benzoate 3d
White crystals. Yield 80%. MP > 300 °C. FT-IR (KBr, υ, cm−1) = 3359, 3065, 2932 (NH's), 1715, 1666 (C=O’s), 1270 (C=S). 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 11.65 (s, 1H, NH), 11.45 (s, 1H, NH) 10.72 (s, 1H, NH), 7.24–8.00 (m, 8H, Ar–H), 7.54–8.08 (dd, 4H, Ar–H), 4.35 (q, 2H, CH2), 1.36 (t, 3H, CH3). 13C-NMR (DMSO): δ 14.72, 60.98, 114.78, 115.78, 120.02, 123.08, 125.11, 128.06 128.87, 129.80, 130.59, 134.96, 135.81, 139.36, 140.34, 144.07, 150.49, 162.62, 165.25, 165.84, 166.16. MS (EI) (RT: 1.91–1.95): m/z = 488.05 (obs.), 488.53 (expected) [M+]. Anal. Calcd for C25H20N4O5S: C, 61.47; H, 4.13; N, 11.47; S, 6.56%. Found C, 61.56; H, 4.29; N, 11.35; S, 6.66%.
N-((4-chlorophenyl)carbamothioyl)-4-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl) benzamide 3e
Pale violet crystals. Yield 81%. MP 250 °C. FT-IR (KBr, υ, cm−1) = 3323, 3248 (NH's), 1724, 1671 (C=O’s), 1245 (C=S). 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 11.67 (s, 1H, NH), 10.60 (s, 1H, NH), 10.36 (s, 1H, NH), 7.29–8.11 (m, 8H, Ar–H), 7.43–7.97 (dd, 4H, Ar–H). 13C-NMR (DMSO): δ 114.74, 115.79, 123.05, 128.02, 128.48, 129.02, 129.41, 129.50, 129.74, 132.17, 134.61, 135.76, 138.78, 140.33, 150.47, 162.60, 167.47, 167.93. MS (EI) (RT: 1.74–1.76): m/z = 450.33 (obs.), 450.91 (expected) [M+], 452.33 (obs.), 452.91 (expected) [M+ + 2]. Anal. Calcd for C22H15ClN4O3S: C, 58.60; H, 3.35; Cl, 7.86; N, 12.43; S, 7.11%. Found C, 58.74; H, 3.46; Cl, 7.91; N, 12.38; S, 7.22%.
4-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-((4-nitrophenyl)carbamothioyl) benzamide 3f.
Brown crystals. Yield 65%. MP 255 °C. FT-IR (KBr, υ, cm−1) = 3393, 3247, 3071 (NH's), 1718, 1667 (C = O's), 1237 (C = S). 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 11.90 (s, 1H, NH), 11.64 (s, 1H, NH), 11.40 (s, 1H, NH), 7.24–7.97 (m, 8H, Ar–H), 7.50–8.06 (dd, 4H, Ar–H). 13C-NMR (DMSO): δ 114.77, 115.79, 120.61, 122.10, 123.08, 125.06, 128.05, 129.69, 129.78, 132.55, 135.80, 135.82, 139.75, 140.34, 150.41, 162.57, 167.75, 182.55. MS (EI) (RT: 5.60–5.85): m/z = 459.00 (obs.), 459.46 (expected) [M+-2], 461.00 (obs.), 461.46 (expected) [M+]. Anal. Calcd for C22H15N5O5S: C, 57.26; H, 3.28; N, 15.18; S, 6.95%. Found C, 57.40; H, 3.40; N, 15.09; S, 7.08%.
4-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-((4-hydroxyphenyl)carbamothioyl) benzamide 3g
Dark brown crystals. Yield 75%. MP 260 °C. FT-IR (KBr, υ, cm-1) = 3386 (OH), 3246, 3067, 2919 (NH's), 1716, 1665 (C=O's), 1268 (C=S). 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 12.44 (s, 1H, OH), 11.64 (s, 1H, NH), 11.62 (s, 1H, NH), 11.40 (s, 1H, NH), 6.81–8.10 (m, 8H, Ar–H), 7.50–8.06 (dd, 4H, Ar–H). 13C-NMR (DMSO): δ 114.78, 115.57, 115.78, 120.04, 123.09, 126.47, 128.05, 128.48, 129.70, 129.78, 132.55, 135.82, 140.33, 150.42, 162.57, 167.76, 168.10, 182.54. MS (EI) (RT: 3.40–3.75): m/z = 432.00 (obs.), 432.45 (expected) [M+]. Anal. Calcd for C22H16N4O4S: C, 61.10; H, 3.73; N, 12.96; S, 7.41%. Found C, 61.22; H, 3.85; N, 12.85; S, 7.55%.
4-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-((4-(N-(pyrimidin-2-yl)sulfamoyl) phenyl)carbamothioyl)benzamide 3h
Pale brown crystals. Yield 80%. MP 300 ºC. FT-IR (KBr, υ, cm−1) = 3397, 3094, 2920 (NH's), 1718, 1666 (C=O’s), 15,835 (C=N), 1337 (O=S=O), 1263 (C=S). 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 12.76 (S, 1H, NH), 11.83 (s, 1H, NH), 11.63 (s, 1H, NH), 11.40 (s, 1H, NH), 7.08–8.55 (m, 11H, Ar–H + pyrimidine protons), 7.50–8.05 (dd, 4H, Ar–H). 13C-NMR (CDCl3): δ 114.77, 115.78, 122.10, 123.09, 123.85, 124.35, 127.57, 128.05, 128.75, 129.71, 129.78, 129.86, 132.55, 135.82, 140.32, 140.35, 142.75, 150.42, 162.57, 167.76, 182.54. MS (EI) (RT: 2.50–2.75): m/z = 573.00 (obs.), 573.54 (expected) [M+], 575.00 (obs.), 575.54 (expected) [M+ + 2]. Anal. Calcd for C26H19N7O5S2: C, 54.44; H, 3.34; N, 17.09; S, 11.18%. Found C, 54.66; H, 3.64; N, 17.00; S, 11.33%.
General procedures for the synthesis of heterylquinazolin-2,4-diones 4a-b
A mixture of 4-(2,4-dioxo-1,4-dihydro-2H-quinazolin-3-yl)-benzoyl chloride 1 (0.005 mol, 1.5 gm) and ammonium thiocyanate (0.005 mol, 0.39 gm) in dioxane (20 ml) and few drops of TEA was refluxed for 2 h. Afterward, the formed ammonium chloride precipitate was removed by filtration, then hetero amines namely 5-amino-[1,3,4]-thiadiazole-2-thiol, and 2-aminobenzothiazole (0.005 mol) were added to the filtrate and the reaction mixture was refluxed for 8–10 h. The formed solids were filtered off, purified by recrystallization using benzene/ethanol to furnish compounds 4a-b, respectively.
4-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-((5-mercapto-1,3,4-thiadiazol-2-yl)carbamothioyl)benzamide 4a
Yellow crystals. Yield 82%. MP > 300 °C. FT-IR (KBr, υ, cm−1) = 3408, 3212, 3064 (NH's), 2723 (SH), 1717, 1655 (C=O’s), 1608 (C=N), 1275 (C=S). 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 13.16 (s, 1H, SH), 11.68 (s, 1H, NH), 11.63 (s, 1H, NH), 11.61 (s, 1H, NH), 7.24–8.08 (m, 4H, Ar–H(quinazoline)), 7.42–7.98 (dd, 4H, Ar–H). 13C-NMR (DMSO): δ 114.76, 115.75, 123.06, 128.05, 128.47, 129.52, 130.28, 131.04, 134.64, 135.77, 138.78, 140.30, 150.43, 162.60, 167.35, 167.94. MS (EI) (RT: 3.30–3.50): m/z = 455.00 (obs.), 455.53 (expected) [M+-1], 456.00 (obs.), 456.53 (expected) [M+]. Anal. Calcd for C18H12N6O3S3: C, 47.36; H, 2.65; N, 18.41; S, 21.07%. Found C, 47.50; H, 2.78; N, 18.32; S, 21.21%.
N-(benzo[d]thiazol-2-ylcarbamothioyl)-4-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)benzamide 4b
Brown crystals. Yield 75%. MP > 300 °C. FT-IR (KBr, υ, cm−1) = 3406, 3234, 3063 (NH's), 1716, 1655 (C=O’s), 1608 (C=N), 1280 (C=S). 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 11.57 (s, 2H, 2NH), 11.40 (s, 1H, NH), 7.23–8.08 (m, 8H, Ar–H), 7.42–7.98 (dd, 4H, Ar–H). 13C-NMR (CDCl3): δ 114.76, 115.75, 123.06, 124.05, 128.05, 128.46, 129.52, 129.71, 129.78, 132.51, 134.63, 135.08, 135.78, 138.78, 140.30, 146.19, 150.42, 150.50, 162.60, 167.76, 167.93. MS (EI) (RT: 1.90–2.10): m/z = 473.00 (obs.), 473.54 (expected) [M+], 474.00 (obs.), 474.54 (expected) [M+ + 1]. Anal. Calcd for C23H15N5O3S2: C, 58.34; H, 3.19; N, 14.79; S, 13.54%. Found C, 58.50; H, 3.29; N, 14.62; S, 13.70%.
Biological evaluation
Supporting information includes all experimental details for the MTT assay, and in vitro enzyme inhibition of VEGFR-2 and c-Met TKs.
In silico studies
Molecular modeling study
Molecular docking and visualizations of 2D and 3D styles were conducted by using MOE 2014.0901 and Discovery Studio visualizer softwares. The structures of co-crystallized TKs were retrieved from protein data bank (VEGFR-2: 4ASD & c-Met: 3LQ8). The structures of compound 3c and 3e were standardized and their energies were minimized. After that, the protein structures were prepared using MOE standard protocol27. Validation of docking approach was performed by redocking process of the original ligands (VEGFR-2: sorafenib & c-Met: foretinib) into the binding sites of both proteins9. Docking of compounds 3c and 3e were performed using default MOE docking setting. The 2D and 3D representations of the most stable poses were selected for further studies.
In silico prediction of physicochemical and pharmacokinetic properties
The freely available SwissADME website was utilized to predict the physicochemical and pharmacokinetics of the target compounds. Supporting information includes how different filters and parameters were calculated9.
Data availability
All data generated or analysed during this study are included in the supplementary information file.
References
Global cancer observatory: Cancer today. International Agency for Research on Cancer, Lyon. https://gco.iarc.fr/ (2023).
Li, J. et al. Design, synthesis, and biological evaluation of thieno[2,3-d]pyrimidine derivatives as novel dual c-Met and VEGFR-2 kinase inhibitors. Bioorg. Med. Chem. 25, 6674–6679 (2017).
Liu, X. et al. Design, synthesis, and biological evaluation of [1,2,4]triazolo[4,3-a] pyrazine derivatives as novel dual c-Met/VEGFR-2 inhibitors. Front. Chem. 2022, 10 (2022).
Hu, H., Chen, F., Dong, Y., Liu, Y. & Gong, P. Discovery of novel dual c-Met/HDAC inhibitors as a promising strategy for cancer therapy. Bioorg. Chem. 101, 103970 (2020).
Obaid-Arhema-Frejat, F. et al. Novel indazole derivatives as potent apoptotic antiproliferative agents by multi-targeted mechanism: Synthesis and biological evaluation. Bioorg. Chem. 126, 105922 (2022).
Abd-El-Meguid, E. A., Naglah, A. M., Moustafa, G. O., Awad, H. M. & El-Kerdawy, A. M. Novel benzothiazole-based dual VEGFR-2/EGFR inhibitors targeting breast and liver cancers: Synthesis, cytotoxic activity, QSAR and molecular docking studies. Bioorg. Med. Chem. Lett. 58, 128529 (2022).
Zhan, Z. et al. Discovery of anilinopyrimidines as dual inhibitors of c-Met and VEGFR-2: Synthesis, SAR, and cellular activity. ACS Med. Chem. Lett. 5, 673–678 (2014).
Liu, X.-J. et al. Recent development of multi-target VEGFR-2 inhibitors for the cancer therapy. Bioorg. Chem. 133, 106425 (2023).
Hassan, A. et al. Design and biological evaluation of 3-substituted quinazoline-2,4(1H,3H)-dione derivatives as dual c-Met/VEGFR-2-TK inhibitors. J. Enzyme Inhib. Med. Chem. 38, 2189578 (2023).
Yang, F. et al. Design, synthesis and biological evaluation of 4-phenoxy-pyridine/pyrimidine derivatives as dual VEGFR-2/c-Met inhibitors. New J. Chem. 46, 12651–12665 (2022).
Gu, W. et al. Discovery of novel 2-substituted-4-(2-fluorophenoxy) pyridine derivatives possessing pyrazolone and triazole moieties as dual c-Met/VEGFR-2 receptor tyrosine kinase inhibitors. Bioorg. Chem. 72, 116–122 (2017).
Zhang, Q., Zheng, P. & Zhu, W. Research progress of small molecule VEGFR/c-Met inhibitors as anticancer agents (2016–Present). Molecules 25, 2666 (2020).
Wang, M.-S. et al. Synthesis and biological evaluation of new MET inhibitors with 1,6-naphthyridinone scaffold. Eur. J. Med. Chem. 185, 111803 (2020).
Qiang, H. et al. Design, synthesis and biological evaluation of 4-aminopyrimidine-5-cabaldehyde oximes as dual inhibitors of c-Met and VEGFR-2. Bioorg. Med. Chem. 24, 3353–3358 (2016).
Wei, D. et al. Synthesis and anti-tumor activity of [1,4] dioxino [2,3-f] quinazoline derivatives as dual inhibitors of c-Met and VEGFR-2. Bioorg. Chem. 88, 102916 (2019).
Claridge, S. et al. Discovery of a novel and potent series of thieno[3,2-b]pyridine-based inhibitors of c-Met and VEGFR2 tyrosine kinases. Bioorg. Med. Chem. Lett. 18, 2793–2798 (2008).
Wang, W. et al. Synthesis and bioevaluation and doking study of 1H-pyrrolo[2,3-b]pyridine derivatives bearing aromatic hydrazone moiety as c-Met inhibitors. Eur. J. Med. Chem. 145, 315–327 (2018).
Wang, L. X. et al. Discovery of novel pyrrolo-pyridine/pyrimidine derivatives bearing pyridazinone moiety as c-Met kinase inhibitors. Eur. J. Med. Chem. 141, 538–551 (2017).
Shi, W. et al. Exploration of novel pyrrolo[2,1-f][1,2,4]triazine derivatives with improved anticancer efficacy as dual inhibitors of c-Met/VEGFR-2. Eur. J. Med. Chem. 158, 814–831 (2018).
Elkamhawy, A. et al. Design, synthesis, biological evaluation, and molecular dynamics studies of novel lapatinib derivatives. Pharmaceuticals 2013, 16 (2023).
Zhao, Y. et al. Synthesis and evaluation of a series of pyridine and pyrimidine derivatives as type II c-Met inhibitors. Bioorg. Med. Chem. 25, 3195–3205 (2017).
Musumeci, F., Radi, M., Brullo, C. & Schenone, S. Vascular endothelial growth factor (VEGF) receptors: Drugs and new inhibitors. J. Med. Chem. 55, 10797–10822 (2012).
Tang, Q. et al. Discovery of novel 7-azaindole derivatives bearing dihydropyridazine moiety as c-Met kinase inhibitors. Eur. J. Med. Chem. 133, 97–106 (2017).
Smith, B. D. et al. Altiratinib inhibits tumor growth, invasion, angiogenesis, and microenvironment-mediated drug resistance via balanced inhibition of MET, TIE2, and VEGFR2. Mol. Cancer Therapeut. 14, 2023–2034 (2015).
Schroeder, G. M. et al. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J. Med. Chem. 52, 1251–1254 (2009).
Tang, Q. et al. Discovery of novel pyrrolo[2,3-b]pyridine derivatives bearing 1,2,3-triazole moiety as c-Met kinase inhibitors. Bioorg. Med. Chem. Lett. 26, 1680–1684 (2016).
Hassan, A. et al. Design, synthesis, in vitro antiproliferative evaluation and in silico studies of new VEGFR-2 inhibitors based on 4-piperazinylquinolin-2(1H)-one scaffold. Bioorg. Chem. 120, 105631 (2022).
Hassan, A., Badr, M., Hassan, H. A., Abdelhamid, D. & Abuo-Rahma, G. E. D. A. Novel 4-(piperazin-1-yl)quinolin-2(1H)-one bearing thiazoles with antiproliferative activity through VEGFR-2-TK inhibition. Bioorg. Med. Chem. 40, 116168 (2021).
Zhou, S. et al. Discovery andw biological evaluation of novel 6,7-disubstituted-4-(2-fluorophenoxy)quinoline derivatives possessing 1,2,3-triazole-4-carboxamide moiety as c-Met kinase inhibitors. Bioorg. Med. Chem. 22, 6438–6452 (2014).
Cai, Z.-W. et al. Discovery of orally active pyrrolopyridine- and aminopyridine-based Met kinase inhibitors. Bioorg. Med. Chem. Lett. 18, 3224–3229 (2008).
Kung, P.-P. et al. Structure activity relationships of quinoline-containing c-Met inhibitors. Eur. J. Med. Chem. 43, 1321–1329 (2008).
Schroeder, G. M. et al. Identification of pyrrolo[2,1-f][1,2,4]triazine-based inhibitors of Met kinase. Bioorg. Med. Chem. Lett. 18, 1945–1951 (2008).
Fujita, H. et al. The novel VEGF receptor/MET–targeted kinase inhibitor TAS-115 has marked in vivo antitumor properties and a favorable tolerability profile. Mol. Cancer Therapeut. 12, 2685–2696 (2013).
Chen, P. et al. Design, synthesis and anticancer evaluation of 6,7-disubstituted-4-phenoxyquinoline derivatives bearing 1,8-naphthyridine-3-carboxamide moiety as novel multi-target TKIs. Bioorg. Chem. 121, 105672 (2022).
Ghany, L. M. A. A., El-Dydamony, N. M., Helwa, A. A., Abdelraouf, S. M. & Abdelnaby, R. M. Coumarin-acetohydrazide derivatives as novel antiproliferative agents via VEGFR-2/AKT axis inhibition and apoptosis triggering. New J. Chem. 46, 17394–17409 (2022).
Guagnano, V. et al. Discovery of 3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 54, 7066–7083 (2011).
Haredi-Abdelmonsef, A., Eldeeb-Mohamed, M., El-Naggar, M., Temairk, H. & Mohamed-Mosallam, A. Novel quinazolin-2,4-dione hybrid molecules as possible inhibitors against malaria: Synthesis and in silico molecular docking studies. Front. Mol. Biosci. 2020, 7 (2020).
Noser, A. A., El-Naggar, M., Donia, T. & Abdelmonsef, A. H. Synthesis, in silico and in vitro assessment of new quinazolinones as anticancer agents via potential AKT inhibition. Molecules 25, 4780 (2020).
Abdelnaby, R. M. et al. In vitro anticancer activity screening of novel fused thiophene derivatives as VEGFR-2/AKT dual inhibitors and apoptosis inducers. Pharmaceuticals 15, 700–717 (2022).
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hassan, A., Mosallam, A.M., Ibrahim, A.O.A. et al. Novel 3-phenylquinazolin-2,4(1H,3H)-diones as dual VEGFR-2/c-Met-TK inhibitors: design, synthesis, and biological evaluation. Sci Rep 13, 18567 (2023). https://doi.org/10.1038/s41598-023-45687-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-45687-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
