
Abstract
Plastins, also known as fimbrins, are highly conserved eukaryotic multidomain proteins that are involved in actin-bundling. They all contain four independently folded Calponin Homology-domains and an N-terminal headpiece that is comprised of two calcium-binding EF-hand motifs. Since calcium-binding has been shown to be integral to regulating the activity of the three mammalian plastin proteins, we decided to study the properties of the headpiece regions of fimbrins from the model plant Arabidopsis thaliana, the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe and the amoeba Dictyostelium discoideum. Of these protein domains only the FimA headpiece from the amoeba protein possesses calcium binding properties. Structural characterization of this protein domain by multidimensional NMR and site-directed mutagenesis studies indicates that this EF-hand region of FimA also contains a regulatory ‘switch helix’ that is essential to regulating the activity of the human L-plastin protein. Interestingly this regulatory helical region seems to be lacking in the plant and yeast proteins and in fimbrins from all other nonmotile systems. Typical calmodulin antagonists can displace the switch-helix from the FimA headpiece, suggesting that such drugs can deregulate the Ca2+-regulation of the actin-bunding in the amoeba, thereby making it a useful organism for drug screening against mammalian plastins.
Similar content being viewed by others

Introduction
Plastins and fimbrins are a class of proteins that are found in all eukaryotes. They have uncommon, yet conserved, domain structures, containing four independently folded Calponin Homology (CH)-domains that are preceded by two Ca2+-binding EF-hand motifs1. It is well known that two adjacent CH-domains together make up an actin-binding motif. Hence, plastins and fimbrins contain two adjoining actin binding motifs, allowing them to bind two actin filaments simultaneously, leading to overall actin bundling2. The three human plastin proteins are expressed differentially in distinct tissues. In healthy individuals the L-plastin protein is normally only expressed in hematopoietic cells; it is also known as lymphocyte cytosolic protein (LCP-1)3,4. The T-plastin protein is expressed in all solid tissues, while the more recently discovered I-plastin protein is expressed selectively in the intestine and the kidney5. I-plastin is also found abundantly in vestibular hair cell stereocilia6. These three related proteins are expressed from genes that are located on different chromosomes, chromosome 3 (I-plastin), chromosome 13 (L-plastin) and chromosome X (T-plastin), respectively. Based on their extensive amino acid sequence homology and the similar organization of these three genes in introns and exons, it seems likely that the three proteins have arisen from a common ancestor. Interestingly, these three differentially expressed plastins are found amongst all vertebrates1. However, the plastins, which are also known as fimbrins, are conserved from lower eukaryotes to humans. Unique fimbrins have been found and characterized in the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe, that are called Sac6P7,8,9,10 and Fim111,12, respectively. Similar proteins have been found in Aspergillus nidulans13 and in pathogenic fungi such as Candida albicans14 and Cryptococcus neoformans15. Moreover, fimbrins have also been described in various plants, and the roles of the AtFim1, AtFim4 and AtFim5 proteins from the model plant Arabidopsis thaliana, have already been studied in some detail16,17,18,19,20.
The plastin and fimbrin proteins are multidomain proteins that are build-up of distinct independently folded units. They are comprised of two N-terminal calcium-binding EF-hand motifs that are followed by four CH-domains. This unique domain architecture, with four CH-domains, has not been found in any other proteins. CH-domains are independently folded highly helical structures, of about 120 amino acids, that are found approximately 90 times in the human genome. It is well-known that two tandem CH-domains together make up an actin binding motif as is seen in many other multidomain actin-binding proteins, such as actinin, spectrin and dystrophin for example21. Because all the plastins and fimbrins have four CH-domains they all contain two Actin Binding Domains (ABD) and hence they can bridge between two F-actin strands, thereby giving rise to actin bundling. It seems likely that all the plastins and fimbrins from lower eukaryotes to humans must play a role in actin bundling, as the four CH-domain architecture is strictly conserved across all species. Indeed, the crystal structure of the four CH-domains of the yeast and plant proteins shows that they are arranged in a horsehoe-like structure2, where the CH1 and CH4 domains interact with one another. The same compact structure for the four plastin CH-domains is maintained in aqueous solution as well, as demonstrated by small angle X-ray scattering studies22. Previous Cryo-EM studies have shown that this domain orientation could allow the protein to bind two actin strands in a parallel manner, giving rise to actin-bundling23,24,25,26,27. Furthermore, recent studies have proposed regulatory mechanisms for the actin-bundling via calcium-binding and/or protein phosphorylation28,29.
When the similarities between the chicken intestinal fimbrins and the human leukocyte plastins were originally recognized30, the two proteins were described as having domains that are homologous with calmodulin and actin-binding proteins. Calmodulin is a well-known calcium-regulatory protein that can bind four calcium ions and regulate the activity of many target proteins31. As a result, it was not totally unexpected when it was shown that L-plastin could bundle actin in a calcium-dependent manner, where increased calcium levels led to inhibition of the actin-bundling activity32. Although the two EF-hand motifs, that together would make up one half of calmodulin, are preserved in plastins from all species, several mutations can be found in the calcium binding loops of all plant and yeast proteins that would likely abolish effective calcium binding and hence it is predicted that these are not capable of binding calcium33. The solution structure of the EF-hand containing headpiece region of human L-plastin was recently determined by NMR spectroscopy34. The protein domain was folded like a single lobe of calmodulin, and similar to most regulatory EF-hand calcium-binding proteins35. In the apo-state the four helices that are contributed by the two helix-loop-helix EF-hands, are oriented in a parallel manner, and the structure undergoes a typical helix reorientation and overall opening following the binding of calcium. Intriguingly the target for this calcium-binding domain was not located in one of the four CH-domains, as had originally been expected, but it was located in the unstructured region between the two EF-hand domains and the first CH-domain. This region folds into a well-defined α-helix when it binds to the holo-form of the headpiece. Consequently, this regulatory region has been termed the switch helix, as it appears to be involved in switching the actin-bundling on-and-off. Recent studies have shown that the same mechanism, with a regulatory switch helix, also occurs in the human T-plastin headpiece, albeit at slightly different calcium concentrations36,37,38.
In this work we have studied the headpiece regions of plastins and fimbrins obtained from different species, such as yeasts, plants and the amoeba Dictyostelium discodeum33,39. The latter protein was included in our work because earlier reports had suggested that the amoeba FimA protein, like the animal plastins, was also regulated in a calcium-dependent manner40. Using NMR and CD spectroscopy, we could show that none of the four plant and yeast headpieces were responding to calcium, even though all constructs were well folded. In contrast, the headpiece region of the amoeba protein responded to calcium in a manner that resembled the results obtained with the human L-plastin protein. We subsequently analyzed the structure of the FimA headpiece in more detail and studied the interaction with its own switch helix. Taken together our results show that the amoeba FimA protein can be regulated in a similar calcium-dependent manner as the human plastins, making this organism a potentially suitable target for screening drug fragments and potential drugs directed towards inhibiting L-plastin-mediated cancer metastasis.
Results
Nuclear magnetic resonance (NMR) studies of fimbrin headpiece domains
Our sequence alignment shows that among the fimbrin headpiece regions of five different species including Saccharomyces cerevisiae (Sac6p), Schizosaccharomyces pombe (Fim1), Arabidopsis thaliana (AtFim1 and 5), and Dictyostelium discodeum (FimA), only FimA contains functional EF-hand calcium binding sites (Fig. 1)35. For example, the positions X, Y, Z provide side-chain oxygen atoms as ligands for the calcium, which typically come from aspartate residues. The glutamate residue found at the –Z position is also an important Ca2+ ligand. To confirm this notion, we have expressed and purified all five headpiece domains. All constructs are soluble and can be expressed in the cytoplasm of E. coli. 1H,15N HSQC NMR shows that all spectra were well dispersed, indicating the presence of a well-folded protein domain (Fig. 2). As expected from the amino acid sequences, upon addition of a saturating amount of calcium, no substantial chemical shift changes were observed in the HSQC spectra of AtFim1, AtFim5, Sac6p, and Fim1. Our results confirm that these headpiece domains form an independently folded helical domain but that they are not able to bind calcium ions with micromolar affinities and that they do not undergo the typical conformational change seen in most EF-hand proteins.

Sequence alignment of the headpiece domains of various fimbrins and human L-plastin. Identical residues are boxed in red. Similar residues are boxed in yellow and highlighted in red. A schematic drawing of the experimentally determined secondary structure elements of L-plastin is also included. The regions with coils and arrows indicate α-helices and β-sheets, respectively. The helices are labeled as in the text. The position of the key hydrophobic residue (F86) is indicated by an arrow. UniProtKB accession numbers are FimA, P54680; human L-plastin, P13796; Sac6p, C8Z545; Fim1, O59945; AtFim1, Q7G188; and AtFim5, Q9FKI0. The residues that serve as the calcium ligands in the EF-hand Ca2+-binding sites 1 and 2 are indicated by their coordination.

Overlaid 1H, 15N-HSQC spectra of AtFim1 (a), AtFim5 (b), Fim1 (c), Sac6p (d), FimA (e), and FimAN81Ter (f) in the presence (red) and absence (black) of Ca2+.
On the other hand, drastic structural changes, resembling those reported earlier for human L-plastin (LPL) and T-plastin (TPL) headpiece domains34,37, were observed with the headpiece region of FimA (Fig. 2e). Furthermore, our sequence analysis and secondary structure predictions indicate that the FimA handpiece domain, but not other fimbrin domains studied here, is followed by an extra alpha-helix (H5 in Fig. 1), which could correspond to the “switch-helix” region of human LPL that regulates actin-binding in Ca2+-dependent manner34. Considering the helical regulatory region and the two functional calcium-binding motifs, the FimA headpiece resembles the headpiece domain of human L-plastin (LPL-EF). This observation is in line with the similarities between FimA and LPL reported earlier40.
Structural characterization of the headpiece domain of FimA
After this initial characterization, we sought to further investigate the calcium regulatory mechanisms of the headpiece domain of FimA (here in after, FimA-EF). For NMR spectroscopy, we successfully expressed and purified the 13C, 15N-labeled FimA-EF (residues 1–93) and we could unambiguously assign all the backbone amide signals other than one proline residue in the HSQC spectrum of Ca2+-bound form (Fig. 3). In the apo-form, the signal from G25 was also not identified likely due to signal broadening. Note that considerably less signals are observed around 8 ppm in the spectrum of the Ca2+-bound form (Fig. 2e), indicating that the protein gains a more defined structure when Ca2+ is bound. By analyzing the chemical shift index (CSI) values along the protein backbone, we could determine the secondary structures of both the apo- and Ca2+-bound forms (Fig. 4a). FimA-EF has four α-helices (H1-H4) consisting of two helix-loop-helix EF-hand Ca2+-binding motifs in both forms. {1H}-15N heteronuclear NOE data shows that the unstructured C-terminal region is flexible in the apo-form (Fig. 4a). However, upon binding Ca2+, this region forms another stable α-helix (H5). This could also be observed by CD spectroscopy as an increased helical content (Supplementary Fig. S1), which was also reported for LPL-EF37. We then obtained the predicted model structure for FimA-EF from the AlphaFold database (Fig. 4b). The AlphaFold structure of FimA-EF is totally consistent with the secondary structures determined with our NMR data for the Ca2+-form. However, it is not in agreement with the apo-form since the H5 is present and interacts with the hydrophobic pocket of the main structure. It seems that the long H4 present in the apo-form needs to be partially unfolded to achieve this interaction (Fig. 4a), which is an effect that was not observed with LPL-EF34. Figure 4b also shows the superposition of the structures of FimA-EF and Ca2+-bound LPL-EF. Although the global sequence identity between FimA and human LPL is 48%, it is reduced to 27% when only the headpiece domains are compared. However, the positions of all the secondary structure elements are well conserved in the tertiary structures and the backbone RMSD for the regions of FimA-EF (4–57, 62–82, and 83–97) and L-plastin (2–55, 56–76, and 79–93) is only 4.32 Å. The most prominent differences between the two structures would be the orientation of H3 as well as the length of H5. The regulatory H5 helix of FimA (residues 84–90) is much shorter than that of LPL (residues 85–94). This is most likely due to the presence of a Pro residue at position 84 in FimA (Fig. 1). The key hydrophobic residue for the interaction between H5 and the main structure of FimA-EF appears to be the F86 residue that is deeply inserted into the hydrophobic pocket formed by the EF-hand motifs (Fig. 4b), which would correspond to the key hydrophobic F90 residue in human LPL (Fig. 1; Ishida et al.34). After this finding, we decided to generate the FimA-EF-F86A mutant for further investigations.

Assigned 1H, 15N-HSQC spectra of FimA in the presence of EDTA (top) and CaCl2 (bottom). The positions of a few signals that are invisible at this counter level are indicated by asterisks.

(a) Chemical shift index (CSI) and {1H}-15N heteronuclear NOE values are plotted as a function of the residue number in the presence of EDTA and CaCl2. The secondary structures predicted from the CSI are also displayed. (b) The predicted structure of FimA-EF was obtained from the AlphaFold database (left). The regions are colored according to a per-residue confidence score of very high (blue), confident (sky blue), low (yellow), and very low (orange). The sidechain of F86 is show. α-helices are labeled. The structures of FimA-EF (orange) and the headpiece domain of human L-plastin (sky blue; 5JOJ) are superposed (right). The Phe residues that serve as an anchor residue are displayed in both structures.
To further characterize the Ca2+-induced conformational changes of the EF-hand domain of FimA as well as its interaction with the H5 region, we also generated a truncated construct, FimA-EF-N81ter (residues 1–80) that omits the entire H5 region. Similar to FimA-EF, large spectral changes were observed in the HSQC spectrum upon Ca2+-binding (Fig. 2f). We performed the NMR chemical shift assignment for this construct in both the apo- and Ca2+-bound forms. Although the assignment of the Ca2+-bound form was completed without any ambiguity, that of the apo-form could not be completed due to several missing or very weak signals (Supplementary Fig. S2). By analyzing the CSI values, we could confirm that the secondary structures of Ca2+-bound FimA-EF-N81ter retains the same conformation as the FimA-EF despite the absence of H5 (Supplementary Fig. S3).
Ca2+-induced conformational changes monitored by fluorescence spectroscopy
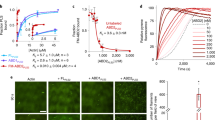
To further evaluate the Ca2+-induced conformational changes of the EF-hand domain, we performed ANS dye fluorescence experiments with FimA-EF, FimA-EF-N81ter, and FimA-EF-F86A. The fluorescence intensity of ANS is enhanced when it moves from aqueous solution to a non-polar environment, and therefore it can be utilized as a probe to detect hydrophobic surfaces of proteins41,42. In the absence of calcium ions, very little enhancement was observed for all three protein domains (Fig. 5). However, in the presence of calcium, the fluorescence intensity with FimA-EF-N81ter was drastically enhanced approx. 60 times, whereas the intensity of FimA-EF remains much like that seen in the presence of EDTA.

Ca2+-induced exposure of the hydrophobic surfaces of EF-hand domain monitored by ANS fluorescence. The steady-state fluorescence emission spectra of ANS in the absence (dotted line) and the presence of FimA-EF (dash-dotted line), FimA-EF-N81Ter (black solid line), FimA-EF-F86A (gray solid line). The spectra in the presence of EDTA are shown in the inset. All experiments were carried out at 25 °C.
From these results, it is reasonable to conclude that the EF-hand domain of FimA transforms from a closed state into an open-conformation in which the previously hidden hydrophobic pocket becomes exposed to the solvent, allowing it in turn to interact with the H5 region of the protein. This sequence of events is analogous to LPL34. No fluorescence enhancement is observed with FimA-EF since the H5 region already occupies the hydrophobic pocket and thereby blocks access for ANS. On the other hand, the FimA-EF-F86A mutant showed a fluorescence enhancement that is only slightly smaller than that of FimA-EF-N81ter regardless of the presence of H5. This result confirms that F86 on the H5 helix plays a major role in anchoring the H5 to the hydrophobic pocket of the FimA EF-hand domain. Clearly, by removing the phenyl ring of F86, we could totally disrupt this interaction.
Interaction between EF-hand domain and H5
Next, we have investigated the interaction between FimA-EF-N81ter and a 14-residue synthetic peptide which encompasses the H5 region of FimA-EF (see Methods section) using NMR techniques (Fig. 6). By titrating 15N-labeled FimA-EF-N81ter with the H5 peptide, we observed substantial chemical shift changes in the HSQC spectrum of FimA-EF-N81ter in a fast exchange manner on the NMR time scale, indicating a relatively weak interaction. We could determine the dissociation constant value by fitting the chemical shift changes to a Kd of 3.0 × 10–4 M, which is much weaker than expected from the previous report for LPL (3.5 × 10–6 M)34. This can likely be attributed to the much shorter H5 of FimA compared to that of LPL. When the chemical shift changes are mapped on the model structure, all influenced residues locate in or around the hydrophobic pocket, supporting the overall model structure (Supplementary Fig. S4).

The interactions between the EF-hand domain of FimA (FimA-EF-N81Ter) and the H5 peptide, W7, and TFP are monitored by 1H, 15N-HSQC. The chemical shift or peak volume changes of well separated signals are fitted to obtain dissociation constant (Kd) of binding.
Isothermal titration calorimetry (ITC) experiments
In order to study the calcium-binding properties of FimA, we used ITC to monitor the binding of calcium to the FimA constructs. Figure 7 shows the ITC isotherms obtained for the FimA-EF, FimA-EF-N81ter, and FimA-EF-F86A protein constructs. The ITC parameters derived from the curve fittings for all three constructs are summarized in Table 1. The isotherm obtained for the FimA-EF could be fit with one set of site model and produced a single Kd value of 0.6 µM and a stoichiometry of 1.7 (Fig. 7 and Table 1). Therefore, the binding of two calcium ions seem to be cooperative and the Kd value agrees with the previous observation that 200 µM calcium can almost fully turn off the actin-bundling activity of this protein40. On the other hand, for the other two constructs a two sets of sites model was required to fit the data. Two distinct calcium binding events are clearly visible in their binding isotherms. The Kd values obtained for FimA-EF-N81ter were 0.5 and 3.7 µM. These results resemble those obtained for human LPL34, and it suggests that the H5 region of FimA also modulates its calcium binding properties. It seems that the H5 is required to achieve the cooperativity between the two binding sites by enhancing the calcium binding affinity of the weaker site to the low uM range. From our current data, it is uncertain which EF-hand motif is the one that is modulated by H5 as both calcium binding loops possess proper amino-acid sequence for calcium binding (Fig. 1 and35). Nonetheless, our NMR data show that the first EF-hand motif (EF1) undergoes substantial structural changes upon H5 peptide binding (Supplementary Fig. S4). This suggests, but does not prove, that EF1 is the weaker site and requires the enhancement by the H5 binding to gain functional calcium affinity. We obtained essentially similar results for the FimA-EF-F86A construct with two distinct Kd values of 0.4 and 3.0 µM. This agrees with our ANS fluorescence results, showing that F86 is very important for the interaction between the H5 and the EF-hand domain.

Calorimetric titration of FimA-EF (a), FimA-EF-N81Ter (b), and FimA-EF-F86A (c). The baseline corrected ITC titrations (top panel) and the fitted binding isotherms (bottom panel) are shown.
Effect of calmodulin antagonists
Since the hydrophobic interaction between EF-hand domain and the H5 of FimA is rather weak, this interaction could potentially be disrupted by inhibitor drugs, which could lead to a deregulation of the Ca2+-sensitive actin-bundling of FimA. Therefore, we have tested the binding of two typical CaM antagonists, including W7 and TFP, to FimA-EF-N81ter. As expected from the similarity to the CaM domain, both drugs could bind to the FimA-EF-N81ter. The patterns of the chemical shift changes on the HSQC spectra upon binding to W7 and TFP are very similar to those observed with the H5 peptide (Fig. 6), indicating that these drugs bind to the same binding interface as the H5 peptide. However, W7 and TFP did bind with Kds of 3.8 × 10–6 M and 1.5 × 10–6 M, respectively (Fig. 6), which is two orders of magnitude stronger than the synthetic H5 peptide. This suggests that these drugs could possibly inhibit the interaction between the connected H5 region and the EF-hand domain of FimA. To confirm this, first, we titrated the H5 peptide into FimA-EF-N81ter in the presence of these drugs. No chemical shift changes were observed, indicating that the H5 peptide could not bind to the EF-hand domain as the binding interface is blocked by the drugs (Supplementary Fig. S5). Next, we titrated the FimA-EF construct with these drugs. Both TFP and W7 caused significant chemical shift changes in the HSQC spectrum of FimA-EF (Fig. 8). Particularly, the signals from the H5 region already disappeared when 0.5 molar equivalent of these drugs were added, indicating that the H5 region was dissociated from the EF-hand domain and no longer adopts a helical conformation. Thus, binding of W7 or TFP can displace the H5 region from the remainder of FimA-EF.

FimA-EF is titrated with TFP (top) or W7 (bottom). 1H, 15N-HSQC NMR spectra of FimA-EF with 0 (black), 0.5 (red), 1.0 (green), and 1.5 (blue) molar excess of TFP (a) or W7 (b) are overlaid. The signals from the H5 region are labeled.
Discussion
In this study, we have demonstrated that Dictyostelium discoideum FimA could function in a similar calcium regulatory manner as human LPL. Indeed, Dictyostelium discoideum has been used as a model organism to study dynamic actin filament organization as it is a highly motile organism and amenable to gene modifications43. Its genome contains 11 actin-bundling proteins44 and among them two are calcium regulatory proteins, namely FimA and a 34 kDa actin binding protein (ABP34). ABP34 also contains an N-terminal EF-hand domain which shows uM calcium affinity45 followed by three non-CH actin-binding domains46,47. Like FimA, Ca2+-binding to ABP34 negatively regulates the actin-bundling activity of the ABP34 protein48,49,50. Previously it has been shown that knockout mutation of one of these proteins did not alter the phenotype of the Dictyostelium discoideum amoeba, but that knocking out both proteins simultaneously resulted in abnormal phenotypes such as a small cell size and the formation of a small fruiting body51. Expressing a FimA construct that lacked the EF-hand domain did not rescue these phonotypes, suggesting that the Ca2+-regulated actin-bundling of FimA is important for these phenotypes. It seems that these two proteins (FimA and ABP34) have complementary roles in calcium regulated actin-bundling in the amoeba.
On the other hand, previous studies of the AtFim1, AtFim5 and yeast fimbrin proteins also indicate that these proteins do not respond to changes in the calcium levels in the micromolar range, and our NMR data confirmed that calcium binding does not occur and that calcium-dependent structural changes do not take place in these fimbrin EF-hand headpiece domains. Furthermore, the secondary structure predictions of these fimbrins as well as that of Tetrahymena fimbrin, which is also known to be insensitive to calcium52, do not find any evidence for the presence of an H5 region, suggesting that the presence of the H5, and the capacity to respond to changes in the calcium concentration go hand in hand. Interestingly, the headpiece domain is conserved in the vast majority of known fimbrins and plastins regardless of their functionality for Ca2+-binding1. Indeed, careful studies by Skau et al. indicate that plastins/fimbrins in yeasts and mammals play rather distinct roles53. Consequently, it seems that this region must play an important biological role. Be that as it may, it remains to be determined at a molecular level, what the exact role is for the non-calcium-binding headpiece regions that are found in several plastin and fimbrin proteins.
Our studies have demonstrated that Dictyostelium FimA can be regulated by calcium-binding to the EF-hand domain followed by the binding to the regulatory switch-helix (H5) in the same manner as the human LPL. Since Dictyostelium, leukocytes, and metastatic cancer cells all use amoeboid migration, this further supports the idea that the switch-helix is essential in a dynamic actin-bundling of fimbrins and plastins by responding to the calcium signalling that leads to the motility. LPL is known to be expressed ectopically during the transformation of a normal cell to a metastatic cancerous cell54,55,56,57,58,59 and it plays an important role in their motility. About 90% of cancer-related deaths is estimated to be caused by metastasis rather than by the original tumor. In recent years, various inhibitors towards fascin-1, another human actin-bundling protein that is also upregulated in metastatic cancerous cells have been synthesized (reviewed in60). On the other hand, to the best of our knowledge, inhibitors directly targeting LPL as a potential anti-metastatic drug have yet to be reported. We therefore suggest that this amoeba may become a valuable test organism for the development of novel drugs that aim to target the LPL involved in cancer metastasis.
Methods
The synthetic gene constructs for the expression of the headpiece regions from fimbrins from Arabidopsis thaliana (AtFim1; residues 1–114, and AtFim5; residues 1–113), Saccharomyces cerevisiae (Sac6p; residues 1–99), Schizosaccharomyces pombe (Fim1; residues 1–102), and Dictyostelium discoideum (FimA; residues 1–93) were obtained from GeneArt (ThermoFisher) with codon optimization for expression in Escherichia coli. The headpiece regions of these fimbrins were determined according to sequence alignments with the headpiece domain of L-plastin (Fig. 1). All constructs were designed to include the two EF-hands as well as the potential regulatory switch-helix region. All the genes were subcloned in the inducible pET15b vector (Invitrogen) using NdeI/XhoI sites, which contains an N-terminal 6His-tag followed by a TEV protease cleavage site. We have also generated two FimA mutants including FimA-EF-N81ter in which the last 12 amino-acids (the putative regulatory domain) are deleted and FimA-EF-F86A using site-directed mutagenesis. A synthetic peptide (Ac-SANASPAFASAVKK-NH2) encompassing the sequence of the putative regulatory domain from FimA was obtained from Genscript; Its purity was over 95% as shown by mass spectrometry and reverse phase HPLC analysis. TEV protease was expressed and purified form the pRK793 plasmid (Addgene) as previously described61.
Protein overexpression and purification
Overexpression of all the FimA constructs including FimA-EF, FimA-EF-N81Ter, and FimA-EF-F86A was achieved in BL21(DE3) E. coli cells, grown in 1 L of Luria Bertani (LB) medium with 100 μg/mL ampicillin at 37 °C. Once an optical density at 600 nm of approximately 0.6 was reached, cultures were induced with 1.0 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) for 4 h. For uniform isotope labelling of the constructs, expression was conducted in M9 minimal media, containing 0.5 g/L 15NH4Cl and 3 g/L 13C-glucose or 12C-glucose. Bacterial cells were harvested by centrifugation, and subsequently resuspended in buffer containing 20 mM Tris (pH 8), 100 mM NaCl, and 50 mM imidazole. The suspension was then homogenized and cells were lysed via French Press. Cell particulate matter was pelleted through centrifugation at 38,000×g for 45 min. The supernatant was then loaded onto an IMAC column (Cytiva), washed with the above resuspension buffer, and the purified protein was washed off with buffer containing 20 mM Tris (pH 8.0), 100 mM NaCl, and 300 mM imidazole. The protein is then dialyzed overnight in 4 L of buffer containing 20 mM Tris (pH 7.5), 100 mM NaCl, 1 mM β-Mercaptoethanol, and 0.5 mM EDTA. The N-terminal His-tag was cleaved using Tobacco Etch Virus (TEV) protease in the presence of 1 mM dithiothreitol (DTT) following dialysis for 1 h at 34 °C. Following cleavage, the mixture is loaded onto a cOmplete column (Roche) equilibrated with 20 mM Tris (pH 7.5) and 100 mM NaCl. The FimA-EF construct is collected in the flow-through, leaving the cleaved His-tag and TEV protease bound to the column. FimA-EF was prone to degradation during purification and necessitated further purification using a reverse-phase high performance liquid chromatography (RP-HPLC) with a Protein-R column (Nacalai Tesque Inc., Kyoto, Japan) in order to separate the intact protein from the degraded species. Purity was confirmed via SDS-PAGE gel (Supplementary Fig. S6). All the protein concentrations were estimated using the molar extinction coefficient of peptide bonds at A214 nm62.
Nuclear magnetic resonance (NMR) spectroscopy
All NMR experiments were performed with a Bruker Avance 600 MHz NMR spectrometer operating at 25 °C. Main-chain NMR signal assignments for the apo and Ca2+-bound FimA-EF and FimA-EF-N81ter constructs were completed with two-dimensional 1H,15N-HSQC spectra as well as multidimensional HNCACB, CBCA(CO)NH, HN(CA)CO, HNCO, HNCA, and HN(CO)CA experiments. {1H}-15N heteronuclear NOE data were obtained using a 5 s train of 120° proton pulses. All NMR samples contained ~ 0.5 mM 15N or 13C,15N-labelled protein, 100 mM KCl, 20 mM Bis–Tris (pH 7.0), 10% D2O, 0.03% NaN3, 10 mM 2H-DTT, and 0.5 mM 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) supplemented with either 1 mM EDTA or 5 mM CaCl2. The DSS was used as the reference peak to obtain the 1H, 15N and 13C chemical shifts. All NMR spectra were processed using NMRPipe/NMRDraw63, and analyzed using NMRView software64. The CSP values were calculated as a weighted average chemical shift difference for the1H and 15N resonances, using the equation CSP = √(DHN)2 + (DN/5)2. Chemical Shift Index (CSI) values were calculated using the Cα and C’ chemical shifts65. The program MOLMOL was used to generate molecular graphics66.
Isothermal titration calorimetry (ITC)
ITC experiments were performed on a MicroCal VP-ITC microcalorimeter (Malvern) at 25 °C. 40 μM (or 100 μM) FimA in calcium-free 20 mM HEPES buffer pH 7.2, 100 mM KCl, and 1 mM TCEP HCl, was titrated with 1.2 mM CaCl2 solution in same buffer and salt components. Prior to each titration, the sample cell of the ITC was soaked in 5 mM EDTA solution, and was rinsed stringently afterwards with calcium free buffer which was prepared via 1 week incubation with Chelex chelating agent. Each protein sample was exchanged into calcium free buffer and passed through a Calcium Sponge S column (Life Technologies). Data was fitted using the one or two site model, as applicable, with the MicroCal Origin software to obtain dissociation constants (Kd), stoichiometry (n), and free enthalpy (ΔH) values.
Fluorescence spectroscopy
Fluorescence spectra were acquired at room temperature on a Varian Cary Eclipse spectrofluorimeter. Fluorescence experiments with 8-Anilinonaphthalene-1-sulfonic acid (ANS) were conducted with samples containing 60 µM ANS with and without 60 µM FimA-EF, FimA-EF-N81Ter, or FimA-EF-F86A at pH 7.5 in the presence and absence of Ca2+. The excitation wavelength was 370 nm and the emissions were recorded from 400 to 600 nm using excitation and emission slit widths of 5 and 10 nm, respectively.
Circular dichroism (CD) spectroscopy
CD spectra were collected on a Jasco J-810 spectropolarimeter (Jasco, Inc., Easton, Md.). All the samples contained 10 μM protein in 10 mM Tris buffer (pH 7.0) and 2 mM DTT. Far-UV CD spectra were collected between 260 and 185 nm in a 1 mm pathlength cuvette, using a 0.5 nm step size and a scanning speed of 100 nm/min. The bandwidth was set to 1 nm and the response time was set to 0.5 s. For each sample, 10 spectra were accumulated and averaged, then data smoothing in the Jasco Spectral Analysis tool was utilized to generate the final spectra.
Data availability
The datasets analyzed in this study are available from the corresponding author on reasonable request.
References
Delanote, V., Vandekerckhove, J. & Gettemans, J. Plastins: Versatile modulators of actin organization in (patho)physiological cellular processes. Acta Pharmacol. Sin. 26, 769–779 (2005).
Klein, M. G. et al. Structure of the actin crosslinking core of fimbrin. Structure 12, 999–1013 (2004).
Wang, C. et al. Actin-bundling protein L-plastin regulates T cell activation. J. Immunol. 185, 7487–7497 (2010).
Morley, S. C. The actin-bundling protein L-plastin: A critical regulator of immune cell function. Int. J. Cell Biol. 2012, 935173 (2013).
Lin, C. S., Shen, W., Chen, Z. P., Tu, Y. H. & Matsudaira, P. Identification of I-plastin, a human fimbrin isoform expressed in intestine and kidney. Mol. Cell Biol. 14, 2457–2467 (1994).
Shin, J. B. et al. Molecular architecture of the chick vestibular hair bundle. Nat. Neurosci. 16, 365–374. https://doi.org/10.1038/nn.3312 (2013).
Adams, A. E., Botstein, D. & Drubin, D. G. Requirement of yeast fimbrin for actin organization and morphogenesis in vivo. Nature 354, 404–408. https://doi.org/10.1038/354404a0 (1991).
Brower, S. M., Honts, J. E. & Adams, A. E. Genetic analysis of the fimbrin-actin binding interaction in Saccharomyces cerevisiae. Genetics 140, 91–101. https://doi.org/10.1093/genetics/140.1.91 (1995).
Adams, A. E. & Botstein, D. Dominant suppressors of yeast actin mutations that are reciprocally suppressed. Genetics 121, 675–683. https://doi.org/10.1093/genetics/121.4.675 (1989).
Drubin, D. G., Miller, K. G. & Botstein, D. Yeast actin-binding proteins: evidence for a role in morphogenesis. J. Cell Biol. 107, 2551–2561. https://doi.org/10.1083/jcb.107.6.2551 (1988).
Nakano, K., Satoh, K., Morimatsu, A., Ohnuma, M. & Mabuchi, I. Interactions among a fimbrin, a capping protein, and an actin-depolymerizing factor in organization of the fission yeast actin cytoskeleton. Mol. Biol. Cell 12, 3515–3526. https://doi.org/10.1091/mbc.12.11.3515 (2001).
Wu, J. Q., Bahler, J. & Pringle, J. R. Roles of a fimbrin and an alpha-actinin-like protein in fission yeast cell polarization and cytokinesis. Mol. Biol. Cell 12, 1061–1077. https://doi.org/10.1091/mbc.12.4.1061 (2001).
Upadhyay, S. & Shaw, B. D. The role of actin, fimbrin and endocytosis in growth of hyphae in Aspergillus nidulans. Mol. Microbiol. 68, 690–705. https://doi.org/10.1111/j.1365-2958.2008.06178.x (2008).
Zhang, B. et al. The Candida albicans fimbrin Sac6 regulates oxidative stress response (OSR) and morphogenesis at the transcriptional level. Biochim. Biophys. Acta 2255–2266, 2016. https://doi.org/10.1016/j.bbamcr.2016.06.002 (1863).
Chang, Y. C., Lamichhane, A. K. & Kwon-Chung, K. J. Role of actin-bundling protein Sac6 in growth of Cryptococcus neoformans at low oxygen concentration. Eukaryot Cell 11, 943–951. https://doi.org/10.1128/EC.00120-12 (2012).
Kovar, D. R., Staiger, C. J., Weaver, E. A. & McCurdy, D. W. AtFim1 is an actin filament crosslinking protein from Arabidopsis thaliana. Plant J. 24, 625–636. https://doi.org/10.1046/j.1365-313x.2000.00907.x (2000).
Kovar, D. R., Gibbon, B. C., McCurdy, D. W. & Staiger, C. J. Fluorescently-labeled fimbrin decorates a dynamic actin filament network in live plant cells. Planta 213, 390–395. https://doi.org/10.1007/s004250000494 (2001).
Wang, Y. S., Motes, C. M., Mohamalawari, D. R. & Blancaflor, E. B. Green fluorescent protein fusions to Arabidopsis fimbrin 1 for spatio-temporal imaging of F-actin dynamics in roots. Cell Motil. Cytoskelet. 59, 79–93. https://doi.org/10.1002/cm.20024 (2004).
Su, H. et al. Fimbrins 4 and 5 Act synergistically during polarized pollen tube growth to ensure fertility in Arabidopsis. Plant Cell Physiol. 58, 2006–2016. https://doi.org/10.1093/pcp/pcx138 (2017).
Ding, X., Zhang, S., Liu, J., Liu, S. & Su, H. Arabidopsis FIM4 and FIM5 regulates the growth of root hairs in an auxin-insensitive way. Plant Signal Behav. 13, e1473667. https://doi.org/10.1080/15592324.2018.1473667 (2018).
Gimona, M., Djinovic-Carugo, K., Kranewitter, W. J. & Winder, S. J. Functional plasticity of CH domains. FEBS Lett. 513, 98–106 (2002).
Shinomiya, H., Shinjo, M., Fengzhi, L., Asano, Y. & Kihara, H. Conformational analysis of the leukocyte-specific EF-hand protein p65/L-plastin by X-ray scattering in solution. Biophys. Chem. 131, 36–42 (2007).
Galkin, V. E., Orlova, A., Cherepanova, O., Lebart, M. C. & Egelman, E. H. High-resolution cryo-EM structure of the F-actin-fimbrin/plastin ABD2 complex. Proc. Natl. Acad. Sci. USA 105, 1494–1498 (2008).
Hanein, D., Matsudaira, P. & DeRosier, D. J. Evidence for a conformational change in actin induced by fimbrin (N375) binding. J. Cell Biol. 139, 387–396 (1997).
Hanein, D. et al. An atomic model of fimbrin binding to F-actin and its implications for filament crosslinking and regulation. Nat. Struct. Biol. 5, 787–792 (1998).
Rost, L. E., Hanein, D. & DeRosier, D. J. Reconstruction of symmetry deviations: a procedure to analyze partially decorated F-actin and other incomplete structures. Ultramicroscopy 72, 187–197 (1998).
Volkmann, N., DeRosier, D., Matsudaira, P. & Hanein, D. An atomic model of actin filaments cross-linked by fimbrin and its implications for bundle assembly and function. J. Cell Biol. 153, 947–956 (2001).
Mei, L. et al. Structural mechanism for bidirectional actin cross-linking by T-plastin. Proc. Natl. Acad. Sci. USA 119, e2205370119. https://doi.org/10.1073/pnas.2205370119 (2022).
Schwebach, C. L. et al. Allosteric regulation controls actin-bundling properties of human plastins. Nat. Struct. Mol. Biol. 29, 519–528. https://doi.org/10.1038/s41594-022-00771-1 (2022).
de Arruda, M. V., Watson, S., Lin, C. S., Leavitt, J. & Matsudaira, P. Fimbrin is a homologue of the cytoplasmic phosphoprotein plastin and has domains homologous with calmodulin and actin gelation proteins. J. Cell Biol. 111, 1069–1079 (1990).
Ishida, H. & Vogel, H. J. Protein-peptide interaction studies demonstrate the versatility of calmodulin target protein binding. Protein Pept. Lett. 13, 455–465 (2006).
Namba, Y., Ito, M., Zu, Y., Shigesada, K. & Maruyama, K. Human T cell L-plastin bundles actin filaments in a calcium-dependent manner. J. Biochem. 112, 503–507 (1992).
Friedberg, F. Single and multiple CH (calponin homology) domain containing multidomain proteins in Arabidopsis and Saccharomyces: an inventory. Mol. Biol. Rep. 38, 213–218. https://doi.org/10.1007/s11033-010-0097-z (2011).
Ishida, H., Jensen, K. V., Woodman, A. G., Hyndman, M. E. & Vogel, H. J. The calcium-dependent switch helix of L-plastin regulates actin bundling. Sci. Rep. 7, 40662. https://doi.org/10.1038/srep40662 (2017).
Gifford, J. L., Walsh, M. P. & Vogel, H. J. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem. J. 405, 199–221 (2007).
Schwebach, C. L., Agrawal, R., Lindert, S., Kudryashova, E. & Kudryashov, D. S. The roles of actin-binding domains 1 and 2 in the calcium-dependent regulation of actin filament bundling by human plastins. J. Mol. Biol. 429, 2490–2508. https://doi.org/10.1016/j.jmb.2017.06.021 (2017).
Miyakawa, T. et al. Different Ca(2)(+)-sensitivities between the EF-hands of T- and L-plastins. Biochem. Biophys. Res. Commun. 429, 137–141 (2012).
Khassan, O., Jensen, K. V., Woodman, A. G., Vogel, H. J. & Ishida, H. Characterization of the EF-hand calcium-binding domains of human plastins. Methods Mol. Biol. 245–260, 2019. https://doi.org/10.1007/978-1-4939-9030-6_16 (1929).
Friedberg, F. & Rivero, F. Single and multiple CH (calponin homology) domain containing multidomain proteins in Dictyostelium discoideum: An inventory. Mol. Biol. Rep. 37, 2853–2862. https://doi.org/10.1007/s11033-009-9839-1 (2010).
Prassler, J. et al. Interaction of a Dictyostelium member of the plastin/fimbrin family with actin filaments and actin-myosin complexes. Mol. Biol. Cell 8, 83–95 (1997).
LaPorte, D. C., Wierman, B. M. & Storm, D. R. Calcium-induced exposure of a hydrophobic surface on calmodulin. Biochemistry 19, 3814–3819. https://doi.org/10.1021/bi00557a025 (1980).
Ishida, H., Huang, H., Yamniuk, A. P., Takaya, Y. & Vogel, H. J. The solution structures of two soybean calmodulin isoforms provide a structural basis for their selective target activation properties. J. Biol. Chem. 283, 14619–14628. https://doi.org/10.1074/jbc.M801398200 (2008).
Schleicher, M. et al. Structure/function studies on cytoskeletal proteins in Dictyostelium amoebae as a paradigm. FEBS Lett. 369, 38–42. https://doi.org/10.1016/0014-5793(95)00579-x (1995).
Eichinger, L. et al. The genome of the social amoeba Dictyostelium discoideum. Nature 435, 43–57. https://doi.org/10.1038/nature03481 (2005).
Furukawa, R. et al. Calcium regulation of actin crosslinking is important for function of the actin cytoskeleton in Dictyostelium. J. Cell Sci. 116, 187–196. https://doi.org/10.1242/jcs.00220 (2003).
Lim, R. W., Furukawa, R., Eagle, S., Cartwright, R. C. & Fechheimer, M. Three distinct F-actin binding sites in the Dictyostelium discoideum 34,000 dalton actin bundling protein. Biochemistry 38, 800–812. https://doi.org/10.1021/bi981392d (1999).
Kim, M. K., Kim, J. H., Kim, J. S. & Kang, S. O. Structure of the 34 kDa F-actin-bundling protein ABP34 from Dictyostelium discoideum. Acta Crystallogr. D Biol. Crystallogr. 71, 1835–1849. https://doi.org/10.1107/S139900471501264X (2015).
Fechheimer, M. & Taylor, D. L. Isolation and characterization of a 30,000-dalton calcium-sensitive actin cross-linking protein from Dictyostelium discoideum. J. Biol. Chem. 259, 4514–4520 (1984).
Fechheimer, M. The Dictyostelium discoideum 30,000-dalton protein is an actin filament-bundling protein that is selectively present in filopodia. J. Cell Biol. 104, 1539–1551. https://doi.org/10.1083/jcb.104.6.1539 (1987).
Chung, J. M., Kim, H. U., Kim, G. J., Jeoung, D. & Jung, H. S. The actin bundling activity of actin bundling protein 34 is inhibited by calcium binding to the EF2. Biochem. Biophys. Res. Commun. 503, 1836–1840. https://doi.org/10.1016/j.bbrc.2018.07.122 (2018).
Pikzack, C., Prassler, J., Furukawa, R., Fechheimer, M. & Rivero, F. Role of calcium-dependent actin-bundling proteins: characterization of Dictyostelium mutants lacking fimbrin and the 34-kilodalton protein. Cell Motil. Cytoskelet. 62, 210–231. https://doi.org/10.1002/cm.20098 (2005).
Shirayama, S. & Numata, O. Tetrahymena fimbrin localized in the division furrow bundles actin filaments in a calcium-independent manner. J. Biochem. 134, 591–598. https://doi.org/10.1093/jb/mvg183 (2003).
Skau, C. T. et al. Actin filament bundling by fimbrin is important for endocytosis, cytokinesis, and polarization in fission yeast. J. Biol. Chem. 286, 26964–26977. https://doi.org/10.1074/jbc.M111.239004 (2011).
Leavitt, J., Goldman, D., Merril, C. & Kakunaga, T. Changes in gene expression accompanying chemically-induced malignant transformation of human fibroblasts. Carcinogenesis 3, 61–70 (1982).
Lin, C. S., Park, T., Chen, Z. P. & Leavitt, J. Human plastin genes. Comparative gene structure, chromosome location, and differential expression in normal and neoplastic cells. J. Biol. Chem. 268, 2781–2792 (1993).
Park, T., Chen, Z. P. & Leavitt, J. Activation of the leukocyte plastin gene occurs in most human cancer cells. Cancer Res. 54, 1775–1781 (1994).
Zheng, J. et al. Steroid hormone induction and expression patterns of L-plastin in normal and carcinomatous prostate tissues. Am. J. Pathol. 150, 2009–2018 (1997).
Foran, E., McWilliam, P., Kelleher, D., Croke, D. T. & Long, A. The leukocyte protein L-plastin induces proliferation, invasion and loss of E-cadherin expression in colon cancer cells. Int. J. Cancer 118, 2098–2104 (2006).
Samstag, Y. & Klemke, M. Ectopic expression of L-plastin in human tumor cells: diagnostic and therapeutic implications. Adv. Enzyme Regul. 47, 118–126 (2007).
Sarantelli, E., Mourkakis, A., Zacharia, L. C., Stylianou, A. & Gkretsi, V. Fascin-1 in cancer cell metastasis: Old target-new insights. Int. J. Mol. Sci. https://doi.org/10.3390/ijms241411253 (2023).
Kapust, R. B. et al. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 14, 993–1000 (2001).
Kuipers, B. J. H. & Gruppen, H. Prediction of molar extinction coefficients of proteins and peptides using UV absorption of the constituent amino acids at 214 nm to enable quantitative reverse phase high-performance liquid chromatography-mass spectrometry analysis. J. Agric. Food Chem. 55, 5445–5451. https://doi.org/10.1021/jf070337l (2007).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Johnson, B. A. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352 (2004).
Wishart, D. S. & Sykes, B. D. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4, 171–180. https://doi.org/10.1007/BF00175245 (1994).
Koradi, R., Billeter, M. & Wuthrich, K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph 14(51–55), 29–32 (1996).
Acknowledgements
This work was supported by a Discovery grant from the Natural Sciences and Engineering Research Council of Canada to HJV. We are indebted to Dr. Leonard T. Nguyen for making some valuable suggestions in the early stages of this work.
Author information
Authors and Affiliations
Contributions
H.J.V. conceived the study and supervised the research. H.I., A.G.W., and N.K. performed the experiments. All authors contributed to the experimental design. H.J.V. and H.I. prepared the manuscript. All authors contributed to correcting the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ishida, H., Woodman, A.G., Kitada, N. et al. The Dictyostelium discoideum FimA protein, unlike yeast and plant fimbrins, is regulated by calcium similar to mammalian plastins. Sci Rep 13, 16208 (2023). https://doi.org/10.1038/s41598-023-42682-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-42682-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
