
Abstract
ER-to-Golgi trafficking partakes in the sorting of misfolded cytoplasmic proteins to reduce their cytological toxicity. We show here that yeast Sec7, a protein involved in proliferation of the Golgi, is part of this pathway and participates in an Hsp70-dependent formation of insoluble protein deposits (IPOD). Sec7 associates with the disaggregase Hsp104 during a mild heat shock and increases the rate of Hsp104 diffusion in an Hsp70-dependent manner when overproduced. Sec7 overproduction increased formation of IPODs from smaller aggregates and mitigated the toxicity of Huntingtin exon-1 upon heat stress while Sec7 depletion increased sensitivity to aẞ42 of the Alzheimer’s disease and α-synuclein of the Parkinson’s disease, suggesting a role of Sec7 in mitigating proteotoxicity.
Similar content being viewed by others
Introduction
The protein quality control (PQC) machinery in the cell is crucial to keep protein synthesis, folding, transport, degradation and sequestration in a functional balance (proteostasis) and prevents toxicity caused by aberrant proteins. It consists of many concerted pathways, which are largely facilitated by the action of molecular chaperones. There is a multitude of stressors that can cause an imbalance in proteostasis, leading to loss or gain of function of proteins and disrupting cellular processes1,2,3,4. Such stressors are for example heat shock and oxidative stress. In addition, a breakdown in proteostasis plays a central role in many age-associated neurodegenerative diseases, including Huntington’s, Alzheimer’s and Parkinson’s disease5,6,7,8; diseases characterized by the accumulation of toxic protein oligomers and aggregates.
Neurodegenerative diseases, breakdown of PQC, and protein aggregation have been linked to defects in intracellular vesicle trafficking occurring during aging in various organisms9,10,11,12,13,14,15,16,17: For example, the Huntingtin protein interacts with proteins involved in vesicle trafficking18,19 and the disease protein causes defects in endocytosis12. Similarly, aß processing from the amyloid precursor protein (APP) is facilitated by endolysosomal trafficking and is thus directly linked to different endomembrane systems that malfunction in Alzheimer’s disease10,11,17,20,21,22. Moreover, α-synuclein of the Parkinson’s disease is known to mediate membrane trafficking and failures in such trafficking play an important role in α-synuclein aggregation and in cell-to-cell disease propagation16,23,24,25,26,27. In S. cerevisiae, there is also evidence that functional membrane trafficking is important to handle α-synuclein aggregates26,28.
In addition to the function of endomembrane trafficking in neurological diseases, a role for endocytotic trafficking in yeast aging, proteostasis, and lifespan control has been described previously29. Yeast vesicle trafficking is known to consist of several highly conserved interconnected pathways30,31,32, which require constant communication and exchange of material. The systems are thus in close proximity and in contact with one another via cellular organelles and connecting proteins. These trafficking routes consist of the exocytosis/secretory (SEC) pathway, which directs proteins towards the plasma membrane or out of the cell, and the VPS and ALP pathways guiding cargo towards the vacuole. Endocytosis (END pathway) allows for uptake of plasma membrane proteins and extracellular medium components, which after internalization into endosomes are sorted and sent to the vacuole for degradation or to the Golgi for recycling (RCY pathway). The pathways participating in endomembrane trafficking rely on a multitude of factors including Rab GTPases and SNARE proteins such as the yeast syntaxin-5 Sed5, which is involved in COPII anterograde trafficking from the ER to Golgi and was recently shown to function as a bottleneck in spatial PQC (sPQC) in yeast33.
The sPQC sorts misfolded proteins into distinct organelle-associated sites within the cell. Upon heat shock, misfolded proteins in the yeast cytoplasm first accumulate at multiple sites called CytoQs, also known as Q-bodies or stress foci, which subsequently coalesce into larger deposition sites commonly referred to as inclusions34,35,36,37. These inclusions are localized to at least three distinct sites known as the juxtanuclear and/or intranuclear quality control site (JUNQ/INQ); the peripheral, vacuole-associated insoluble protein deposit (IPOD); and a site adjacent to mitochondria33,35,38,39,40,41.
In this paper, we report that the yeast guanine nucleotide exchange factor, Sec7, a key component in Golgi trafficking42,43 is an additional bottleneck in Hsp70-dependent sPQC, including the formation of organelle-associated inclusions, and that overproduction of Sec7 boosts spatial proteostasis during mild heat shock and aging and mitigates the toxicity of Huntingtin exon-1.
Results and discussion
Sec7 is a limiting factor in the formation of IPOD inclusions
In a recent study, we uncovered the role of vesicle trafficking and specifically the yeast syntaxin-5 in ensuring proper sPQC upon mild heat shock by screening the library of yeast mutants deleted for non-essential genes for defects in inclusion formation33. Here, we applied a similar screen including the essential genes of yeast, i.e. performing high content-microscopy after a mild heat shock and determining which temperature sensitive (ts) mutant strains show three or more Hsp104-GFP aggregates per cell (type 3 mutant) rather than the typical one to two inclusions. The main functional categories enriched among the essential genes involved in inclusion formation (SAFE analysis44; Fig. 1a) are cell polarity, vesicle trafficking, chromatin/transcription, and protein turnover (Fig. 1a). The role of cell polarity in sPQC has been studied in detail previously45,46,47. Vesicle trafficking was also enriched as a functional category among hits when screening the non-essential mutant library33 and confirms this category as involved in sPQC of endogenous protein aggregates upon heat shock. Among the identified genes were known factors required for proper sPQC such as the essential syntaxin-5 and components of the COG complex (supplementary table 133,). In addition to analyzing the data from heat shock after 110 min as an endpoint, we set out to find ts alleles, which cause prolonged failure in aggregate coalescence by monitoring the number of aggregates per cell also at later time points, 220 and 330 min. By comparing the late (330 min) to the early time point (110 min) of heat shock, we pinpointed mutations that caused a drastic failure in sPQC (supplementary table 2).

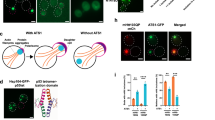
Sec7, among other vesicle trafficking components, was identified to maintain proper spatial PQC upon heat shock. (a) SAFE analysis, image modified from TheCellMap.org, hits for type 3 cells upon continuous heat shock for 110 min using the essential gene yeast mutant library. (b) Fraction of type 3 in WT, and sec7-1 mutant cells upon continuous heat shock (38 °C, 90 min). (c) Aggregate phenotype of WT and sec7-1 cells (38 °C, 90 min), representative cells from (b). (d) Fraction of type 3 upon continuous heat shock (38 °C, 90 min) in WT cells overproducing SEC7 compared to the vector control (using 316-SEC7-mRFP plasmid (ADH1 promoter)). (e) Aggregate phenotype of WT cells without and with overproduction of Sec7 (38 °C, 90 min, representative cells from (d). Images are maximum Z-projections of relevant slices with brightness/contrast adjustment consistently across the strains within the figure. Scale bar 2 μm.
The essential SEC7, with a role in Golgi maturation/proliferation and intra-Golgi trafficking, was selected for further analysis among the hits due to its type 3 phenotype and since it was previously found as a physical interactor of Hsp104 at permissive temperature and even more so during a mild heat shock29. Conditional sec7-1 mutant cells displayed severe defects in the formation of IPOD inclusions and instead harbored multiple (three or more), smaller aggregates (Fig. 1b,c) and only reduced the number of Hsp104-GFP foci-free cells during continued heat shock by 17%. It was thus among the stronger hits of mutants with compromised ability to alleviate Hsp104-associated aggregation during prolonged heat stress. We tested if Sec7 is limiting for IPOD formation by overproducing it on a plasmid (Fig. 1d,e) and by exchanging its native promoter to the strong, constitutive, GPD promoter (supplementary Fig. 1a). We found that such overproduction drastically increased IPOD formation from small aggregates, i.e. reduced the number of type 3 cells, indicating that Sec7 is limiting in wild type cells for sPQC. A link between Sec7 and secretion in sPQC was further confirmed by treating cells with brefeldin A, an inhibitor of Sec7 and other secretion factors33,48,49 (supplementary Fig. 1b). To determine if Sec7 and Sed5 act through the same pathway in sPQC, we performed aggregate clearance assays with the Sec7 overproduction strain (supplementary Fig. 1c). In contrast to the stark increase in clearance observed in SED5 overexpression cells33, no such effect was found when overexpressing SEC7, indicating that overproduction of each of the two factors may not enhance the same sPQC pathways, or that quantitatively different thresholds for achieving an effect in aggregate clearance exist for the two proteins. In addition, we overproduced, via plasmids, Sec7 in sed5-1 and Sed5 in sec7-1 conditional mutant cells and found that the defects observed in inclusion formation in each mutant could not be suppressed by such overproduction (supplementary Fig. 1d). This suggests that Sed5 and Sec7 are not functioning in parallel redundant pathways in boosting sPQC as they depend on the presence of each other for their function. Another explanation would be that Sed5 and Sec7 work together to increase inclusion formation.
Next, we wondered if the beneficial effects of Sec7 overproduction on inclusion formation could be explained by its essential role in autophagy50, as autophagy contributes to degradation of aberrant proteins. To address this, we tested if the increase in inclusion formation displayed by Sec7 overproduction cells depends on presence of the essential autophagy factor Atg1. SEC7 overexpression was able to boost inclusion formation also in atg1∆ background (supplementary Fig. 1e). Additionally, we visualized vacuole morphology in WT, sec7-1 and SEC7 overexpressing cells containing Vph1-GFP before and after heat shock. In line with previous data51, vacuole area in WT cells increased during heat shock with a simultaneous decrease in vacuole number. While the sec7-1 conditional mutant displayed an increase in number of small vacuoles upon heat shock, indicating defects in vacuolar dynamics, the Sec7 overproduction strain was unaffected (supplementary Fig. 1f). Collectively, the data indicate that Sec7 action in sPQC is independent of its function in autophagy.
Sec7 increases the dynamic movement of the disaggregase Hsp104
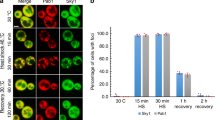
It has been demonstrated previously that elevated IPOD formation is associated with increased movement of the disaggregase Hsp104 (decreased time of diffusion 33;), which specifically binds to protein aggregates and is required for their proper disaggregation52,53,54. Therefore, we investigated if Sec7 overproduction affected Hsp104 diffusion using fluorescence correlation spectroscopy (FCS). We found that the diffusion time was reduced when Sec7 was overproduced (Fig. 2a). Moreover, this effect of Sec7 on Hsp104 movement required the presence of either one of the major cytosolic Hsp70 chaperones, Ssa1 and Ssa2 (Fig. 2b,c), which is interesting as Hsp70s are required for the functions of Hsp104 in protein quality control55,56. Sec7 forms foci on Golgi-associated coated vesicles57 and we found that such Sec7 foci and Hsp104-associated aggregates transiently co-localized upon mild heat shock33 and that this co-localization was dependent on both Ssa1 and Ssa2 (Fig. 2d,e, supplementary Fig. 2). In addition, Ssa1 or Ssa2 was required for Sec7 overproduction to increase IPOD formation from smaller aggregates (Fig. 2f,g).

Sec7 overproduction increases the dynamic movement of Hsp104 in a Hsp70-dependent manner. (a) Diffusion time of soluble Hsp104-GFP in WT cells without and with overproduction of Sec7 (38 °C). (b) Diffusion time of soluble Hsp104-GFP in ssa1Δ cells without and with overproduction of Sec7 (38 °C). (c) Diffusion time of soluble Hsp104-GFP in ssa2∆ cells without and with overproduction of Sec7 (38 °C). For (a–c) The bottom and top of the box represent the first and third quartiles. The circle shows the mean and whiskers indicate the variability of diffusion times outside the upper and lower quartiles for 15–25 cells. (d) Fraction of cells displaying co-localization of Hsp104-GFP with Sec7-RFP upon heat shock in WT cells (38 °C). Cells with a visible signal in both channels (green and red) were scored for co-localization. (e) Co-localization of Hsp104-GFP with Sec7-RFP after 30 min of heat shock in WT cells. (f) Fraction of type 3 upon continuous heat shock (38 °C, 90 min) in ssa1∆ cells overexpressing SEC7 compared to the vector control. (g) Fraction of type 3 upon continuous heat shock (38 °C, 90 min) in ssa2Δ cells overexpressing SEC7 compared to the vector control.
Sec7 overproduction reduces aggregation and the presence of multiple aggregates during aging
Since Sec7 appeared limiting for inclusion formation during a mild heat shock, we next investigated if the same was true upon replicative aging. Protein aggregates have been shown to accumulate during replicative aging of yeast mother cells58,59 and have a tendency to form multiple aggregates rather than discrete inclusions in old mother cells60,61. We isolated old mother cells (average 13 bud scars) and compared their aggregate content with and without Sec7 overproduction. We found, first, that Sec7 overproduction reduced the number of mother cells displaying any type of aggregate (Fig. 3a), and secondly, that old cells that harbored aggregates displayed fewer aggregates when Sec7 was overproduced (Fig. 3b,c). However, we did not find any effect of Sec7 overproduction on the lifespan of cells (Fig. 3d).

Sec7 overproduction boosts spatial PQC during replicative aging. (a) Fraction of aged cells displaying Hsp104-GFP aggregates in WT cells without and with overproduction of Sec7. (b) Fraction of type 3 cells in WT without and with overproduction of Sec7. (c) Number of aggregates per aged cell of the WT (counted in 449 cells), and SEC7 OE strains (counted in 544 cells). (d) Replicative lifespan of WT, and SEC7 OE.
Sec7 affects sensitivity towards human disease proteins
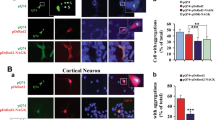
The Huntingtin exon-1, Htt103Q poly-glutamine peptide, of the Huntington disease has been shown to be toxic also in yeast cells18,62,63. Given the effect of Sec7 on sPQC, we tested if Sec7 overproduction affected Htt103Q toxicity and found this to be the case both in liquid and plate growth assays (Fig. 4a,b). The liquid growth profile shows that Sec7 overproduction rescues the growth rate during the exponential phase of growth to vector control levels at an elevated temperature, even if the SEC7 overexpression cells do not reach the same yield as the vector control when monitoring growth for 24 h (Fig. 4a). Additionally, Sec7 overproducing cells display an increased cellular fitness compared to wild type cells containing Htt103Q in growth assays on plates, especially at higher temperatures (Fig. 4b), indicating that overproduction of Sec7 markedly reduces the toxicity of this disease protein. However, sec7-1 cells are not more sensitive to Htt103Q expression than WT cells (supplementary Fig. 3a). We did not observe any obvious changes in aggregation of Htt103Q in the SEC7 overexpression cells or in the sec7-1 conditional mutant compared to WT (supplementary Fig. 3b).

Sec7 affects toxicity of neurodegenerative disease proteins. (a) Growth profile in liquid media of WT and SEC7 OE cells overproducing Htt103Q via the GPD promoter (103Q) compared to the vector control (pRS416) measured using an automated plate reader. (b) Fitness of WT and SEC7 OE cells overproducing Htt103Q via the GPD promoter (Htt103Q) compared to the vector control (pRS416). (c) Fitness of WT and sec7-1 mutant cells overproducing aβ42 (pRS416-GPD- aβ42) compared to the vector control (pRS416). (d) Fitness of WT and sec7-1 mutant cells in the absence (pYX242) or presence of α-synuclein (WT SYN, A30P).
We tested several constructs of two other human disease proteins, aβ42 and α-synuclein of the Alzheimer’s and Parkinson’s disease, respectively. Firstly, we used variants of the proteins that are not, or only very modestly, toxic in yeast cells17,33 and found that cells with reduced Sec7 activity, in contrast to wild type cells, displayed sensitivity towards both these proteins (Fig. 4c,d). Secondly, additional constructs of these disease proteins with known toxicity in wild type cells were tested in SEC7 overexpression cells. α-synuclein and its point mutation A53T version are both toxic when constitutively expressed at high levels via 2μ plasmids64 and we found that Sec7 overproduction could not ameliorate toxicity of these constructs and did not affect aggregation of the model proteins (supplementary Fig. 3c,d). Similarly, we used GAL-inducible constructs of wild type aβ42, which is moderately toxic, and its highly toxic point mutant version, G37C65. Increased levels of Sec7 were unable to mitigate toxicity when disease protein expression was induced by growing cells on galactose (supplementary Fig. 3e). We tested if overexpression of SEC7 could mitigate any potentially arising toxicity of constitutively overexpressed aβ42 during cellular aging in stationary phase cultures. However, no difference was observed between WT and Sec7 overproducing cells containing aβ42, in contrast to those expressing Htt103Q (supplementary Fig. 3f). We speculate that increased Sec7 levels may be unable to aid α-synuclein and aβ42 detoxification as both these disease proteins interact with, and impede, the secretory pathway64,65, which is the pathway Sec7 is expected to boost when overproduced. In contrast to these disease proteins, mutant Huntingtin may not inhibit secretion in such a manner, allowing Sec7 overproduction to act on the pathway and provide resistance against this protein.
In conclusion, recent data has highlighted the role of ER to Golgi trafficking in directing misfolded proteins to the surface of vacuoles and mitochondria and that vesicles of the trafficking pathway may serve as platforms on which misfolded and aggregated proteins can hitchhike towards specific deposition sites29,33,66 including IPODs. Such deposition of aggregates at the surface of mitochondria speeds up their clearance from the cell and this deposition also relies on proteins known to make contacts sites between vacuoles and mitochondria33. In addition, it is known that deficiencies in endocytosis, and vesicle trafficking in general, renders cells more sensitive to damaged proteins, including neurological disease proteins12,19. Herein, we demonstrate that Sec7, a guanine-nucleotide exchange factor required for maturation and proliferation of the Golgi as well as intra-Golgi transport, is a key factor in directing misfolded proteins to the IPOD deposition site. Moreover, Sec7 appears to be a limiting factor in such sPQC as ectopically elevating the levels of the protein is not only boosting IPOD formation but is also mitigating the toxicity of Huntingtin exon-1 during heat stress. It remains to be investigated whether Sec7 Golgi-related functions are mechanistically responsible for the observed effects on sPQC or whether Sec7 may act on sPQC via other pathways. Thus, Sec7, and associated factors involved in Golgi maintenance, might be interesting targets for therapeutic intervention of age-related proteopathies.
Materials and methods
Yeast Saccharomyces cerevisiae
As described previously33, strains with HSP104-GFP-LEU2 in the S228C SGA (synthetic genetic array) background were used in the inclusion body formation screen as well as in manual aggregate assays and stem from a cross performed according to SGA technology67 between the Y7092 SGA query strain with HSP104-GFP-LEU2 and the essential gene yeast library version 5 unless indicated that the BY4741 background was used. The SEC7 overexpression in BY4741 was generated using pYM-N1568. ATG1 was deleted using pFA6-hphNT168. Vph1-GFP-HIS3 C-terminal region was amplified from genomic DNA of the GFP collection strain69 via PCR and integrated into the WT, sec7-1 and SEC7 OE genome. Detailed information about strains and plasmids are listed in supplementary table S3.
Yeast cells were grown in synthetic drop out media supplemented with 2% glucose and corresponding antibiotics or in YPD. Cells were cultured at 30 °C except for temperature sensitive strains, which were kept at 22 °C.
High content-microscopy screen
A genome-wide SGA screen was performed as described previously67, using a BM3-BC colony handling robot (S&P Robotics Inc.).
Cells from the temperature sensitive mutant collection version 5 (Boone lab, Toronto, Canada) were crossed with the Y7092 query strain containing HSP104-GFP-LEU2. The new array was grown in synthetic complete media to an OD600 ̴ 0.5 at 22 °C and subsequently shifted to 38 °C in a water bath. Cells were collected and imaged live at time points 110 and 330 min of heat shock using the ImageXpress Micro XLS system (Molecular Devices) with a 100 × objective (CFI L Plan EPI cc 0 mm to 0.7 mm) and GFP filter (excitation 472/30 nm, emission 520/35 nm, dichroic mirror 495 nm). The images were analyzed for number of cells and number of aggregates using an algorithm built with MetaXpress software (version 6.54.532, Molecular Devices), which segments the images and keeps masks of shapes to exclude areas outside of cells when determining aggregate number. Mutants were considered hits when cells contained an average of 3 or more Hsp104-GFP aggregates per cell (type 3 phenotype) after 110 min heat insult. The hits were analyzed for functional enrichments (SAFE44) using the in-browser tool TheCellMap.org 70 by uploading the gene list of hits and setting the significance cutoff to 5e-2. Mutants with prolonged failure in sPQC were identified by determining the percentage of aggregates per cell detectable after 330 min of heat shock in comparison to the initial 110 min time point.
Protein aggregation analysis
Cells were grown in respective media supplemented with 2% glucose at permissive temperature (30 °C/22 °C) until mid-exponential phase (OD600 ̴ 0.5). Protein aggregation was induced by shifting the cells to 38 °C for 90 min. Cells were fixed in 3.7% final concentration formaldehyde and washed with PBS. Z-stack images were acquired using a conventional fluorescence microscope, Zeiss Axio Observer .Z1 inverted microscope equipped with Axiocam 506 camera, and a Plan-Apochromat 100x/1.40 NA Oil DIC M27 objective. Images were quantified by manually counting fraction of type 3 cells (containing three aggregates or more) in ImageJ software. Vph1-GFP containing cells and those expressing disease protein plasmids were imaged directly without formaldehyde fixation.
All figure data are based on the average of at least three individual experiments, error bars representing SD. Data were tested for significance using an unpaired two-tailed t-test, with p-values < 0.05 considered significant, depicted as * < 0.05, ** < 0.005, *** < 0.0005.
Clearance assay
Aggregate clearance assay was performed as described previously33.
Brefeldin A treatment
The treatment was performed as described previously33.
Fluorescence correlation spectroscopy
Fluorescence correlation spectroscopy (FCS) was performed as described previously33. In brief, measurements were performed on a ConfoCor2 FCS unit attached to an inverted LSM 880 confocal microscope (Zeiss) equipped with a C-Apochromat 40x/1.20 W Corr objective. To find the measurement position in the cytoplasm of each cell, GFP (excited at 488 nm, emission detected at 500–530 nm) and the transmission channel were simultaneously imaged using an open pinhole. Measurements were performed for 10 × 8 s with excitation at 488 nm and detection at 500–550 nm, pinhole 1 AU. Cells were grown until mid-exponential phase (OD600 ~ 0.5), heat shocked for 20 min at 38 °C and attached to ConA treated glass bottom culture dishes while the incubation chamber was at 38 °C. 15–25 cells were analyzed in each condition. 488 nm laser power was set to an AOTF transmission of 0.005%. Fitting of the FCS autocorrelation curves was done using a single component free diffusion fit in the ConfoCor2 software.
Co-localization analysis
Cells with indicated reporters were grown in respective media supplemented with 2% glucose at permissive temperature (30 °C/22 °C) until mid-exponential phase (OD600 ~ 0.5). Protein aggregation was then induced at 38 °C. Co-localization was investigated during or post heat shock at the depicted time points.
Replicative lifespan analysis
The replicative lifespan analysis was performed as described previously71. In brief, early logarithmic phase cells were plated and virgin cells were arranged according to a grid on YPD plates using the micromanipulator MSM400 (Singer Instruments). These cells were the designated new mother cells whose daughter cells were continuously removed and scored. The plates were incubated at 30 °C and cells were dissected at room temperature until they ceased dividing. Overnight, plates were kept at 4 °C. Each analysis was performed independently two times.
Growth assay on plates
Strains with indicated plasmids were grown in respective media supplemented with 2% glucose at permissive temperature (30 °C/22 °C) until mid-exponential phase (OD600 ~ 0.5). The culture ODs were adjusted to 0.5 and serial dilutions at a factor of 10 were performed in multiwell plates. The cell suspensions were spotted onto the respective drop out media plates and incubated at the indicated temperatures for 2 days except when noted otherwise.
Stationary phase growth assay
Strains with indicated plasmids were grown in -Ura drop out media supplemented with 2% glucose at permissive temperature (30 °C/22 °C) overnight and re-diluted in 15 ml fresh media to OD600 = 0.1. The cultures were kept at 30 °C for growth into stationary phase. After 3 days and 10 days of growth, 150 μl were sampled from the cultures, serially diluted at a factor of 10 and spotted onto the respective drop out media plates. Plates were incubated at 30 °C for 2 days and imaged.
Liquid growth assay
Cells were grown at permissive temperature, 22 °C until reaching logarithmic phase. They were re-diluted to an OD600 = 0.01 and arranged as 2–3 technical replicates (error bars are SD) on 24 well-plates. The plate was incubated at 37 °C in the BioTek Synergy 2 plate reader for 24 h with fast shaking while taking measurements of the OD600 every 15 min.
Old cell isolation
Old cells were isolated using the magnetabind biotin-streptavidin method29,72,73. Biotin labeled cells were isolated after growing overnight until OD600 < 1. The average replicative age of the cells was determined by manually counting bud scars after fixing cells in 3.7% formaldehyde (final concentration) and staining with Wheat Germ Agglutinin Alexa Fluor 555 conjugate (10 mg/ml final concentration, Life Technologies).
Data availability
All data generated or analyzed during this study are contained in this article and its supplementary data files. Requests for strains and materials should be directed to the corresponding author.
References
Balchin, D., Hayer-Hartl, M. & Hartl, F. U. In vivo aspects of protein folding and quality control. Science 353, 4354–4354 (2016).
Hartl, F. U. & Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 16, 574–581 (2009).
Hartl, F. U., Bracher, A. & Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 (2011).
Lansbury, P. T. & Lashuel, H. A. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 443, 774–779 (2006).
Balch, W. E., Morimoto, R. I., Dillin, A. & Kelly, J. W. Adapting proteostasis for disease intervention. Science 319, 916–919 (2008).
Hipp, M. S., Park, S.-H. & Hartl, F. U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 24, 506–514 (2014).
Ross, C. A. & Poirier, M. A. Protein aggregation and neurodegenerative disease. Nat. Med. 10, 10–17 (2004).
Vilchez, D., Saez, I. & Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 5, 5659 (2014).
Ahmadpour, D., Babazadeh, R. & Nystrom, T. Hitchhiking on vesicles: A way to harness age-related proteopathies?. FEBS J. 287, 5068–5079 (2020).
Bettayeb, K. et al. δ-COP modulates Aβ peptide formation via retrograde trafficking of APP. Proc. Natl. Acad. Sci. USA 113, 5412–5417 (2016).
Bettayeb, K. et al. Relevance of the COPI complex for Alzheimer’s disease progression in vivo. Proc. Natl. Acad. Sci. USA 113, 5418–5423 (2016).
Meriin, A. B. et al. Aggregation of expanded polyglutamine domain in yeast leads to defects in endocytosis. Mol. Cell. Biol. 23, 7554–7565 (2003).
Apetri, M. M. et al. Direct observation of α-synuclein amyloid aggregates in endocytic vesicles of neuroblastoma cells. PLoS ONE 11, e0153020 (2016).
Calafate, S., Flavin, W., Verstreken, P. & Moechars, D. Loss of Bin1 promotes the propagation of tau pathology. Cell Rep. 17, 931–940 (2016).
Spencer, B. et al. α-Synuclein interferes with the ESCRT-III complex contributing to the pathogenesis of Lewy body disease. Hum. Mol. Genet. 25, 1100–1115 (2016).
Tang, F.-L. et al. VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for α-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J. Neurosci. 35, 10613–10628 (2015).
Xu, W. et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Investig. 126, 1815–1833 (2016).
Berglund, L. L., Hao, X., Liu, B., Grantham, J. & Nyström, T. Differential effects of soluble and aggregating polyQ proteins on cytotoxicity and type-1 myosin-dependent endocytosis in yeast. Sci. Rep. 7, 11328 (2017).
Kim, Y. E. et al. Soluble oligomers of PolyQ-expanded huntingtin target a multiplicity of key cellular factors. Mol. Cell 63, 951–964 (2016).
Ginsberg, S. D. et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol. Psychiatry 68, 885–893 (2010).
Szaruga, M. et al. Alzheimer’s-causing mutations shift Aβ length by destabilizing γ-secretase-Aβn interactions. Cell 170, 443-456.e14 (2017).
Takahashi, R. H. et al. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 161, 1869–1879 (2002).
Cole, N. B. et al. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J. Biol. Chem. 277, 6344–6352 (2002).
Singh, P. K. & Muqit, M. M. K. Parkinson’s: a disease of aberrant vesicle trafficking. Annu. Rev. Cell Dev. Biol. 36, 237–264 (2020).
Yoshida, S. et al. Parkinson’s disease-linked DNAJC13 mutation aggravates alpha-synuclein-induced neurotoxicity through perturbation of endosomal trafficking. Hum. Mol. Genet. 27, 823–836 (2018).
Zabrocki, P. et al. Phosphorylation, lipid raft interaction and traffic of alpha-synuclein in a yeast model for Parkinson. Biochim. Biophys. Acta 1783, 1767–1780 (2008).
Zimprich, A. et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 89, 168–175 (2011).
D’Angelo, F. et al. A yeast model for amyloid-β aggregation exemplifies the role of membrane trafficking and PICALM in cytotoxicity. Dis. Model. Mech. 6, 206–216 (2013).
Hill, S. M. et al. Asymmetric inheritance of aggregated proteins and age reset in yeast are regulated by Vac17-dependent vacuolar functions. Cell Rep. 16, 826–838 (2016).
Conibear, E. Converging views of endocytosis in yeast and mammals. Curr. Opin. Cell Biol. 22, 513–518 (2010).
Feyder, S., De Craene, J.-O., Bär, S., Bertazzi, D. L. & Friant, S. Membrane trafficking in the yeast Saccharomyces cerevisiae model. Int. J. Mol. Sci. 16, 1509–1525 (2015).
Muñiz, M. & Zurzolo, C. Sorting of GPI-anchored proteins from yeast to mammals–common pathways at different sites?. J. Cell Sci. 127, 2793–2801 (2014).
Babazadeh, R. et al. Syntaxin 5 is required for the formation and clearance of protein inclusions during proteostatic stress. Cell Rep. 28, 2096–2110 2098 (2019).
Escusa-Toret, S., Vonk, W. I. M. & Frydman, J. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat. Cell Biol. 15, 1231–1243 (2013).
Miller, S. B. et al. Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J. 34, 778–797 (2015).
Specht, S., Miller, S. B. M., Mogk, A. & Bukau, B. Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J Cell Biol 195, 617–629 (2011).
Spokoini, R. et al. Confinement to organelle-associated inclusion structures mediates asymmetric inheritance of aggregated protein in budding yeast. Cell Rep. 2, 738–747 (2012).
Böckler, S. et al. Fusion, fission, and transport control asymmetric inheritance of mitochondria and protein aggregates. J. Cell Biol. 216, 2481–2498 (2017).
Kaganovich, D., Kopito, R. & Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 454, 1088–1095 (2008).
Ruan, L. et al. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 543, 443–446 (2017).
Zhou, C. et al. Organelle-based aggregation and retention of damaged proteins in asymmetrically dividing cells. Cell 159, 530–542 (2014).
Achstetter, T., Franzusoff, A., Field, C. & Schekman, R. SEC7 encodes an unusual, high molecular weight protein required for membrane traffic from the yeast Golgi apparatus. J. Biol. Chem. 263, 11711–11717 (1988).
Wolf, J., Nicks, M., Deitz, S., van Tuinen, E. & Franzusoff, A. An N-end rule destabilization mutant reveals pre-Golgi requirements for Sec7p in yeast membrane traffic. Biochem. Biophys. Res. Commun. 243, 191–198 (1998).
Baryshnikova, A. Systematic functional annotation and visualization of biological networks. Cell Syst. 2, 412–421 (2016).
Liu, B. et al. The polarisome is required for segregation and retrograde transport of protein aggregates. Cell 140, 257–267 (2010).
Liu, B. et al. Segregation of protein aggregates involves actin and the polarity machinery. Cell 147, 959–961 (2011).
Song, J. et al. Essential genetic interactors of SIR2 required for spatial sequestration and asymmetrical inheritance of protein aggregates. PLoS Genet. 10, e1004539 (2014).
Peyroche, A. et al. Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: Involvement of specific residues of the Sec7 domain. Mol. Cell 3, 275–285 (1999).
Sata, M., Donaldson, J. G., Moss, J. & Vaughan, M. Brefeldin A-inhibited guanine nucleotide-exchange activity of Sec7 domain from yeast Sec7 with yeast and mammalian ADP ribosylation factors. Proc. Natl. Acad. Sci. 95, 4204–4208 (1998).
van der Vaart, A., Griffith, J. & Reggiori, F. Exit from the Golgi is required for the expansion of the autophagosomal phagophore in yeast Saccharomyces cerevisiae. Mol. Biol. Cell 21, 2270–2284 (2010).
Keuenhof, K. S. et al. Large organellar changes occur during mild heat shock in yeast. J. Cell Sci. 135, (2022).
Glover, J. R. & Lindquist, S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82 (1998).
Parsell, D. A., Kowal, A. S., Singer, M. A. & Lindquist, S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature 372, 475–478 (1994).
Mogk, A., Bukau, B. & Kampinga, H. H. Cellular handling of protein aggregates by disaggregation machines. Mol. Cell 69, 214–226 (2018).
Lee, J. et al. Heat shock protein (Hsp) 70 is an activator of the Hsp104 motor. Proc. Natl. Acad. Sci. USA 110, 8513–8518 (2013).
Rosenzweig, R., Moradi, S., Zarrine-Afsar, A., Glover, J. R. & Kay, L. E. Unraveling the mechanism of protein disaggregation through a ClpB-DnaK interaction. Science 339, 1080–1083 (2013).
Franzusoff, A., Redding, K., Crosby, J., Fuller, R. S. & Schekman, R. Localization of components involved in protein transport and processing through the yeast Golgi apparatus. J. Cell Biol. 112, 27–37 (1991).
Aguilaniu, H. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 299, 1751–1753 (2003).
Erjavec, N., Larsson, L., Grantham, J. & Nyström, T. Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p. Genes Dev. 21, 2410–2421 (2007).
Oling, D., Eisele, F., Kvint, K. & Nystrom, T. Opposing roles of Ubp3-dependent deubiquitination regulate replicative life span and heat resistance. EMBO J. 33, 747–761 (2014).
Yang, J., Hao, X., Cao, X., Liu, B. & Nyström, T. Spatial sequestration and detoxification of Huntingtin by the ribosome quality control complex. eLife 5, 11792 (2016).
Meriin, A. B. et al. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol. 157, 997–1004 (2002).
Duennwald, M. L., Jagadish, S., Muchowski, P. J. & Lindquist, S. Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc. Natl. Acad. Sci. USA 103, 11045–11050 (2006).
Outeiro, T. F. & Lindquist, S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302, 1772–1775 (2003).
Fruhmann, G. et al. The impact of ESCRT on Aβ(1–42) induced membrane lesions in a yeast model for Alzheimer’s disease. Front. Mol. Neurosci. 11, 406 (2018).
Kumar, R., Nawroth, P. P. & Tyedmers, J. Prion aggregates are recruited to the insoluble protein deposit (IPOD) via myosin 2-based vesicular transport. PLOS Genet. 12, e1006324 (2016).
Tong, A. H. et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294, 2364–2368 (2001).
Janke, C. et al. A versatile toolbox for PCR-based tagging of yeast genes: New fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962 (2004).
Huh, W.-K. et al. Global analysis of protein localization in budding yeast. Nature 425, 686–691 (2003).
Usaj, M. et al. TheCellMap.org: A web-accessible database for visualizing and mining the global yeast genetic interaction network. G3 Genes Genomes Genet. 7, 1539–1549 (2017).
Kaeberlein, M., McVey, M. & Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580 (1999).
Sinclair, D. A. & Guarente, L. Extrachromosomal rDNA circles—A cause of aging in yeast. Cell 91, 1033–1042 (1997).
Smeal, T., Claus, J., Kennedy, B., Cole, F. & Guarente, L. Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell 84, 633–642 (1996).
Acknowledgements
We thank Joris Winderickx, Tiago Outeiro, Per Widlund, Dina Petranovic, Srishti Chawla and Charles Boone for kindly sharing yeast strains and plasmids. We thank the Centre for Cellular Imaging at the University of Gothenburg and the National Microscopy Infrastructure, NMI (VR-RFI 2019-00022, NMI00246) for assistance with FCS. This work was supported by grants from the Swedish Natural Research Council (VR) and the Knut and Alice Wallenberg Foundation (KAW; Grant and Wallenberg Scholar) to TN, from the German research foundation (DFG) to AF and from the Swedish Cancer Fund (Cancerfonden) [CAN 2017/643 and 19 0069] and the Swedish Natural Research Council (Vetenskapsrådet) [VR 2015-04984 and VR 2019-03604] to BL.
Funding
Open access funding provided by University of Gothenburg.
Author information
Authors and Affiliations
Contributions
R.B., K.L.S., A.F., X.H., B.L. and T.N. designed the experiments. R.B., K.L.S., A.F., X.H. and B.L. performed experiments. R.B., K.L.S., A.F. and T.N. analyzed the experiments. T.N. and K.L.S. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Babazadeh, R., Schneider, K.L., Fischbach, A. et al. The yeast guanine nucleotide exchange factor Sec7 is a bottleneck in spatial protein quality control and detoxifies neurological disease proteins. Sci Rep 13, 14068 (2023). https://doi.org/10.1038/s41598-023-41188-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-41188-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
