
Abstract
A novel catalyst was fabricated in this study based on zirconium MOF modified with pyridine carboxaldehyde in a solvothermal reaction, embedded with cerium. In order to confirm the catalyst structure, various characterization techniques, including FTIR, Far IR, EDX, XRD, TGA, FE-SEM, ICP, and BET analyses, were employed. The results indicated that the UiO-66-Pyca-Ce (III) catalyst had a Langmuir surface area of 501.63 m2/g, a pore volume of 0.28 cm3/g, and a pore size of 2.27 nm. To study catalytic activity, a sequential approach of Knoevenagel condensation and Michael addition was used to synthesize various polyhydroquinoline derivatives. The reaction took place at ambient temperature. The UiO-66-Pyca-Ce (III) catalyst demonstrated high efficacy (90%) and reusability in asymmetric synthesis of polyhydroquinoline derivatives for several reasons, including the possession of three Lewis acid activation functions.
Similar content being viewed by others


Introduction
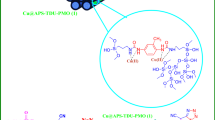
Recently, multi-component reactions have been employed to develop new methods of Synthesis strategies with abundant molecular diversity1,2. These strategies have resulted high atom economy and simple methods for synthesizing heterocyclic compounds3 which leading to biological active compounds like antituberculosis, anticancer, antipain, and antidiabetic agents. As a result, a wide variety of studies have been carried out on the popularization of the synthesis of these heterocyclic compounds using both heterogeneous and homogeneous catalysts4,5,6,7. In addition, the development of synthetic methods to fabricate N-heterocyclic polyhydroquinoline has been highlighted by researchers8. To prepare bioactive molecules, different types of catalysts, such as LiBr9, CTAB10, CuBr11, GO nanoparticles12, l-proline13, CTACl14 and salicylic acid15, have been utilized. Many of these reactions suffer from disadvantages like harsh reaction conditions, low product yields, and slow and tedious procedures, which all considered negative points. To solve these problems, scientists have developed and offered novel synthetic techniques. Recently; metal–organic frameworks (MOFs) that include metal-exo clusters and organic linkers have received more attention16,17,18. These MOFs are utilized in various applications because of their high porosity, highly effective surface, and chemical stability. These applications include drug delivery19,20, food storage21, gas absorption or storage22, hazardous gas storage23, chemical sensing24 and optoelectronics25. MOFs either have catalytic activity or used as a suitable support for catalysts which make them a good choice in catalysis applications26,27,28,29,30,31,32. Post-synthetic modification (PSM) method is used to functionalize MOFs which can improve their catalytic activity33. PSM can be performed without causing any negative effects on the framework’s stability. these modifications are resulted by adding new active sites in MOFs structure34 that enhance catalytic activities of these compounds. Another enhancement leads to easy separation and recovery of heterogeneous catalysts which highlighted MOFs as a reusable catalysts35,36,37,38,39,40,41,42,43,44. Cerium (III) chloride heptahydrate (CeCl3·7H2O), known as a promoter in organic synthesis, has attracted much attention due to its various applications45. Cerium halides have advantages such as being water resistance, user friendly, non-toxicity, and affordability. Furthermore, they can be reused multiple times without any purification46. They are recognized as effective Lewis acid catalysts47. In current study, catalytic activity of zirconium MOF from the UiO-66 family was investigated. Pyridine carbaldehyde was used to modify UiO-66-NH2 using a post-synthesis approach. The cerium was subsequently incorporated into UiO-66-Pyca. The asymmetric Hantzsch reaction was employed to evaluate the catalytic activity of UiO-66-Pyca-Ce (III). The catalyst showed recyclability and multiple reusability, which is very favorable for both the environment and the economic standpoint. The sufficiency and catalytic performance of this new UiO-66-Pyca-Ce (III) catalyst were assessed in the asymmetric Hantzsch condensation reaction for the synthesis of polyhydroquinoline derivatives (5a-f) (Fig. 1). It should be noted that the response conducted as part of this research is environmentally safe and no toxic waste is produced.

Preparation of MOF UiO-66-Pyca-Ce (III) correction reaction in 2 steps.
Experimental
Materials and physical techniques
All reagents for synthesis and analysis were commercially available from Aldrich and Merck Companies and were used without further purification. The infrared (IR) spectra were recorded using a Thermo Nicolet IR 100 FT-IR spectrometer. A field emission scanning electron microscope (FESEM), specifically the German-made ZEISS SIGMA VP model with a gold coating, was used to analyze the samples. Utilizing monochromatic Co-Kα (1.78897 Å) radiation and a Philips X’pert diffractometer, measurements of X-ray powder diffraction (XRD) were made. The N2 desorption/adsorption isotherms of the synthesized samples were obtained using the BET technique with a Microtrac Bel Corp Belsorp mini II instrument. The N2 adsorption isotherm at 77 K was measured using a Micromeritics ASAP 2030 surface area analyzer. Thermogravimetry (TGA) was performed to determine the thermal stability using an SDTQ600 V20.9 analyzer with a heating rate of 10 °C/min under airflow. The metal cerium ion loading of the catalyst was determined by inductively coupled plasma (ICP) analysis using a Vavian 715-ES instrument. Proton nuclear magnetic resonance (1H NMR) spectroscopy was conducted on a Varian UnityPlus 400 instrument.
Synthesis of UiO-66-Pyca
The UiO-66-NH2 crystals were prepared according to the literature procedure48,49,50. The prepared UiO-66-NH2 (0.3 g) was dispersed in 30 mL of ethanol in an orbicular-bottomed flask and stirred for 30 min. Thereafter, 0.9 mL of pyridine carboxaldehyde was added to the mixture, and the sealed flask was transferred to an oil bath. The solution was stirred at 80 °C for 20 h. The precipitate was separated by centrifugation after the yellow solution was cooled to room temperature. The acquired products were washed with abundant ethanol. Finally, the obtained UiO-66-Pyca was dried in a vacuum oven at 50 °C for 24 h.
Synthesis of UiO-66-Pyca-Ce (III)
UiO-66-Pyca (10 mg) was dispersed into 20 mL of ethanol, and the suspension was sonicated for 10 min. Then, 40 mg of CeCl3 was added, and the mixture was stirred at room temperature for four days in darkness. The mixture was refluxed at 80 °C for 24 h. The yellow precipitated material was separated using centrifugation, and it was washed with ethanol and acetone. Finally, the material was dried at 50 °C for 24 h. This compound was named UiO-66-Pyca-Ce (III).
Catalyst reaction
General method for one-pot synthesis of polyhydroquinoline derivatives (5a-f), to assess the catalytic activity of the UiO-66-Pyca-Ce (III)
To investigate the catalytic activity of UiO-66-Pyca-Ce (III) in the one-pot synthesis of polyhydroquinoline derivatives under ambient temperature conditions, several substituted aldehydes (1 mmol), dimedone (1 mmol), ammonium acetate (2 mmol), and ethyl acetoacetate (1 mmol) were stirred inside a flask in the presence of UiO-66-Pyca-Ce (III) (0.001 g) as a catalyst and ethanol as a green solvent. A 5:1 n-hexane/ethyl acetate ratio was used to monitor the reaction’s progress using the thin layer chromatography (TLC). Once the reaction was complete, the catalyst was separated through filtration and then washing. Additionally, to purify the received product, each individual product was recrystallized in ethanol.
Results and discussion
The Zr6O4(OH)4(BDC-NH2)6 clusters that make up the UiO-66-NH2 metal–organic framework which resulting a 3D structure, has special properties like high surface area (1187 m2/g–1), good stability in wide variety of solvents, acidic and basic aqueous media (pH values ranging from 1 to 9), and thermal stability51. Through post-synthetic modification (PSM), it is possible to create functionalized MOFs with multiple active sites34,52. The high activity and good recyclability of these heterogeneous catalysts have shown that MOFs are an appropriate catalytic support. In this study, zirconium (IV) chloride, 2-amino-1,4-benzene dicarboxylic acid, and N, N-dimethylformamide (DMF) were used in a solvothermal reaction to create UiO-66-NH2. Three Lewis acid activating functions as a catalyst’s special quality, help to promote the asymmetric synthesis of polyhydroquinoline derivatives at ambient temperature. The structural confirmation of UiO-66-Pyca-Ce (III) were interpreted using various types of analysis. The functional groups present in the compound structure were identified using the FTIR spectrum. The morphology and elemental composition were determined using FE-SEM images and EDX analysis, respectively. The crystalline phase of the material was analyzed using XRD, and its thermal stability was assessed through TGA analysis. In order to examine the surface and porosity of UiO-66-Pyca-Ce (III), BET analysis was also carried out. Additionally, ICP analysis was performed to identify the elements present in the composition and determine their concentration.
Analysis
XRD patterns of UiO-66-NH2, UiO-66-Pyca, and UiO-66-Pyca-Ce (III)
One benefit of crystalline materials is that the crystallinity and structural integrity can be assessed using XRD after post-synthesis modification. Figure 3a–c shows the XRD patterns of UiO-66-NH2, UiO-66-Pyca, and UiO-66-Pyca-Ce as they were synthesized using the solvothermal method. The UiO-66-NH2 metal–organic framework's XRD pattern is consistent with those mentioned in references49,53. The compound's high crystallinity is also indicated by the presence of sharp peaks. The XRD patterns of UiO-66- Pyca and UiO-66-Pyca-Ce (Fig. 3b and c) show that the post-synthesis modification process did not alter the UiO-66- NH2 structure.
FTIR analysis of UiO-66-NH2, UiO-66-Pyca, and UiO-66-Pyca-Ce (III)
Figure 2 displays the FTIR spectra of the synthetic UiO-66-NH2, UiO-66-Pyca, and UiO-66-Pyca-Ce (III) compounds. The bands associated with the bending vibrations of N–H, the asymmetric and symmetric stretching vibrations of carboxylate (COO–), and the C–N stretching vibration of aromatic amines are found at 1568, 1629, 1386, and 1256 cm–1, respectively, in the FTIR spectrum of UiO-66-NH2 (Fig. 2a), which is consistent with the knowledge gleaned from prior studies49. A new band is seen at 1687 cm–1 in the FTIR spectra of UiO-66-Pyca and UiO-66-Pyca-Ce (III) (Fig. 2b and c), which can be attributed to the stretching vibration of the C=N bond caused by imine condensation of amino groups with the carbonyl group of pyridinecarbaldehyde, indicating the successful modification of the structure49. In the FTIR spectrum of UiO-66-Pyca-Ce (III), the bands associated with the asymmetric and symmetric stretching vibrations of N-Ce are observed at 360 and 460 cm–1, respectively, which are in agreement with the reported values in the literature54. To observe the peaks related to cerium, the spectrum in the range below 400 cm–1 should be checked. The closest vibration, VCe-Cl, appears at (356, 339, 343, and 336 cm–1) (m), while the tensile vibration VCe-N and the flexural vibration due to Cl are in the same band at (145, 148, 150, and 142 cm–1) (Fig. 2d)54.

FTIR spectra of (a) the as-synthesized UiO-66-NH2, (b) UiO-66-Pyca, (c) UiO-66-Pyca-Ce (III) and (d) The Far IR spectrum of UiO-66-Pyca-Ce (III).
ICP and TGA of UiO-66-Pyca-Ce (III)
Two main weight losses were discovered during the thermal analysis of the UiO-66-Pyca-Ce (III) catalyst, which were supported by TGA-DTA curves (Fig. 3d). The first mass loss started when the temperature reached 120 °C. The solvent molecules trapped in the holes and the water molecules absorbed on the surface of the MOF are responsible for this 20% decrease, which results in a broad endothermic peak in this region of the DTA curve. The second 20% drop was noticed in the 120 to 500 °C temperature range. The presence of aliphatic groups, unreacted molecules, the breakdown of organic compounds, and cerium species are thought to be the causes of this reduction. Water molecules that were guests in big cages have also relaxed, which is another reason for the decrease. The effect of this reduction can also be seen in the DTA curve and the strength of its exothermic peak. Decomposition of the framework occurs at a temperature higher than 550 °C, whereas UiO-66-Pyca-Ce (III) exhibits thermal stability up to 500 ℃. Inductively coupled plasma-atomic emission spectrometry (ICP-AES) was also performed for UiO-66-Pyca-Ce (III), which revealed that the resulting compound contained 4.350 mol% Ce and 30.372 mol% Zr.

XRD patterns of (a) UiO-66-NH2, (b) UiO-66-Pyca, (c) UiO-66-Pyca-Ce (III) and (d) TGA-DTA curves of the UiO-66-Pyca-Ce (III) catalyst.
SEM images and elemental mapping analysis
SEM images of UiO-66-Pyca-Ce (III) reveal that the catalyst’s structure remains unchanged after the modification during the functionalization process. Figure 4 demonstrates that the crystal size distribution appears uniform (Fig. 4a). Additionally, energy-dispersive X-ray (EDX) analysis confirms the presence of cerium in UiO-66-Pyca-Ce (III) (Fig. 4b). Figure 4c demonstrates the elemental mapping analysis of the UiO-66-Pyca-Ce (III) catalyst. This figure further confirms the continuous dispersion of Ce, Zr, N, O, and C elements on the catalyst.

(a) SEM images of UiO-66-Pyca-Ce (III, (b) EDX analysis of UiO-66-Pyca-Ce (III) and (c) The elemental mapping analysis of the UiO-66-Pyca-Ce (III).
BET analysis
The nitrogen adsorption–desorption isotherm of UiO-66-Pyca-Ce (III) is shown in Fig. 5. As seen in the Figure; each sample exhibits a type I isotherm. The Langmuir surface area of UiO-66-Pyca-Ce (III) was determined to be 501.63 m2/g, with a pore volume of 0.28 cm3/g and a pore size of 2.27 nm. These results indicate that a relatively high surface area and the presence of pores are necessary for constructing an effective catalyst.

BET analysis of UiO-66-Pyca-Ce (III).
Catalytic application evaluation of UiO-66-Pyca-Ce (III)
Optimization of factors affecting the one-pot synthesis of polyhydroquinolines derivatives
To identify the ideal reaction conditions, a number of variables affecting the one-pot, four-component condensation reaction of polyhydroquinoline derivatives were examined (Table 1, entries 1–10). The model reaction consisted of a mixture of benzaldehyde (1 mmol), ethyl acetoacetate (1 mmol), dimedone (1 mmol), and ammonium acetate (2 mmol), performed at ambient temperature. Initially, without UiO-66-Pyca-Ce (III) present, no significant progress was observed in the reaction (Table 1, entry 1). Subsequently, the effect of different parameters such as solvent type, reaction time, reaction conditions, and the amount of UiO-66-Pyca-Ce (III) was investigated (Table 1, entries 2–10). Under fixed conditions with UiO-66-Pyca-Ce (III), the model reaction was conducted at room temperature and under reflux conditions (Table 1, entries 2–3). It was found that the ambient temperature provided optimal reaction conditions, resulting in shorter reaction times and higher efficiency (Table 1, entry 3). Additionally, using ethanol instead of water as the reaction's solvent produced noticeably better results (Table 1, entry 3). The model reaction was performed with varying amounts of UiO-66-Pyca-Ce (III), and based on the results, the most optimal amount was determined to be 0.01 g (Table 1, entry 3), considering that a lower value is preferable. Therefore, based on these investigations, it was determined that conducting the model reaction at room temperature, using ethanol as the solvent, and using 0.01 g of UiO-66-Pyca-Ce (III) were the best conditions.
To generalize the optimal reaction conditions and evaluate the performance and yield of UiO-66-Pyca-Ce (III) as a catalyst, an extensive range of substituted aldehydes (1 mmol) with both electron-donating and electron-withdrawing substituents, along with dimedone (1 mmol), ethyl acetoacetate (1 mmol), and ammonium acetate (2 mmol), were used (Table 1, entries a-f). Diverse substituted polyhydroquinoline derivatives (5a-f) were synthesized using 0.01 g of UiO-66-Pyca-Ce (III) at room temperature. The outcomes shown in Table 2 resulted that the desired products were synthesized in a brief amount of time with a high yield. Based on the relevant results, it can be deduced that the UiO-66-Pyca-Ce (III), at room temperature, resulted a successful synthesis of products with a high yield in a short amount of time.
Assessment of UiO-66-Pyca-Ce (III) in the synthesis of polyhydroquinoline derivatives in comparison with other introduced researches
In Tables 1 and 2, the obtained results are presented to evaluate the efficiency and performance of UiO-66-Pyca-Ce (III) in the synthesis of polyhydroquinoline derivatives by replacing a wide range of aldehydes (electron-withdrawing or electron-donating). In order to evaluate the effectiveness and performance of UiO-66- Pyca-Ce (III), Table 3 compares UiO-66- Pyca-Ce (III) with other prior catalysts for the synthesis of polyhydroquinoline derivatives. For this, a number of variables have been taken into account, such as the quantity of catalyst, type of solvent, temperature during the reaction, reaction time, and the proportion of isolated products. The results are summarized in Table 3, entries 1–5. In addition to the advantages, the reported data have certain disadvantages, including the high cost of the catalyst used, toxic and volatile solvents, harsh reaction conditions such as long reaction times and high temperatures, and low product yields (Table 3, entries 1–4). However, compared to prior studies, UiO-66-Pyca-Ce (III) demonstrates unique and distinct advantages. Its proficiency allows for synthesizing polyhydroquinoline derivatives with a lower catalyst dosage, shorter reaction times, and high efficiency (Table 3, entry 5).
Proposed mechanism for synthesis of polyhydroquinoline derivatives
This study identified UiO-66-Pyca-Ce (III) as the primary catalytic agent driving the synthesis reaction of polyhydroquinoline derivatives forward. The proposed mechanism shown in Fig. 6 can be explained in two different ways, according to earlier studies. In the first method, intramolecular interactions between the substituted aldehyde and the carbonyl group of ethyl acetoacetate activate each other. This leads to a Knoevenagel condensation reaction between the activated substituted aldehyde (1) and the enol dimedone (3), resulting in the formation of intermediate (I). Simultaneously, intermediate (II) is formed from the reaction between activated ethyl acetoacetate (2) and ammonium acetate (4). Finally, a Michael addition reaction occurs between intermediates (I) and (II), followed by cyclization and elimination of water, leading to the desired polyhydroquinoline derivatives (5a-f). In the second method, intermediate (III) is initially created in the Knoevenagel reaction between the active aldehyde (1) and the enol form of ethyl acetoacetate (2). Intermediate (IV) is also produced by the reaction between activated dimedone (3) and ammonium acetate. Similarly, to the first method, a Michael addition reaction occurs between intermediates (III) and (IV), followed by cyclization and elimination of water, resulting in the desired polyhydroquinoline derivatives (5a-f). For further explanation, it can be said that the synthesized catalyst is an acidic catalyst, which generally activates electrophiles. In the case of aldehydes, either cerium or zirconium with vacant orbitals will perform this activation. They coordinate with the oxygen of the aldehyde, weakening the carbonyl double bond. Alternatively, the oxygen of the aldehyde forms a hydrogen bond with COOH groups, leading to the weakening of the carbonyl double bond. The weakened carbonyl then becomes vulnerable to attack. These events happen for ethyl acetoacetate and dimedone in both the first and second routes, and are followed by a tautomerization procedure that adds a double bond. The double bond has the potential to engage in nucleophilic attack on the activated aldehyde. In the subsequent steps, the dimedone and/or ethyl acetoacetate undergo similar activation processes, and ammonium acetate attacks the activated species.

Proposed mechanism for the synthesis of polyhydroquinoline derivatives in the presence of the UiO-66-Pyca-Ce (III).
Reusability assessment of UiO-66-Pyca-Ce (III) after several catalytic performances
Recyclability and reuse of catalysts, whether in industrial applications or the discussion of green chemistry, are essential and vital for developing of chemical reactions. Therefore, in this study, the reusability of UiO-66-Pyca-Ce (III) in the synthesis of polyhydroquinoline derivatives was investigated. After each reaction, the catalyst was detached through washing and centrifugation, and then dried. The results are depicted in Fig. 7, and they show that the catalyst can used as a qualified catalyst even after being used five times without significantly reducing the efficiency of the desired products.

Reusability study of UiO-66-Pyca-Ce (III).
Conclusions
In this study, a catalyst was fabricated using a solvothermal reaction based on zirconium MOF that had been modified with pyridine carboxaldehyde and cerium metal. UiO-66-Pyca-Ce (III) was created and introduced as an effective catalyst with noteworthy features for the synthesis of heterocyclic compounds. The structural properties of this catalyst were assessed through various analyses, including XRD, TGA, FE-SEM, ICP, and BET. Verification of the presence of functional groups, analysis of its main constituent elements, and their dispersion were performed through FT-IR, Far IR, and EDX analysis, respectively. This catalyst demonstrated high efficiency in producing the desired product in the shortest amount of time at room temperature, without requiring special conditions, for a number of reasons, including three Lewis acid activating functions. These features show promising potential for the manifestation of a highly efficient catalyst, with a yield of 90%, particularly in the asymmetric synthesis of polyhydroquinoline derivatives. In addition to the benefits above, considering the importance of catalyst recovery, reusability in the industry, and its impact on the environment, the efficiency of the produced catalyst was evaluated multiple times. The results demonstrated the catalyst's pleasant stability and performance in reuse.
Data availability
The datasets generated and/or analyzed during the current study are available at the http://www.crystallography.net/cod/4348132.cif.
References
Domling, A., Wang, W. & Wang, K. Chemistry and biology of multicomponent reactions. Chem. Rev. 112, 3083–3135 (2012).
Maleki, A., Panahzadeh, M. & Eivazzadeh-keihan, R. Agar: A natural and environmentally-friendly support composed of copper oxide nanoparticles for the green synthesis of 1, 2, 3–triazoles. Green Chem. Lett. Rev. 12, 395–406 (2019).
Zhu, J. & Bienaymé, H. Multicomponent Reactions (Wiley, 2006).
Maleki, A., Taheri-Ledari, R., Eivazzadeh-Keihan, R., de la Guardia, M. & Mokhtarzadeh, A. Preparation of carbon-14 labeled 2-(2-mercaptoacetamido)-3-phenylpropanoic acid as metallo-beta-lactamases inhibitor (MBLI), for coadministration with beta-lactam antibiotics. Curr. Org. Synth. 16, 765–771 (2019).
Eivazzadeh-Keihan, R. et al. Synthesis of core-shell magnetic supramolecular nanocatalysts based on amino-functionalized calix [4] arenes for the synthesis of 4H-chromenes by ultrasonic waves. ChemistryOpen 9, 735–742 (2020).
Eivazzadeh-Keihan, R., Bahrami, S., Ghafori Gorab, M., Sadat, Z. & Maleki, A. Functionalization of magnetic nanoparticles by creatine as a novel and efficient catalyst for the green synthesis of 2-amino-4H-chromene derivatives. Sci. Rep. 12, 1–12 (2022).
Hajizadeh, Z., Radinekiyan, F., Eivazzadeh-Keihan, R. & Maleki, A. Development of novel and green NiFe2O4/geopolymer nanocatalyst based on bentonite for synthesis of imidazole heterocycles by ultrasonic irradiations. Sci. Rep. 10, 1–11 (2020).
Nasr-Esfahani, M., Hoseini, S. J., Montazerozohori, M., Mehrabi, R. & Nasrabadi, H. Magnetic Fe3O4 nanoparticles: Efficient and recoverable nanocatalyst for the synthesis of polyhydroquinolines and Hantzsch 1, 4-dihydropyridines under solvent-free conditions. J. Mol. Catal. A Chem. 382, 99–105 (2014).
Yadav, D., Patel, R., Srivastava, V., Watal, G. & Yadav, L. LiBr as an efficient catalyst for one-pot synthesis of Hantzsch 1, 4-dihydropyridines under mild conditions. Chin. J. Chem. 29, 118–122 (2011).
Xia, J.-J. & Zhang, K.-H. Synthesis of N-substituted acridinediones and polyhydroquinoline derivatives in refluxing water. Molecules 17, 5339–5345 (2012).
Sarkar, R. & Mukhopadhyay, C. Cross-dehydrogenative regioselective Csp 3–Csp 2 coupling of enamino-ketones followed by rearrangement: An amazing formation route to acridine-1, 8-dione derivatives. Org. Biomol. Chem. 14, 2706–2715 (2016).
Kagne, R. P., Nikam, G. H., Kalalawe, V. G., Niwadange, S. N. & Munde, D. R. An efficient protocol for synthesis of 1, 4-dihydropyridine derivatives by using graphene oxide nano particles as a catalyst. J. Chem. Chem. Sci. 7, 1064–1070 (2017).
Hantzsch, A. Synthese von thiazolen und oxazolen. Ber Dtsch. Chem. Ges. 21, 942–946 (1888).
Wu, M., Feng, Q., Wan, D. & Ma, J. CTACl as catalyst for four-component, one-pot synthesis of pyranopyrazole derivatives in aqueous medium. Synth. Commun. 43, 1721–1726 (2013).
Khodja, I. A. et al. Solvent-free synthesis of dihydropyridines and acridinediones via a salicylic acid-catalyzed Hantzsch multicomponent reaction. Synth. Commun. 44, 959–967 (2014).
Yaghi, O. M. et al. Reticular synthesis and the design of new materials. Nature 423, 705–714 (2003).
Kitagawa, S., Kitaura, R. & Noro, S. I. Functional porous coordination polymers. Angew. Chem. Int. Ed. 43, 2334–2375 (2004).
Férey, G. Hybrid porous solids: Past, present, future. Chem. Soc. Rev. 37, 191–214 (2008).
Mueller, U. et al. Metal–organic frameworks—Prospective industrial applications. J. Mater. Chem. 16, 626–636 (2006).
Horcajada, P. et al. Metal–organic frameworks as efficient materials for drug delivery. Angew. Chem. Int. Ed. 118, 6120–6124 (2006).
Allendorf, M. D., Bauer, C. A., Bhakta, R. & Houk, R. Luminescent metal–organic frameworks. Chem. Soc. Rev. 38, 1330–1352 (2009).
Jiang, L. et al. Structures and fluorescence properties of two novel metal-organic frameworks based on the bis (2-benzimidazole) and aromatic carboxylate ligands. Inorg. Chem. Commun. 14, 1077–1081 (2011).
Lu, G. & Hupp, J. T. Metal−organic frameworks as sensors: A ZIF-8 based Fabry−Pérot device as a selective sensor for chemical vapors and gases. J. Am. Chem. Soc. 132, 7832–7833 (2010).
Wang, H. et al. The moisture-triggered controlled release of a natural food preservative from a microporous metal–organic framework. Chem. Commun. 52, 2129–2132 (2016).
Britt, D., Tranchemontagne, D. & Yaghi, O. M. Metal-organic frameworks with high capacity and selectivity for harmful gases. Proc. Natl. Acad. Sci. U.S.A. 105, 11623–11627 (2008).
Zadehahmadi, F. et al. Catalytic CO2 fixation using tin porphyrin supported on organic and inorganic materials under mild conditions. J. Mol. Catal. A Chem. 398, 1–10 (2015).
Burrows, A. D. et al. Subtle structural variation in copper metal-organic frameworks: Syntheses, structures, magnetic properties and catalytic behaviour. Dalton Trans. 47, 6788–6795 (2008).
Burrows, A. D., Fisher, L. C., Richardson, C. & Rigby, S. P. Selective incorporation of functional dicarboxylates into zinc metal–organic frameworks. Chem. Commun. 47, 3380–3382 (2011).
Zadehahmadi, F. et al. Manganese (III) tetrapyridylporphyrin-chloromethylated MIL-101 hybrid material: A highly active catalyst for oxidation of hydrocarbons. Appl. Catal. A Gen. 477, 34–41 (2014).
Kardanpour, R. et al. Highly dispersed palladium nanoparticles supported on amino functionalized metal-organic frameworks as an efficient and reusable catalyst for Suzuki cross-coupling reaction. J. Organomet. Chem. 761, 127–133 (2014).
Bhattacharjee, S., Lee, Y.-R., Puthiaraj, P., Cho, S.-M. & Ahn, W.-S. Metal–organic frameworks for catalysis. Catal. Surv. Asia 19, 203–222 (2015).
Zadehahmadi, F. et al. Highly efficient protection of alcohols and phenols catalysed by tin porphyrin supported on MIL-101. Appl. Organomet. Chem. 29, 209–215 (2015).
Wang, Z. & Cohen, S. M. Postsynthetic modification of metal–organic frameworks. Chem. Soc. Rev. 38, 1315–1329 (2009).
Tanabe, K. K. & Cohen, S. M. Postsynthetic modification of metal–organic frameworks—A progress report. Chem. Soc. Rev. 40, 498–519 (2011).
Das, S., Kim, H. & Kim, K. Metathesis in single crystal: Complete and reversible exchange of metal ions constituting the frameworks of metal−organic frameworks. J. Am. Chem. Soc. 131, 3814–3815 (2009).
Li, W., Li, G. & Liu, D. Synthesis and application of core–shell magnetic metal–organic framework composites Fe3O4/IRMOF-3. RSC Adv. 6, 94113–94118 (2016).
Jiang, D. et al. Size selectivity of a copper metal-organic framework and origin of catalytic activity in epoxide alcoholysis. Chem. A Eur. J. 15, 12255–12262 (2009).
Natarajan, S. & Mahata, P. Metal–organic framework structures–how closely are they related to classical inorganic structures?. Chem. Soc. Rev. 38, 2304–2318 (2009).
Lee, J. et al. Metal–organic framework materials as catalysts. Chem. Soc. Rev. 38, 1450–1459 (2009).
Shultz, A. M., Farha, O. K., Hupp, J. T. & Nguyen, S. T. A catalytically active, permanently microporous MOF with metalloporphyrin struts. J. Am. Chem. Soc. 131, 4204–4205 (2009).
Lu, Y. et al. A cobalt (II)-containing metal-organic framework showing catalytic activity in oxidation reactions. Z. Anorg. Allg. Chem. 634, 2411–2417 (2008).
Proch, S. et al. Pt@ MOF-177: Synthesis, room-temperature hydrogen storage and oxidation catalysis. Chem. Eur. J. 14, 8204–8212 (2008).
Ingleson, M., Barrio, J., Guilbaud, J. & Khimyak, Y. MJ 85 Rosseinsky. Chem. Commun. 23, 2680 (2008).
Wu, C.-D., Hu, A., Zhang, L. & Lin, W. A homochiral porous metal− organic framework for highly enantioselective heterogeneous asymmetric catalysis. J. Am. Chem. Soc. 127, 8940–8941 (2005).
Li, W.-D.Z. & Peng, Y. An effective and highly stereoselective julia olefination of cyclopropyl carbinol mediated by CeCl3⊙ 7H2O/NaI. Org. Lett. 7, 3069–3072 (2005).
Bellucci, B. G. B. M., Marcantoni, E., Sambri, L. & Torregiani, E. Cerium(III) chloride catalyzed michael reaction of 1,3-dicarbonyl compounds and enones in the presence of sodium iodide under solvent-free conditions. Eur. J. Org. Chem. 1999, 617–620 (1999).
Kidwai, M. & Anwar, J. Cerium chloride (CeCl3·7H2O) as a highly efficient catalyst for one-pot three-component Mannich reaction. J. Braz. Chem. Soc. 21, 2175–2179 (2010).
Luan, Y., Qi, Y., Gao, H., Zheng, N. & Wang, G. Synthesis of an amino-functionalized metal–organic framework at a nanoscale level for gold nanoparticle deposition and catalysis. J. Mater. Chem. 2, 20588–20596 (2014).
Hou, J. et al. Synthesis of UiO-66-NH2 derived heterogeneous copper (II) catalyst and study of its application in the selective aerobic oxidation of alcohols. J. Mol. Catal. A Chem. 407, 53–59 (2015).
Pintado-Sierra, M., Rasero-Almansa, A. M., Corma, A., Iglesias, M. & Sánchez, F. Bifunctional iridium-(2-aminoterephthalate)–Zr-MOF chemoselective catalyst for the synthesis of secondary amines by one-pot three-step cascade reaction. J. Catal. 299, 137–145 (2013).
Kandiah, M. et al. Synthesis and stability of tagged UiO-66 Zr-MOFs. Chem. Mater. 22, 6632–6640 (2010).
Nguyen, J. G. & Cohen, S. M. Moisture-resistant and superhydrophobic metal−organic frameworks obtained via postsynthetic modification. J. Am. Chem. Soc. 132, 4560–4561 (2010).
Lakehal, I., Halaimia, F., Lakehal, S., Boukhari, A. & Djerourou, A. New cerium (III) complex of Schiff base (E)-N-benzylidene-4-methoxyaniline: synthesis and density functional theoretical study of vibrational spectra. Orient. J. Chem. 32, 903–919 (2016).
Sadeghi, S., Jafarzadeh, M., Abbasi, A. R. & Daasbjerg, K. Incorporation of CuO NPs into modified UiO-66-NH 2 metal–organic frameworks (MOFs) with melamine for catalytic C–O coupling in the Ullmann condensation. New J. Chem. 41, 12014–12027 (2017).
Asgharnasl, S., Eivazzadeh-Keihan, R., Radinekiyan, F. & Maleki, A. Preparation of a novel magnetic bionanocomposite based on factionalized chitosan by creatine and its application in the synthesis of polyhydroquinoline, 1, 4-dyhdropyridine and 1, 8-dioxo-decahydroacridine derivatives. Int. J. Biol. Macromol. 144, 29–46 (2020).
Maleki, A., Hassanzadeh-Afruzi, F., Varzi, Z. & Esmaeili, M. S. Magnetic dextrin nanobiomaterial: An organic-inorganic hybrid catalyst for the synthesis of biologically active polyhydroquinoline derivatives by asymmetric Hantzsch reaction. Mater. Sci. Eng. C 109, 110502 (2020).
Singh, H., Garg, N., Arora, P. & Rajput, J. K. Sucrose chelated auto combustion synthesis of BiFeO3 nanoparticles: Magnetically recoverable catalyst for the one-pot synthesis of polyhydroquinoline. Appl. Organomet. Chem. 32, e4357 (2018).
Saha, M. & Pal, A. K. Palladium (0) nanoparticles: An efficient catalyst for the one-pot synthesis of polyhydroquinolines. Tetrahedron Lett. 52, 4872–4877 (2011).
Hajjami, M. & Tahmasbi, B. Synthesis and characterization of glucosulfonic acid supported on Fe3O4 nanoparticles as a novel and magnetically recoverable nanocatalyst and its application in the synthesis of polyhydroquinoline and 2, 3-dihydroquinazolin-4 (1H)-one derivatives. RSC Adv. 5, 59194–59203 (2015).
Acknowledgements
The authors gratefully appreciate the partial support from the Research Council of the Iran University of Science and Technology.
Author information
Authors and Affiliations
Contributions
R.E.-K.: Substantial contributions to the conception, Design of the work, have drafted the work, Writing—Review & Editing, Analysis and interpretation of data and wrote the main manuscript. F.M.: Substantial contributions to the conception, Design of the work, have drafted the work, Writing—Review & Editing, Analysis and interpretation of data and wrote the main manuscript. Z.S.: Have drafted the work, Analysis and interpretation of data, substantively revised it. Wrote the main manuscript and prepared figures A.M.: The corresponding (submitting) author of current study, Substantial contributions to the conception, Design of the work, have drafted the work, Writing—Review & Editing, substantively revised it. A.T.: The corresponding author of current study, Substantial contributions to the conception, have drafted the work, substantively revised it.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moghadaskhou, F., Eivazzadeh-Keihan, R., Sadat, Z. et al. Fabrication and characterization of a novel catalyst based on modified zirconium metal-organic-framework for synthesis of polyhydroquinolines. Sci Rep 13, 16584 (2023). https://doi.org/10.1038/s41598-023-43869-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-43869-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
