
Abstract
The microbiota of the built environment is linked to usage, materials and, perhaps most importantly, human health. Many studies have attempted to identify ways of modulating microbial communities within built environments to promote health. None have explored how these complex communities assemble initially, following construction of new built environments. This study used high-throughput targeted sequencing approaches to explore bacterial community acquisition and development throughout the construction of a new build. Microbial sampling spanned from site identification, through the construction process to commissioning and use. Following commissioning of the building, bacterial richness and diversity were significantly reduced (P < 0.001) and community structure was altered (R2 = 0.14; P = 0.001). Greater longitudinal community stability was observed in outdoor environments than indoor environments. Community flux in indoor environments was associated with human interventions driving environmental selection, which increased 10.4% in indoor environments following commissioning. Increased environmental selection coincided with a 12% reduction in outdoor community influence on indoor microbiomes (P = 2.00 × 10–15). Indoor communities became significantly enriched with human associated genera including Escherichia, Pseudomonas, and Klebsiella spp. These data represent the first to characterize the initial assembly of bacterial communities in built environments and will inform future studies aiming to modulate built environment microbiota.
Similar content being viewed by others

Introduction
Humans spend roughly 87% of their time in indoor environments1. Substantial proportions of the remaining 13% of time is spent in other built environment (e.g. commuting in towns/cities). Recent studies have explored the composition of microbial communities in these built environments, specifically focusing on indoor air2,3 as well as common household and public surfaces4,5,6,7. These studies have shown distinct microbial communities across different built environments, even within individual buildings. Microbial colonization of these environments is related to space utilization8, materials used9 and ventilation strategies10.
As a product of these new indoor environments, the multitudes of microbes living around and inside of humans differ from those of our ancestors or those living in more rural environments11,12, which can impact on health. As such, the microbiome of the built environment has been widely studied in relation to long-term health13,14,15 and nosocomial infections16,17,18,19. In the same way our microbial neighbors can influence human health, human (or non-human) occupancy may also influence the microbial communities of the built environment10,20.
The concept of the microbiome as a malleable entity, which can be altered to reduce likelihood of disease onset, is not new8,21. However, to make informed interventions on built environment microbiotas to promote human health we must first develop an understanding of how these complex networks react to environmental change. Like any living system, microbial environments favour homeostasis22. Similarly to humans, the microbial communities of buildings develop over time. Unlike humans, buildings are not created in a sterile environment23. While others have explored the development of microbiota in hospital environments following public access24, no studies to date have explored the acquisition and longitudinal development of the built environment microbiota across the entire construction and commissioning process of a new building. Instead, studies have often focused on cross-sectional analyses between one built environment and another, identifying greater diversity in infrequently cleaned homes or homes where canines lived alongside humans5. While purely cross-sectional studies are well suited to discount the influence of environmental variables such as occupancy at sampling or cleaning regularity, they fail to capture the influence of seasonality on the microbial communities explored25. Community temporality is especially important when exploring the longitudinal development of built environment microbiota.
The OME is an experimental building in Newcastle upon Tyne, England. It was built as part of a Research England Expanding Excellence in England, funded Hub for Biotechnology in the Built Environment26, and facilitates microbial experimentation and architectural exhibition. In this study we employed longitudinal sampling to define timeframes of microbial acquisition in the built environment by conducting a temporal study at high sampling depth within a single, purpose-built building, The OME. We primarily aimed to quantify the time taken for stable microbial communities to engraft in newly erected structures and assess how commissioning the building influenced the microbial communities as specialist locations (e.g. kitchens & hallways) were used for their specific purpose.
Results
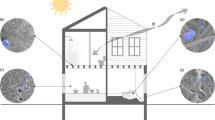
Over a study period of 36 months, 439 microbial community samples were collected from The OME site (Supplementary Table 1). Samples were taken from 8 different objects (soil, floor, shelving, doorframes, windowsills, doorpushes, splashbacks and sinks) spanning 7 sampling locations including outdoor (shrubs, borders and car park) and indoor (foyer, hallway, kitchen and bathroom) locations (Fig. 1A). Interrogating microbial communities longitudinally facilitated this study to account for changes in temperature, humidity and occupancy (using CO2 level as a proxy) throughout the construction process and over seasons. This enabled quantification of the true impact of increasing occupancy within the building after the build was commissioned (Fig. 1B).

illustrates the locations (A) and longitudinal nature (B) of sampling employed in this study. A 3D render of the OME building as well as ground and first floor floorplans illustrate sampling locations interrogated during this study (A). Longitudinal environmental data including relative humidity (blue), CO2 levels (green) and temperature (red) records collected as well as sample types taken at each sampling timepoint are also illustrated (B). Shaded areas around humidity, CO2 and temperature trendlines represent the 95% confidence interval associated with each environmental record (top panel). Each point on the lower panel represents an individual sampling timepoint, coloured by object and arranged by location on the y axis. The impact of Covid-19 on sampling continuation is apparent in the scarcity between spring 2020 and summer 2021. Humidity, temperature and CO2 records were only taken during sampling timepoints and were therefore not recorded during this same period.
High quality bacterial community sequences can be gained from swabbing surfaces
Sequencing of the V4 region of the 16S rRNA gene yielded a median 1.76 × 105 bacterial reads per sample (IQR: 4.64 × 103–2.58 × 105) and identified 568 different bacterial taxonomic units. Microbial samples had significantly greater library sizes than swab (median = 4.5; IQR = 3.25–5.75; P = 0.01), extraction kit (median = 2.9 × 103; IQR = 1.2 × 103–1.3 × 104; P = 0.003), and sequencing control samples (median = 38; IQR = 37–76: P = 0.0007) (Supplementary Fig. 1). Overall community composition of samples and controls were also significantly different (R2 = 0.45, P = 0.001).
Built environment bacterial communities show seasonality
Temperature, relative humidity and CO2 levels were significantly associated with alpha diversity measures of bacterial richness and Shannon diversity in microbial samples (P < 0.05; supplementary Fig. 1). Parabolic relationships were observed between temperature, humidity and alpha diversity. Lowest alpha diversity in microbial communities occurred at temperatures between 10 and 20 °C and relative humidity between 50 and 60%. Additionally, date of sampling was significantly associated with rarefied bacterial richness (P = 0.004) and diversity (P = 0.008).
Taken together, these phenomena could be due to seasonal changes in temperature and humidity, although differences in alpha diversity associated with temperature and humidity changes could be attributed to location. Microbial community compositions were determined from all sampling locations (Fig. 2A). Outdoor samples were taken at lower temperature and were similarly more diverse than indoor samples (Fig. 2B; Table 1). Unsurprisingly, bacterial diversity was highest in temperatures of 30 °C and 80% humidity, which are closest to ideal growth conditions for many host-associated microbes.

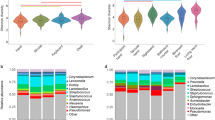
illustrates the overall community compositions of samples analysed in this study. Each tile in the heatmap represents an individual bacterial genus (A). Only genera with mean abundance across all samples > 0.5% were included in each panel and arranged along y axis by phylum. Samples were arranged on the x axis according to the sampling timepoint with earliest samples collected on the left and latest on the right within each panel. Intensity of tile colour indicates the relative abundance of the genus in that sample. Sample diversity and richness are illustrated as violin plots across locations (B) and timepoints (C), where individual samples are represented by points and violin width represents density of samples at that richness/diversity value. Sampling location is illustrated by the hue of points or heatmap tiles with outdoor sampling locations represented by darker purple hues and indoor locations represented by lighter yellow hues, as annotated in the heatmaps.
The impact of occupancy and materials is greater than seasonality on the built environment microbiota
Temperature and humidity changes between seasons may explain some of the longitudinal differences in bacterial richness and diversity observed. However, the impact of commissioning the building on both bacterial richness and diversity (P < 0.001), were greater than those of seasonality. Significantly reduced richness and diversity was observed after the building was commissioned (Fig. 2C).
In addition to seasonal environmental factors and commissioning of the building, sampling locations and object type (P < 0.001) were also significantly associated with rarefied bacterial richness (Fig. 2B). Outdoor locations were significantly richer and more diverse than indoor locations (Table 1). Bacterial communities of shrubs and borders specifically, were significantly more diverse than all indoor locations (P < 0.001).
Several bacterial genera commonly found in soils including Solirubrobacterales, Kaistobacter and Nocardioidaceae spp. were more abundant in outdoor environments than indoor environments, which harbored more human associated bacteria such as Klebsiella, Pseudomonas and Escherichia spp. (Fig. 2A).
Outdoor communities show greater stability over time
Compositions of bacterial communities were significantly associated with sampling location (R2 = 0.31; F = 31.99; P = 0.001) and timepoint (R2 = 0.16; F = 26.70; P = 0.001). The greatest community dissimilarity across locations was observed between outdoor and indoor sampling locations, all of which were significantly dissimilar (R2 > 0.32; P < 0.005; Fig. 3A,C).

Illustrates overall community structure of samples analysed in this study. Bray–curtis dissimilarity was used to ordinate samples across each of the sampling timepoints (A). Each point represents an individual sample and is coloured by location. Longitudinal sample dissimilarity is represented by line graphs (B) coloured by the same logic. Lines represent best-fit for inter-sample community similarity at each location with shaded areas representing the 95% confidence interval. Community dissimilarity between communities from all locations and over time are illustrated in bubble plots (C). Size of bubbles indicated dissimilarity between two comparators with significance indicated by colour of bubble outline. Raup-Crick similarity between samples was used to define impacts of community determinism on overall community structure (D). Bars represent the proportional impact of each phenomenon on samples collected, stratified by locations and timepoints. Influence of outdoor environments on the community structure of indoor communities was identified by SourceTracker (E). Sampling timepoint: Pre-comm. = Pre-commissioning; Post-comm. = Post-commisioning.
Indoor bacterial communities shared high similarity immediately following construction (Median = 0.96, IQR = 0.94–0.97; Fig. 3A,B) but became increasingly divergent over time, with many showing very little intra-location similarity following commissioning (Median = 0.05, IQR = 0.01–0.17; Fig. 3A,B). Conversely, outdoor communities showed far more temporal stability. While they were not as similar as all indoor communities immediately following construction, outdoor communities remained consistently similar throughout the sampling period (Median = 0.73, IQR = 0.62–0.81; Fig. 3A,B). The influence of sampling location and commissioning the building are further reflected in the within-group dissimilarity between sampling locations, where indoor bacterial communities were significantly more dissimilar than outdoor locations (P < 0.0001) (Supplementary Table 2).
Community differences between indoor and outdoor surfaces were largely due to the community flux observed in indoor communities over time. As such, indoor communities became increasingly dissimilar to outdoor communities following completion of construction and subsequent commissioning of the building (R2 = 0.47; P = 0.001; Fig. 3C). Prior to commissioning, bacterial genera were widely dispersed between outdoor and indoor sampling locations (defined as homogenising dispersal). Homogenising dispersal accounted for 74% of community determinism before commissioning but reduced to 34% after commissioning. By contrast, environmental factors shaping microbial communities (defined as environmental selection) increased from 0.3 to 10.5% following commissioning of the building (Fig. 3D). This was paired with a significant reduction in the mean proportional influence of outdoor microbiota on indoor communities from 17 to 0.004% (P = 2.04 × 10–15; Fig. 3E). Increased environmental selection can be attributed to human-associated factors which escalated after commissioning as locations around the building began to be used for their individual, specific purposes.
Soil-associated metabolic generalist bacteria are replaced by specialists and human associated bacteria as environmental selection increases
Increased environmental selection brings external factors which shape bacterial communities. Environmental selection increased after construction was completed and the building was commissioned in this study. Increased footfall in indoor environments along with introduction of specific cleaning and usage regimes had measurable effects on the taxonomic compositions observed across sampling locations (Fig. 4A,B). Klebsiella spp. are common, human-associated, enteric commensals that were significantly associated with indoor environments and pre-commissioning timepoints (Fig. 4B,C). Soil-associated Actinobacteria such as Solirubrobacterales and Gaiellaceaea spp. as well as the nitrogen fixing Bradyrhizobiaceae and the archaea Nitrososphaeraceae spp. were all significantly associated with outdoor environments while halotolerant Oceanospirillales spp. were significantly associated with pre-commissioning communities.

Significant associations of bacterial genera with locations and sampling timepoints. Genera included were significantly associated (Maaslin2 qval < 0.25) with at least one of the environmental variables tested. Only genera with a coefficient of variance > 0.005 are included in each panel. Tile colour indicates the Maaslin2 effect size of each environmental variable on genus proportional abundance (reduced = red; increased = blue). Sampling locations were included as fixed effects with ‘Shrubs’ as the comparator variable (A). Indoor communities were compared to outdoor communities (B) and pre-commissioning communities were compared to post-commissioning communities (C).
Discussion
This study represents the first to our knowledge to characterize the original acquisition and subsequent factors influencing community structure of any built environment microbiota to such depth. The study deliberately included a longitudinal sampling aspect to account for seasonality of microbial communities which we found to be significantly linked with temperature, humidity and CO2 concentrations. Furthermore, by collecting microbial samples within the built environment site prior to construction starting, we were able to benchmark any subsequent microbial communities with that of the indigenous or well-established microbes that preceded them. The importance of doing so became increasingly apparent upon analysing the data, where we highlight the relative stability of outdoor, pre-established, microbial communities by comparison to the highly turbulent indoor communities developing following completion of the structure.
A previous study highlighted the importance of space utilization on bacterial community composition5. Surprisingly, Chase et al., found surface material has minimal influence on microbial community composition25, which is consistent with the findings of this study. We found microbial communities across multiple surfaces in different indoor locations to cluster according to the room they were in and the usage patterns, rather than according to material surface. These results may be a product of the insensitivity to microbial viability of the sampling methods used to assay these environments27,28 as other studies have found surface material to be important in shaping built environment microbial communities29. To facilitate high-throughput processing of the microbiomes interrogated here no viability determinism was performed, therefore the presence of relic DNA from non-viable bacteria cannot be discounted in this, or previous, studies. Approaches that can differentiate viable from non-viable microbes such as culturing30,31 or use of viability dyes32 may serve to answer these questions in the future.
Roughly three months after construction of the built environment investigated in this study, a regular cleaning regime was established. The introduction of biocidal agents at this time matched well with the timepoints at which indoor microbial communities begin to specialise, converging at the commissioning date in communities displaying very high degrees of environmental selection. This is not the first time this phenomenon has been observed. Flores et al. showed specific communities of bio-film forming Gram-negatives associated with regularly cleaned areas6, while others have shown homes in more urban areas, which are cleaned more frequently, have reduced diversity12,33. We found increased abundances of Escherichia, and multiple Bacilli following commissioning and establishment of cleaning regimes within the built environment we explored here. Multiple antimicrobial resistant strains of Escherichia are reported in clinical studies while Bacilli are known to produce glycosphingolipids34 which stabilise the bacterial membrane and confer resistance to polymixin class antibiotics35. These data add to the growing evidence base that where communities are regularly exposed to biocidal interventions, the intended sterility may not be achieved. Instead, frequent cleaning may simply act as a selective pressure for microbes resistant to the killing mechanisms employed36, resulting in more robust survivor communities.
This study found indoor microbiota to become increasingly dissimilar to the outdoor communities that seeded them over time. A longitudinal study of the home microbiota of 7 families over 6 weeks found bacteria in indoor spaces to be predominantly of human origin37. The same study also found that the human associated microbial signatures in indoor environments diminished as quickly as three days after a human vacated their home. In the study by Lax and colleagues, however, the influence of human microbes had already been established in each home. Our study identified that indoor microbiota were still developing as late as 34 weeks after completion of the construction of the building. This phenomenon may be specific to commercial properties and related to a lag period between completion of construction and commissioning of the building. As such, these findings may not be consistent with those observed in residential properties, which are generally occupied sooner following completion. Likewise, in family homes the influence of one individual on microbial community structures may be greater due to them spending substantially more time in their own home compared to visitors to the completed structure investigated here. Nonetheless, as in the study by Lax et al. we found the indoor microbiota to be significantly enriched with multiple commonly human-associated microbes including Klebsiella and Escherichia spp. after commissioning37.
Covid-19 lockdowns impacted our ability to consistently collect longitudinal microbial samples and record environmental variables throughout this study. As a result, numbers of samples and observations available at each timepoint are unbalanced. While the statistical methods employed were specifically chosen to address these limitations, by comparing ranked means rather than actual values, it must be noted that the strength of conclusions drawn on temporal microbiota development could be strengthened via a balanced study design. It can be difficult to identify contaminant bacterial features in low-biomass environmental samples during targeted amplicon sequencing, especially since many common contaminants observed in microbial community analyses originate from soils and watercourses38,39, both of which are sampled locations in this study. To address this we have sequenced blank swabs, kit negative and sequencing reagent controls as recommended by previous studies assessing the influence of contamination. Sample library size and compositions were significantly different to controls, demonstrating the legitimacy of bacterial features identified in this study.
Through high-throughput, longitudinal sampling of a single, newly-built structure, this study demonstrates the impact of human interaction and interventions on the microbiota of the built environment. A transient community of bacteria, initially dominated by those from outdoor environments and showing high degrees of homogenizing dispersal, precedes more location specific communities once humans begin to inhabit and interact with spaces in the built environment. These microbial community changes are likely due to increasing environmental selection.
Methods
Bacterial sample collection
The OME is an experimental building constructed and commissioned as part of the Hub for Biotechnology in the Built Environment. Throughout the construction process between December 2019 and November 2021, and until the official launch in June 2022, a total of 439 microbial samples were collected from multiple surfaces on the OME site. The sites included both indoor and outdoor surfaces, consisting of natural and synthetic materials orientated both vertically and horizontally.
The longitudinal nature of this study enabled sampling of The OME site before ground was broken during the construction process. At this point, only outdoor samples were taken, as no building existed to take samples inside. Supplementary Table 1 describes the location, material and construction timepoints spanned during sampling at each site.
Samples were collected by swabbing a 10 cm2 area for 30 s using Microgen Path-check swabs (Novacyt, UK). Immediately following each collection, batches of swabs were transported back to laboratories at Northumbria University and processed for bacterial nucleic acid isolation. At each sampling timepoint temperature, humidity, and CO2 levels were recorded using an ELKLIV SR-SIO Indoor CO2 meter. Recordings were taken every 90 s for a minimum of 15 min across outdoor, foyer, kitchen and hallway locations. Readings from the ELKLIV SR-SIO meter were validated by cross-referencing against those from a ParticlesPlus Model 7501 Remote Particle counter.
Nucleic acid isolation, library preparation and sequencing
Swab tips were cut off using sterile scissors and suspended in 1 × PBS (1 ml) in sterile microfuge tubes (2 ml). Tubes were vortexed horizontally at high speed for 25 min to release biological material before swab tips were discarded and remaining solution was centrifuged (5000 xG, 30 min) to pellet bacterial cells. The supernatant at this stage was discarded and bacterial pellets were resuspended in DNeasy bead solution (750 µL, (QIAGEN, DE)). Nucleic acids were isolated from resulting bacterial suspension using DNeasy PowerLyzer PowerSoil Kit (QIAGEN, DE) according to manufacturer’s instruction save for an extended bead beating time of 25 min to ensure full lysis of gram positive bacteria and 15 min incubation of filters at room temperature after addition of elution buffer to increase nucleic acid yields. An isolation kit negative control (1 × PBS, 1 ml) was processed with each batch of swab samples.
Sequencing was performed by NUOMICS DNA Sequencing Facility (Northumbria University, Newcastle, UK). Bacterial communities were determined by targeted sequencing of the V4 region of the 16S rRNA gene using primers 515F and 806R40. Prepared genomic libraries were sequenced on the Illumina MiSeq using V2 2 × 250 bp chemistry (Illumina, UK). A sequencing negative control (nuclease free water) was processed with each plate of samples during library preparations.
Raw sequencing output consisted of fastq files which were filtered to include only those with quality phred scores of > Q30. Taxonomic classification of resulting fastq files was performed in QIIME241. Briefly, paired end reads were merged42, trimmed to a maximum 253 bp43 and clustered at 97% similarity42. Chimeric reads were screened and removed44 before taxonomy was assigned using custom-trained classifiers, aligned to the Greengenes2 database (v2022.3)45. Features classified at sub-phylum level taxonomy were culled and taxonomic features were merged at the genus level before biom files were exported46 and compositional analyses were performed in R47 using the phyloseq package48.
Statistical analyses
Comparisons between means of continuous data were performed by Kruksal-Wallis (KW) rank sum test. Bacterial feature abundances were normalized by converting raw counts to proportional abundances per sample. Alpha and beta diversity were calculated using vegan49. Alpha diversity metrics were calculated from raw feature counts as rarefied (10 k reads) bacterial richness and from proportionally normalized abundances as Shannon diversity. Beta diversity was calculated from proportionally normalized abundances as Bray–Curtis dissimilarity and from raw feature counts as rarefied (10 k reads) Raup-Crick similarity between samples.
Generalised linear mixed models (glm) were built to determine associations of environmental factors including (as fixed effects): temperature, humidity, CO2 levels, building location, object type, commissioning and sampling date on alpha diversity50. Least-square means of significantly associated environmental factors were compared51 with multiple pairwise comparisons and corrected where appropriate using Tukey’s method.
PERMANOVA was performed to identify significant associations between Bray–Curtis dissimilarity and environmental factors including location and sampling timepoint. Where significant associations were observed, pairwise PERMANOVA52 was used to identify classes of each variable with significantly different community composition. Comparisons were corrected for multiple hypothesis testing using Bonferroni’s method. Within group dispersal for sampling locations and timepoints was assessed using PERMDISP. ANOVA was used to compare mean distances from individual sample compositions to group Euclidean centroids and Tukey’s HSD identified significant differences in dispersal between location/timepoint groups.
Community determinism was compared by rescaling Raup-Crick similarity to values between − 1 and + 1. Environmental selection, where environmental factors have large determinative effects on the community composition of samples, was defined as samples with Raup-Crick similarity values < − 0.95. Alternatively, homogenising dispersal, describes situations where environmental pressures have a minimal impact on community composition, with multitudes of shared features between samples. This was defined as samples with Raup-Crick similarity values > 0.95. Raup-Crick similarity values between − 0.95 and 0.95 were defined as ecological drift, being the case where environmental factors influenced microbial populations but did not prevent transfer of bacteria between them.
The influence of outdoor on indoor communities was determined by SourceTracker53 with outdoor locations defined as “sources” and indoor locations as “sinks”. Differential bacterial features between sampling locations and timepoints were identified using Maaslin254. Results were visualized with ggplot255.
Data availability
Data are available on the European Nucleotide Archive under accession PRJEB58762.
References
Klepeis, N. E. et al. The National Human Activity Pattern Survey (NHAPS): A resource for assessing exposure to environmental pollutants. J. Expo. Anal. Environ. Epidemiol. https://doi.org/10.1038/sj.jea.7500165 (2001).
Wilkins, D., Leung, M. H. Y. & Lee, P. K. H. Indoor air bacterial communities in Hong Kong households assemble independently of occupant skin microbiomes. Environ. Microbiol. 18, 1754–1763 (2016).
Gaüzère, C. et al. ‘Core species’ in three sources of indoor air belonging to the human micro-environment to the exclusion of outdoor air. Sci. Total Environ. 485–486, 508–517 (2014).
Gibbons, S. M. et al. Ecological succession and viability of human-associated microbiota on restroom surfaces. Appl. Environ. Microbiol. 81, 765–773 (2015).
Dunn, R. R., Fierer, N., Henley, J. B., Leff, J. W. & Menninger, H. L. Home life: Factors structuring the bacterial diversity found within and between homes. PLoS ONE 8, e64133 (2013).
Flores, G. E. et al. Diversity, distribution and sources of bacteria in residential kitchens. Environ. Microbiol. 15, 588–596 (2013).
Hewitt, K. M., Gerba, C. P., Maxwell, S. L. & Kelley, S. T. Office space bacterial abundance and diversity in three metropolitan areas. PLoS ONE 7, e37849 (2012).
Kembel, S. W. et al. Architectural design influences the diversity and structure of the built environment microbiome. ISME J. 6, 1469–1479 (2012).
Meadow, J. F. et al. Bacterial communities on classroom surfaces vary with human contact. Microbiome 2, 7 (2014).
Meadow, J. F. et al. Indoor airborne bacterial communities are influenced by ventilation, occupancy, and outdoor air source. Indoor Air 24, 41–48 (2014).
Ruiz-Calderon, J. F. et al. Walls talk: Microbial biogeography of homes spanning urbanization. Sci. Adv. 2, e1501061 (2022).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012).
Lax, S., Nagler, C. R. & Gilbert, J. A. Our interface with the built environment: Immunity and the indoor microbiota. Trends Immunol. 36, 121–123 (2015).
Stefka, A. T. et al. Commensal bacteria protect against food allergen sensitization. Proc. Natl. Acad. Sci. 111, 13145–13150 (2014).
Fujimura, K. E. et al. House dust exposure mediates gut microbiome Lactobacillus enrichment and airway immune defense against allergens and virus infection. Proc. Natl. Acad. Sci. 111, 805–810 (2014).
Lax, S. & Gilbert, J. A. Hospital-associated microbiota and implications for nosocomial infections. Trends Mol. Med. 21, 427–432 (2015).
Hartz, L. E., Bradshaw, W. & Brandon, D. H. Potential NICU environmental influences on the neonateʼs microbiome. Adv. Neonatal Care 15, 324–335 (2015).
Bokulich, N. A., Mills, D. A. & Underwood, M. A. Surface microbes in the neonatal intensive care unit: Changes with routine cleaning and over time. J. Clin. Microbiol. 51, 2617–2624 (2013).
Brooks, B. et al. Strain-resolved analysis of hospital rooms and infants reveals overlap between the human and room microbiome. Nat. Commun. 8, 1814 (2017).
Afshinnekoo, E. et al. Geospatial resolution of human and bacterial diversity with city-scale metagenomics. Cell Syst. 1, 72–87 (2015).
Marchesi, J. R. et al. The gut microbiota and host health: A new clinical frontier. Gut https://doi.org/10.1136/gutjnl-2015-309990 (2015).
Butler, S. & O’Dwyer, J. P. Stability criteria for complex microbial communities. Nat. Commun. 9, 2970 (2018).
Perez-Muñoz, M. E., Arrieta, M.-C., Ramer-Tait, A. E. & Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome 5, 48 (2017).
Lax, S. et al. Bacterial colonization and succession in a newly opened hospital. Sci. Transl. Med. 9, eaah6500 (2017).
Chase, J. et al. Geography and location are the primary drivers of office microbiome composition. mSystems 1, e00022-16 (2016).
Bridgens, B. How biotechnology can transform delivery and operation of the built environment. Proc. Inst. Civ. Eng. Civ. Eng. 173, 13 (2020).
Rogers, G. B. et al. The exclusion of dead bacterial cells is essential for accurate molecular analysis of clinical samples. Clin. Microbiol. Infect. 16, 1656–1658 (2010).
Nocker, A. & Camper, A. K. Selective removal of DNA from dead cells of mixed bacterial communities by use of ethidium monoazide. Appl. Environ. Microbiol. 72, 1997–2004 (2006).
Tong, X. et al. Metagenomic insights into the microbial communities of inert and oligotrophic outdoor pier surfaces of a coastal city. Microbiome 9, 213 (2021).
Franco, L. C. et al. A microbiological survey of handwashing sinks in the hospital built environment reveals differences in patient room and healthcare personnel sinks. Sci. Rep. 10, 8234 (2020).
Sharpe, T. et al. Influence of ventilation use and occupant behaviour on surface microorganisms in contemporary social housing. Sci. Rep. 10, 11841 (2020).
Young, G. R. et al. Reducing viability bias in analysis of gut microbiota in preterm infants at risk of NEC and sepsis. Front. Cell. Infect. Microbiol. 7, 237 (2017).
Parajuli, A. et al. Urbanization reduces transfer of diverse environmental microbiota indoors. Front. Microbiol. 9, 84 (2018).
Stankeviciute, G., Guan, Z., Goldfine, H. & Eric, A. K. Caulobacter crescentus adapts to phosphate starvation by synthesizing anionic glycoglycerolipids and a novel glycosphingolipid. MBio 10, e00107-19 (2019).
Vaz-Moreira, I., Nunes, O. C. & Manaia, C. M. Diversity and antibiotic resistance patterns of sphingomonadaceae isolates from drinking water. Appl. Environ. Microbiol. 77, 5697–5706 (2011).
Gilbert, J. A. & Stephens, B. Microbiology of the built environment. Nat. Rev. Microbiol. 16, 661–670 (2018).
Lax, S. et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 345, 1048–1052 (2014).
Weyrich, L. S. et al. Laboratory contamination over time during low-biomass sample analysis. Mol. Ecol. Resour. 19, 982–996 (2019).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 1–12 (2014).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. Isme J. 6, 1621–1624 (2012).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90 (2018).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
Amnon, A. et al. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2, e00191-16 (2017).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
McDonald, D. et al. Greengenes2 enables a shared data universe for microbiome studies. bioRxiv https://doi.org/10.1101/2022.12.19.520774 (2022).
McDonald, D. et al. The Biological Observation Matrix (BIOM) format or: How I learned to stop worrying and love the ome-ome. Gigascience 1, 2047 (2012).
R_Core_Team. R: A Language and Environment for Statistical Computing (2014).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013).
Oksanen, J. et al. vegan: Community Ecology Package. (2015).
Brooks, M. E. et al. glmmTMB balances speed and flexibility among packages for zero-inflated generalized linear mixed modeling. R J. 9, 379–400 (2017).
Searle, S. R., Speed, F. M. & Milliken, G. A. Population marginal means in the linear model: An alternative to least squares means. Am. Stat. 34, 216–221 (1980).
Martinez, A. P. pairwiseAdonis:Pairwise multilevel comparison using adonis. R Packag. version 0.0.1. (2017).
Knights, D. et al. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 8, 761–763 (2011).
Mallick, H. et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 17, e1009442 (2021).
Whickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, 2009).
Acknowledgements
The authors would like to thank Lindsay Bramwell at Northumbria University for loaning and describing use of CO2 monitors. Oliver Perry and Ben Bridgens at Newcastle University were instrumental in gaining access to the OME site prior to commissioning of the building. Ben Bridgens also assisted with access to renders of the OME produced by Sadler Brown Group and the Hub for Biotechnology in the Built Environment and used to illustrate sampling locations in Fig. 1A. Andrew Nelson and Clare McCann of NUOMICS DNA Sequencing Facility, Northumbria University, performed 16S rRNA gene sequencing.
Funding
Funding was provided by Research England (Grant number: Expanding Excellence in England (E3), Hub for Biotechnology in the Built Environment).
Author information
Authors and Affiliations
Contributions
G.Y., D.S. conceptualized the study, G.Y. designed, validated and carried out sampling. A.S. assisted with sampling. G.Y. performed laboratory and computational analysis then wrote the manuscript. D.S. supervised the project. All authors reviewed the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Young, G.R., Sherry, A. & Smith, D.L. Built environment microbiomes transition from outdoor to human-associated communities after construction and commissioning. Sci Rep 13, 15854 (2023). https://doi.org/10.1038/s41598-023-42427-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-42427-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
