
Abstract
Across the three domains of life, circadian clock is known to regulate vital physiological processes, like, growth, development, defence etc. by anticipating environmental cues. In this work, we report an integrated network theoretic methodology comprising of random walk with restart and graphlet degree vectors to characterize genome wide core circadian clock and clock associated raw candidate proteins in a plant for which protein interaction information is available. As a case study, we have implemented this framework in Ocimum tenuiflorum (Tulsi); one of the most valuable medicinal plants that has been utilized since ancient times in the management of a large number of diseases. For that, 24 core clock (CC) proteins were mined in 56 template plant genomes to build their hidden Markov models (HMMs). These HMMs were then used to identify 24 core clock proteins in O. tenuiflorum. The local topology of the interologous Tulsi protein interaction network was explored to predict the CC associated raw candidate proteins. Statistical and biological significance of the raw candidates was determined using permutation and enrichment tests. A total of 66 putative CC associated proteins were identified and their functional annotation was performed.
Similar content being viewed by others

Introduction
Earth’s rotation around its axis and the sun generates repetitive changes in the environmental conditions, like, the dark-light cycle, the rhythmicity of light intensity, temperature, seasons etc. Guided by these environmental changes, living organisms possess biological rhythms called circadian rhythms, which are endogenously generated, self-sustained, and temperature compensated and have a period approximately equal to 24 h in free-running conditions1,2,3,4. Circadian rhythms directly influence the physiological processes of all living beings, including plants, that are helpful in the diurnal modulation of biological events5. The involvement of the circadian clock in regulating vital plant processes, like, nutrient biogenesis, root growth, photosynthesis, hormonal signalling, sugar metabolism, flowering time, and plant immunity, has been documented in many studies6,7,8,9,10. These biological rhythms are entrained with their external environment by an internal timekeeper called a circadian clock that keeps track of environmental fluctuations and allows temporal separation of internal physical events accordingly11. The circadian clock is crucial for optimizing a plant’s adaptive responses, in different physiological processes, including growth, development and defence, to varying environmental conditions and improving the overall fitness of the plant8,12.
Circadian rhythms in most life forms are governed by a set of 20–24 genes called core circadian clock (CC) genes which regulate vital life-sustaining processes through another set of genes called core circadian clock associated (CCa). Thus the complete circadian pathway comprises the core clock genes and genes involved in downstream processes13. It has been shown that circadian oscillators are ubiquitous among all three domains of life with minimal elemental (at gene level) conservation across them, however, their architectures and mechanisms are remarkably convergent across domains14,15,16. Recent advancements in high-throughput next generation sequencing platforms have revolutionised the practice of modern biology by generating a massive volume of molecular sequence data17 that can be leveraged to characterize core circadian clock (CC) and clock associated genes at systems scale18. The dynamics of core circadian clock genes under different stress conditions have recently been characterised in Camelia sinensis19, however, comprehensive network theoretic methodology to characterise various core circadian clock associated genes is still lacking. Knowing the identities of genes involved in important downstream pathways will allow us to optimally utilize the cellular circuitry for crop improvement and increase the production of essential oils. Since the number of genes involved in downstream pathways is very large, functioning independently in several distinct sub-modules, studying them together is highly difficult through a reductionist approach. Network science provides a way to explore these interactions at a systems scale and draw conclusions from them.
Topologically similar proteins in protein interaction networks (PINs) have been shown to perform similar functions20. Several approaches have shown that the architectural properties of proteins in an interactome complement their structural and sequence properties and could be successfully utilized for inferring the functional annotations of interacting proteins21,22,23. The present study reports a novel integrated network framework that entirely relies on network topology to characterize rhythmic genes in plants. Various statistical approaches, including LS24, ARSER25, JTK_CYCLE26, RAIN27, MetaCycle28, BIO_CYCLE29 for characterizing oscillatory genes along with their estimated rhythmic parameters i.e. phase, amplitude and waveform have been proposed30. All these approaches utilize temporal transcriptomic data to characterize circadian regulated genes and estimate their rhythmic parameters. Dealing with transcriptomic data is time and labour-intensive and demands extensive computational capacity that is only sometimes feasible. On the other hand, our approach is highly time and memory efficient and requires minimum resources.
Ocimum tenuiflorum (Tulsi) is a member of family Lamiaceae, an aromatic herb that is widely recognized for array of health benefits it offers31. We find references of the holy basil in Ayurveda, an ancient medicinal system, for its usage in treating variety of diseases and disorders either independently or as a formulation in combination with several other herbs32. The plant is reared for its oils and every part of this plant produces a number of secondary metabolites which possess significant pharmaceutical properties33. Recently, genome34,35, tanscriptome36 and proteome37 wide studies have reported a draft catalogue of genes, transcripts and proteins present in Tulsi along with important mechanistic insights about the secondary metabolite biosynthetic pathways38,39. However, an exhaustive systems scale study presenting core circadian clock and clock associated genes along with complex interplay among them at molecular level is still lacking in O. tenuiflorum. This motivated us to exploit the topology of the Tulsi protein interaction network37 to identify circadian regulated genes. In this study, we propose a systems scale approach to mine and characterize core circadian clock proteins giving rise to these rhythms in O. tenuiflorum. We leveraged random walk with restart (RWR) and graphlet degree vector (GDV) methods to explore the local topology of protein interaction network of Tulsi (TulsiPIN) and identified raw candidate proteins associated with the core oscillator. Furthermore, statistical and biological significance of the raw candidates was determined and their functional annotations were determined.
Materials and methods
Data collection
From the literature, 24 core circadian clock (CC) proteins of Arabidopsis thaliana10,13 were identified, and their sequences were retrieved from the TAIR database40. Proteome data of 56 template plants, for which genome information is available, was downloaded from the UniProt database https://www.uniprot.org/. All the CC proteins were then queried against each template proteome using Blast-P to identify homologous proteins. Top hits were selected for each CC sequence having E value \(\le 10^{-5}\), with at least \(40 \%\) sequence identity and \(50 \%\) query coverage. CC sequence specific multiple sequence alignments were then obtained using Clustal Omega41. HMMER http://hmmer.janelia.org/ was used to construct Hidden Markov models for each CC protein. These HMMs were then used to scan O. tenuiflorum proteome obtained from TulsiDB http://caps.ncbs.res.in/Ote/ to identify putative CC proteins in Tulsi. Each identified CC protein was subjected to InerProScan v5.48-83 https://www.ebi.ac.uk/interpro/ to identify their respective domains19. Protein-protein interaction network of O. tenuiflorum called TulsiPIN, developed in our previous work37, was considered for network scale studies. TulsiPIN is an exhaustively constructed network consisting of 13,604 nodes and 327,374 edges with scale free and small world properties.
Mining core circadian clock associated (CCa) proteins using local topological measures
Two local network topology based algorithms, namely, random walk with restart (RWR) and graphlet degree vector (GDV) were leveraged to identify core circadian clock associated proteins.
Random walk with restart (RWR) based mining
RWR is a ranking algorithm that has been successfully applied to estimate the likelihood of genes/proteins being associated with a given biological process (X) based on their proximity to already known proteins belonging to X in the input network42. The algorithm initiates a specific number of walks from one or more seed nodes (CC proteins) to some random nodes in the network and returns the probability of each protein being a CCa protein. This study applied the RWR algorithm on TulsiPIN using 9 CC proteins (which could be mapped to TulsiPIN) as seed nodes. Initially, a vector \(P_0\), containing 13, 604 elements, each corresponding to a node in TulsiPIN, was constructed in which each CC protein was initialized with a number \(\frac{1}{9}\) and other proteins with zero. The vector is then updated using the equation \(P_{i+1} = (1-r) A^T P_i + rP_0\) for ith iteration until \(P_i\) is stabilized. Here, r represents restart probability that is set as (0.8), and A is TulsiPIN’s adjacency matrix every column of that sums up to one. The iterations stop when Manhattan distance between \(P_i+1\) and \(P_i\) is less than \(10^{-6}\) i.e. \(||P_{i+1} - P_i||_{L_1} < 10^{-6}\). This yielded a probability vector containing the probability values corresponding to each protein of TulsiPIN, quantifying their likelihood of being CC associated. Accordingly, a threshold value of \(10^{-5}\) was adopted and all the proteins having probabilities higher than the selected threshold, called raw CCa proteins, were considered for further study43.
Graphlets degree vector (GDV) based mining
Small, non-isomorphic, induced sub-graphs of a large network are defined as graphlets44. Based on the node position within a graphlet, each sub-graph can be differentiated into symmetrically equivalent sets of nodes (symmetry groups) called automorphism orbits45. A total of 15 automorphism orbits are possible for up to 4 node graphlets. Each node of an undirected network can be represented with a vector of length 15, each element of which represents the degree of the corresponding orbit, called graphlet degree vector (GDV). Finally, a vector of 11 non-redundant orbits46 containing orbit degrees, representing wiring around that node, was computed for every node in the network. Since current PPI networks have been shown to be largely incomplete and lower degree nodes are more likely to be present in those incomplete parts, we remove proteins with degrees lower than three22. Among the 24 CC proteins, only 10 could be mapped to TulsiPIN. Since one had a degree lower than three, so removed from the further study. For each of 9 CC proteins, a pairwise similarity (\(S_{GDV}\)) was computed with every TulsiPIN protein having a degree more than three that is defined as
where u and v represent GDV of CC proteins and TulsiPIN proteins respectively, while i represents the ith orbit. All proteins with similarity above \(94 \%\) with the protein of interest were considered CCa proteins. Previously a signature similarity threshold within the range 90–95% has been shown to be optimal22. So to select the signature similarity threshold, we started with a threshold value of \(96\%\) and decreased the threshold value until we obtained at least one TulsiPIN protein similar with each CC protein other than themselves.
Statistical and functional validation of CCa proteins
As these algorithms exploit the topology of the network to estimate the likelihood, this may output some false positive proteins as CCa proteins e.g. hubs will be ranked higher than those having degree lower than the average degree of network43. To reduce these false positive proteins and assess the reliability of each prediction, two methods, namely, permutation test and enrichment test were applied.
Permutation or randomisation test
To test the statistical significance of CCa proteins identified by RWR algorithm, we sampled 1000 random sets of 9 proteins each from 13,604 TulsiPIN proteins. RWR algorithm was then performed on TulsiPIN using each of these samples as a seed. A p value or permutation FDR for every CCa protein predicted was computed based on the following equation \(p (g) = \frac{\theta _{RWR}}{1000}\), where \(\theta _{RWR}\) is the number of random seeds for which probability of g, a CCa protein p(g), is higher than p(g) obtained using CC proteins as seed to RWR algorithm. On the other hand, to test the statistical significance of CCa proteins obtained from the GDV algorithm, we first constructed an ensemble of 1000 random networks with the same number of nodes and similar degree sequence47 and then computed graphlet degree vectors for each protein of every network. Then pairwise \(S_{GDV}\) similarity scores were computed for each CC protein with other proteins in TulsiPIN. Similar to RWR, a permutation FDR for every CCa protein predicted by the GDV algorithm was computed using \(p (g) = \frac{\theta _{GDV}}{1000}\), where \(\theta \) is the number of random networks for which the similarity score of protein pair between g, a CCa gene, and some CC gene is higher than the similarity score obtained for same pair in the real network. Higher p value of a gene indicates the gene is less likely to be associated with CC. Thus CCa proteins with p value less than 0.05 were selected from both methods, and the selected proteins were called candidate CCa proteins for convenience.
Enrichment test
The enrichment test is based on the premise that interacting proteins are more likely to share common functions37,48. So, in this section, we used PPI information from TulsiPIN and Gene Ontology (GO) and KEGG Orthology (KO) annotation information of interacting proteins to infer the validity of candidate CC associated proteins. It has been shown earlier that similar proteins may perform similar molecular functions, localize in the same cellular milieu and may be involved in similar biological processes37,48. Thus candidate CCa proteins having similar functional annotations as that of CC proteins may be the novel CCa proteins. To quantify the relationship between a protein and an annotation term (GO or KEGG pathways), an ontology enrichment score, the negative log of probability value obtained from the hypergeometric test was computed49. Given N total TulsiPIN proteins, among which M are annotated by one annotation term (A), if we randomly draw a set of proteins (H(g)) containing any protein g and its immediate neighbours, then the probability that at least m of them are annotated with A is given by
where n is the number of proteins present in set H(g) and m is the number of H(g) proteins annotated by A. A vector ES(g) can be created by computing S for all the annotation terms of g. These vectors can be used to quantify the similarity between two genes g and \(g^{\prime }\) as follows
Large value of \(\Gamma \) indicate high functional similarity between the two genes g and \(g^{\prime }\). We compute maximum annotation similarity (MAS), which represents the largest \(\Gamma \) value among all possible pairs between a candidate protein p and a set of CC genes. A threshold value of 0.75 was used to select candidate CCa proteins.
Functional annotation of CCa proteins
Each protein identified to be associated with the core circadian clock of O. tenuiflorum was then queried against NCBI non-redundant (nr), UniProt50 and TAIR51 databases using Blast-P52 program to assign putative functions. Default parameters except E value, which is set to be \(10^{-5}\), were used to assign homology. AgriGo53 and WEGO54 were used to predict Gene Ontology (GO) terms for each CCa protein. KEGG ontology (KO) terms were predicted for each CCa protein using KEGG55 database. All the 13 programs aggregated in InterProScan v5.48-83 were utilised to predict protein family, functional domains and sites in each CCa protein56.
Results and discussion
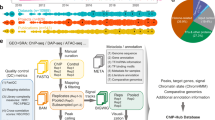
O. tenuiflorum plants have been reported to produce secondary metabolites (essential oils) which are mixtures of diverse aromatic compounds. These chemical compounds possess enormous medicinal properties that have been exploited continuously since ancient times31. Plants are constantly exposed to biotic and abiotic stress under varying environmental conditions, which impede plant growth and development and substantially impact oil production57,58. Circadian clock is the main regulatory mechanism that controls the growth and development of any plant8. A precisely tuned circadian clock plays a substantial role in increasing the fitness and survival of a plant by temporally coordinating various vital processes, like, metabolism, physiology etc. as per the external conditions8,10 Thus, we attempt to identify the protein set comprising the core circadian clock and its associated proteins in O. tenuiflorum through an exhaustive network science based computational framework presented in Fig. 1.

Overall methodology for genome wide identification, characterization of core circadian clock (CC) and core clock associated (CCa) proteins in O. tenuiflorum.
Mining and functional characterization of core circadian clock (CC) proteins
We enlisted 24 core circadian clock proteins and attempted to identify them in Ocimum tenuiflorum. Proteomes of 56 template plants were scanned using Blast-P for putative orthologs of selected clock proteins that resulted in 2, 543 top hits at an E value \(< 10^{-5}\). Further, top hits with sequence identity \(< 40\%\) and query coverage \(< 50\%\) were filtered out, resulting in 963 hits. Separate HMM profiles for each of the CC protein were built by extracting orthologous sequences of that protein from the respective template plants, which satisfy the above criteria. These HMMs were used to scan O. tenuiflorum proteome, and based of top hits, all the 24 candidate CC proteins were identified (Table S1). Transcription factors CIRCADIAN CLOCK ASSOCIATED 1 (CCA1) and LONG ELONGATED HYPOCOTYL (LHY) are partially redundant59. Also, the transcription regulator LIGHT-REGULATED WD1 (LWD1) is partially redundant with LWD2, LUX ARRHYTHMO (LUX) with NOX, REVEILLE 6 (RVE6) with RVE813,60, so the best hits returned the same proteins for them. These genes are important because they express during different times of the day or form complexes to regulate the expression of other genes. Their functions are synergistic60,61, so we selected the second best hit for LWD2, RVE4, PRR5, RVE6, NOX, PSEUDO-RESPONSE REGULATOR 3 (PRR3) and LHY proteins. Functions of all the 24 CC proteins were predicted by querying them against NCBI non-redundant, UniProt and TAIR databases using Blast-P. The results confirmed the identities of predicted clock proteins and reveled that most proteins possess their respective functions (Table S2). Furthermore, protein family and domain databases integrated by InterProScan also confirmed our results (Table S2).
Functional annotation of CC proteins is followed by their gene ontology (GO) annotations (Table 1). A total of 86 GO terms were successfully identified for these CC proteins, with 67 terms classified into 13 categories of biological processes, 2 terms classified into 2 categories of cellular components, and 17 GO terms associated with 2 categories of molecular functions. ‘Regulation of biological process’, ‘Cellular process’ and ‘Response to stimulus’ are highly enriched among biological processes, followed by ‘signalling’ and ‘rhythmic process’. Among molecular functions, ‘binding’ and ‘transcription regulator activity’ were the most enriched categories (Fig. 2). Successful assignment of “rhythmic process” associated gene ontology (GO) terms to the identified CC proteins further confirms the role of these proteins as the core circadian clock of O. tenuiflorum. Furthermore, for finding the CC proteins regulated pathways, the KEGG database was used that predicted KO ids of 16 proteins enriching 8 pathways, of these ‘Circadian rhythms in plants’ is found to be most enriched (Table S2). Other pathways include the ‘Ubiquitin system’ followed by ‘Plant hormone signal transduction’ and ‘Transcription factors’.

Gene ontology annotations of core circadian clock (CC) proteins of O. tenuiflorum.
Prediction and functional characterization of core circadian clock (CCa) associated proteins
It is known that different centrality measures address different topological aspects of a network and therefore capture the snapshot of a network in terms of selected architectural metric62. For example, connectivity-based measures assign higher weights to high degree nodes, whereas middle nodes connecting communities will be ranked higher by measures employing betweenness centrality. Similarly, many graph theoretic methods assign functions to unannotated proteins based on their closeness or shared neighbourhood in the PINs20,21,63. However, recently it has been shown that functional similarities among proteins in a PIN are independent of their network location i.e. sharing the same local neighbourhood rather it depends on similarities of the wiring patterns around them22. So, in this work, two local network topology based algorithms, namely, random walk with restart (RWR) and graphlet degree vector (GDV), have been leveraged to identify CCa proteins. The GDV based approach does not consider only the current node rather, considers the wiring pattern around that node i.e. it incorporates the information about local neighbourhood45. The CC proteins were mapped to the genome-wide interologous protein interaction network of Tulsi (TulsiPIN) to identify circadian clock-associated proteins37. A total of 10 proteins were successfully mapped to TulsiPIN proteins which were found to interact with 248 other proteins and form 317 interactions. Since one protein has a degree lower than 3, so removed from the study. The remaining nine CC proteins were used as seed nodes to the RWR algorithm applied on TulsiPIN. This produced a probability value corresponding to each protein representing its likelihood of being a novel circadian clock associated protein. All the proteins having probability values larger than \(10^{-5}\) were considered for further study resulting in 733 nodes called \(\text {raw}_{RWR}\) proteins (Table S3). Because all RWR proteins can not be circadian clock related, some have been selected due to network architecture alone. We employed permutation test to filter them out and computed a p value called the permutation FDR for every RWR protein. All the nodes having p values less than 0.05 were selected, resulting in 251 candidate CCa proteins called \(\text {candidate}_{RWR}\) proteins and are presented in Table S3. Similarly, we computed pairwise GDV similarity between CC proteins and other TulsiPIN proteins. This resulted in 146 proteins called \(\text {raw}_{GDV}\) proteins, having \(S_{GDV} > 94\), predicted to be CC associated by the GDV algorithm. Pairwise similarities were obtained from an ensemble of 1000 random networks preserving the degree sequence of TulsiPIN. A total of 146 proteins were found to be statistically significant, having p value \(< 0.05\) and were called \(\text {candidate}_{GDV}\) proteins (Table S3). Furthermore, to add another layer of confidence and select only those proteins with similar functions to CC proteins, we leveraged Gene Ontology and pathways information to compute the functional similarity (\(\Gamma \)) between each CC protein and every candidate protein. Among 251 \(\text {candidate}_{RWR}\) proteins, a total of 43 were found to have \(\Gamma > 0.75\) in both GO and pathways information based methods. On the other hand, 25 out of 146 \(\text {candidate}_{GDV}\) proteins were found to have \(\Gamma \) score 0.75 or above in both methods (Table S3). We combined the proteins obtained from the two methods (RWR and GDV) and found 66 unique candidate proteins, among which 2 were common to both methods. We constructed the circadian connectome of Tulsi by mapping the CC proteins and CCa proteins to TulsiPIN and found that these 90 proteins regulate 2412 other proteins and participate in a total of 8017 interactions (Fig. 3a,b).

Sub network of proteins obtained by combining core circadian clock (CC) and core circadian associated (CCa) proteins (including first interactors). Yellow colored nodes represent CCa proteins, red colored nodes are CC proteins.
Like to CC proteins, all the 66 CCa proteins were queried against NCBI non-redundant, UniProt and TAIR databases using the Blast-P program. The functional annotation of CCa proteins revealed that most proteins are involved in various aspects of growth, development and defence of O. tenuiflorum (Table S4). We broadly classified them into five categories, namely, Gene expression (23), metabolism (14), cell signalling (14), transport (9), post translational modification (5) and protein folding (1). It has been shown that approximately one-third of genes in Arabidopsis thaliana and \(\approx 89 \%\) of its transcripts are regulated by varying environmental conditions, like, temperature, light etc.64,65. These studies revealed a tight circadian regulation of A. thaliana genes at transcriptional level64, which is consistent with our results that 20 of CC associated proteins are involved in gene expression and regulation (Fig. 4).

Sub network of circadian clock circuitry including both core circadian clock (CC) and core circadian associated (CCa) proteins only (excluding first interactors). Nodes are colored according to the broad functional class of each protein where hot pink represents proteins involved in gene expression, green (signaling), blue (metabolism), yellow (post translational modification) and cyan represents transport proteins.
Circadian regulatory pathway of O. tenuiflorum
The basic circadian regulation model comprises a core oscillator that is a complex web of multiple coupled transcriptional and translational feedback loops (TTFL). The core oscillator perceives external cues through input pathways and temporally organises various physiological processes of the plant66.
The input module of core clock
Proteins Ote100059320041, Ote100047200201 and Ote10021351017 have been predicted to be light perceiving photoreceptors, namely Phytochrome A, Phytochrome B, Adagio protein 1 (ADO1) or ZEITLUPE (ZTL) protein. It is known that phytochromes primarily perceive red and far red light, while cryptochromes and ZEITLUPE (ZTL) family members are blue light photoreceptors. Among the five phytochromes (PHYA-E) in A. thaliana, when subjected to continuous red light, PHYA and PHYB are primary contributors to photo perception and entrainment of circadian rhythms67. Further, these signals are transduced to the core oscillator through an intermediate protein CONSTITUTIVE PHOTOMORPHOGENIC 1 (COP1)68. Proteins Ote237772210011 and Ote100260480091 have been characterised as an E3 ubiquitin ligase COP1. It triggers light gated proteasomal degradation of core clock components ELF3 and GI, directly linking the clock and light perceiving photoreceptors68,69. The expression of ELONGATED HYPOCOTYL5 (HY5), FAR RED ELONGATED HYPOCOTYL3 (FHY3) and FAR RED IMPAIRED RESPONSE1 (FAR1) are under tight regulation of PHYA signalling. During the day they bind directly to the ELF4 promoter and induce its expression70. The ADO1 and the other two members of this family possess an F-Box domain, a PAS-like LOV domain and six kelch repeats. F-Box domain links target protein for proteasomal degradation by an E3 Ubiquitin ligase complex called skp-cullin-F-Box that is comprised of interactions between F-Box protein with SKP1, CULLIN, and RBX171. It has been shown that ADO1 receive the input signal by interacting with carboxy terminus of blue light photoreceptors phyB and cry172 and mediates blue light dependent degradation of TOC1 and PRR5 through GI mediated stabilization73. Ote100194510162 has been characterized as a serine/threonine phosphatase-5 protein. It has been shown to dephosphorylate a specific serine residue on pfr phytochromes. This dephosphorylation enhances phytochrome stability and increases its ability to bind downstream signal transducers and thus enhancing phytochrome mediated photoresponses74. Proteins Ote237887910022 and Ote100030530111 have been characterized as serine/threonine phosphatase-7 that has been shown to implicate in cryptochromes mediated signalling75. Arabidopsis’s serine/threonine phosphatase-7 (AtPP7) has been shown to act as a nuclear localization signalling molecule that dephosphorylates nuclear signalling intermediates (probably unknown) when exposed to blue light. It acts downstream of the cryptochrome and responds to blue light, ensuring stable signalling75.
The core circadian clock
Proteins CCA1 (Ote100217520081) and LHY (Ote100050210101) represent MYB-related transcription factors that heterodimerize at dawn and repress the expression of morning phased PRR family genes, including PRR9 (Ote100275800021), PRR7 (Ote100120330051) and PRR5 (Ote100011940071). In addition, they repress expression of evening phased genes, including TOC1 (Ote100167620091) and the evening complex (EC) members LUX (Ote100252500041), ELF3 (Ote100215050121), ELF4 (Ote100004890041)76,77. The PRR family members are two-component response regulators similar to the ARR family His-Asp phosphorelay systems, except that the aspartate site that accepts the phosphate group is missing. Circadian control at the transcriptional level of all the members of the PRR family has been documented, and they have shown to express sequentially in order of PRR9, PRR7, PRR5, PRR3, and PRR1 within a period of 24h78. At dawn, CCA1 and LHY negatively regulate PRR1, CCA1 HIKING EXPEDITION (CHE) (Ote237864970011) and their own expression. But during the day, CHE levels rise to repress CCA1, and finally, in the evening TOC1 reset the clock cycle by repressing the expression of CHE77. Similarly, PRR7 has also been shown to repress CCA1 and LHY expressions and regulate the expression of other clock components79. It acts as the master regulator of various physiological processes of plants, including response to abiotic stress80, photoperiodic flowering and temperature compensation79. TOC1 (PRR1) also repress the expression of PRR5, LUX, EARLY FLOWERING 4 (ELF4) and GIGANTEA (GI) (Ote100199680211)79. EC component express from evening onwards throughout night and are required for rhythmic expression of CCA1 and LHY. LUX downregulates the expression of PRR9 and itself by binding the LUX binding sites (LBS) in their promoters. EC is believed to indirectly upregulate the expression of CCA1 and LHY by repressing the expression of PRR9, PRR7 and TOC181. REVEILLE family members, including RVE8 (Ote100018670061), RVE4 (Ote100114610051) along with coactivators NIGHT LIGHT-INDUCIBLE AND CLOCK-REGULATED 1 (LNK1) (Ote100153880201) and LNK2 (Ote100241100111) induce the expression of PRR5, TOC1, LUX and ELF482,83. Figure 5 presents the core clock identified in Tulsi.

The core clock module of O. tenuiflorum.
The output pathways regulated by core clock
Ote100001960091 and Ote100189530101 have been identified as Ethylene receptor-1 and ethylene response sensor-1, respectively. Both encode histidine kinases similar to members of the two-component regulator family and have been shown to be integral components in ethylene signalling84. The gaseous hormone has many implications, like, fruit-ripening, senescence, stress response, seed germination and many more, and its signalling has been shown to be under circadian control85. Protein Ote100058660011 has been predicted as Gibberellin (GA) receptor GID1B that acts as a soluble gibberellin receptor. GA is a diterpenoid hormone that regulates various stages of plant growth and development, like, seed germination, development of fruit, pollen tube and flowering time. GID1 binds with biologically active GAs like GA1, GA3, and GA4 and targets a GRAS family protein DELLA that negatively regulates GA signalling for proteasomal degradation86. Proteins Ote100237240051 and Ote100260840121 are predicted to be UDP-N-acetylglucosamine–peptide N-acetylglucosaminyltransferase SPINDLY (SPY) that negatively regulate Gibberellin signalling to inhibit hypocotyl elongation. It has been shown that SPY and GI function together, and GI acts as a light gated negative regulator of SPY function to control various physiological processes like elongation of hypocotyl, flowering time and cotyledon movements87. Ote100003860081, predicted as a two-component response regulator ARR3 that behaves like a bacterial two-component system that negatively regulates cytokine signalling and affects the phosphorylation state of ARRs by transducing the signal through a multi-step His-Asp phosphotransfer process88. Ote100257670011 has been characterized as a Histidine kinase-3 protein that acts as a positive regulator of cytokine signalling89. Reactive oxygen species (ROS) production, signalling and scavenging have been shown to be linked with circadian clock output and plant response to oxidative stress90. Ote100224900091 has been characterized as an ascorbate oxidase that oxidizes ascorbate to MDA and reduces \(\text {O}_{2}\) to \(\text {H}_2\text {O}\)91. Protein Ote100214260021 has been characterized as a regulator of nonsense transcripts UPF2 that participates in nonsense mediated decay (NMD) pathway and regulates the degradation of mRNA containing premature termination codons92. It has been shown to be involved in photoperiod dependent transcriptional regulation of jasmonic acid and salicylic acid induced defence genes LOX2 and VSP2 in response to biotic and abiotic stresses92.
Summary and conclusions
Considering the importance of circadian rhythms in regulating the physiological processes of the plant, the current study reports a novel network based methodology that exploits the topological information of a network to identify circadian connectome, including the core clock (CC) proteins and clock associated proteins. Although we have implemented the proposed framework in O. tenuiflorum, but we can easily adapt it to any other organism having an endogenous clock. It will increase our understanding of various components of circadian circuitry responsible for the circadian rhythms and the underlying mechanisms sustaining these rhythms, which are largely unknown in Tulsi. This study proposes a methodology to identify 24 known core circadian clock genes in Tulsi that leverage in house built HMMs from 56 template plants. Further, we propose a hybrid methodology that combines RWR and GDV local topological measures to identify novel proteins associated with the core clock. Based on RWR, GDV, and two statistical significance tests 70 putative core clock associated genes proteins were obtained. Functional annotation of the candidate proteins was performed, and we found that many proteins are involved in the regulation of gene expression. We obtained crucial proteins transducing red, far-red and blue light signals to the core oscillator. Furthermore, we obtained proteins involved in different physiological processes, like, gibberellin signalling, ethylene signalling, cytokine signalling, abiotic and biotic stress response etc. We hope that the proposed framework for reporting genome-wide identification and characterization of the core circadian clock and associated proteins in O. tenuiflorum shall pave the way for prioritizing the CC or any other biological process associated proteins in plants.
Data availability
All data is available in the manuscript or the supplementary materials and codes used are available at GitHub https://github.com/vikramsinghlab/Pcrp.
References
Alabadí, D. et al. Reciprocal regulation between toc1 and LHY/CCA1 within the Arabidopsis circadian clock. Science 293(5531), 880–883 (2001).
RobertsonMcClung, C. Plant circadian rhythms. Plant Cell 18(4), 792–803 (2006).
Singh, R. B., Singh, V. & Ramaswamy, R. Stochastic synchronization of circadian rhythms. J. Syst. Sci. Complex. 23(5), 978–988 (2010).
Harmer, S. L. et al. Orchestrated transcription of key pathways in Arabidopsis by the circadian clock. Science 290(5499), 2110–2113 (2000).
Greenham, K. & McClung, C. R. Integrating circadian dynamics with physiological processes in plants. Nat. Rev. Genet. 16(10), 598–610 (2015).
Covington, M. F., Maloof, J. N., Straume, M., Kay, S. A. & Harmer, S. L. Global transcriptome analysis reveals circadian regulation of key pathways in plant growth and development. Genome Biol. 9(8), 1–18 (2008).
Yakir, E., Hilman, D., Harir, Y. & Green, R. M. Regulation of output from the plant circadian clock. FEBS J. 274(2), 335–345 (2007).
Dodd, A. N. et al. Plant circadian clocks increase photosynthesis, growth, survival, and competitive advantage. Science 309(5734), 630–633 (2005).
Wang, W. et al. Timing of plant immune responses by a central circadian regulator. Nature 470(7332), 110–114 (2011).
Lu, H., RobertsonMcClung, C. & Zhang, C. Tick tock: Circadian regulation of plant innate immunity. Annu. Rev. Phytopathol. 55, 287–311 (2017).
Hotta, C. T. et al. Modulation of environmental responses of plants by circadian clocks. Plant Cell Environ. 30(3), 333–349 (2007).
Yerushalmi, S. & Green, R. M. Evidence for the adaptive significance of circadian rhythms. Ecol. Lett. 12(9), 970–981 (2009).
Nohales, M. A. & Kay, S. A. Molecular mechanisms at the core of the plant circadian oscillator. Nat. Struct. Mol. Biol. 23(12), 1061–1069 (2016).
Robertson McClung, C. Beyond Arabidopsis: The circadian clock in non-model plant specie, in Seminars in Cell Developmental Biology Vol. 24, 430–436 (Elsevier, 2013).
Eelderink-Chen, Z. et al. A circadian clock in a nonphotosynthetic prokaryote. Sci. Adv. 7(2), eabe2086 (2021).
Edgar, R. S. et al. Peroxiredoxins are conserved markers of circadian rhythms. Nature 485(7399), 459–464 (2012).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 49(D1), D10 (2021).
Hughes, M. E. et al. Guidelines for genome-scale analysis of biological rhythms. J. Biol. Rhythms 32(5), 380–393 (2017).
Singh, G. & Singh, V. Systems scale characterization of circadian rhythm pathway in camellia sinensis. Comput. Struct. Biotechnol. J. 20, 598–607 (2022).
Davis, D., Yaveroğlu, Ö. N., Malod-Dognin, N., Stojmirovic, A. & Pržulj, N. Topology-function conservation in protein-protein interaction networks. Bioinformatics 31(10), 1632–1639 (2015).
Nabieva, E., Jim, K., Agarwal, A., Chazelle, B. & Singh, M. Whole-proteome prediction of protein function via graph-theoretic analysis of interaction maps. Bioinformatics 21(suppl–1), i302–i310 (2005).
Milenković, T. & Pržulj, N. Uncovering biological network function via graphlet degree signatures. Cancer Inform. 6, CIN-S680 (2008).
Sharan, R. & Ideker, T. Modeling cellular machinery through biological network comparison. Nat. Biotechnol. 24(4), 427–433 (2006).
Glynn, E. F., Chen, J. & Mushegian, A. R. Detecting periodic patterns in unevenly spaced gene expression time series using lomb-scargle periodograms. Bioinformatics 22(3), 310–316 (2006).
Yang, R. & Zhen, S. Analyzing circadian expression data by harmonic regression based on autoregressive spectral estimation. Bioinformatics 26(12), i168–i174 (2010).
Hughes, M. E., Hogenesch, J. B. & Kornacker, K. Jtk_cycle: An efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J. Biol. Rhythms 25(5), 372–380 (2010).
Thaben, P. F. & Westermark, P. O. Detecting rhythms in time series with rain. J. Biol. Rhythms 29(6), 391–400 (2014).
Gang, W., Anafi, R. C., Hughes, M. E., Kornacker, K. & Hogenesch, J. B. Metacycle: An integrated r package to evaluate periodicity in large scale data. Bioinformatics 32(21), 3351–3353 (2016).
Agostinelli, F., Ceglia, N., Shahbaba, B., Sassone-Corsi, P. & Baldi, P. What time is it? Deep learning approaches for circadian rhythms. Bioinformatics 32(12), i8–i17 (2016).
Mei, W. et al. Genome-wide circadian rhythm detection methods: Systematic evaluations and practical guidelines. Brief. Bioinform. 22(3), bbaa135 (2021).
MarcMaurice, C. Tulsi - Ocimum sanctum: A herb for all reasons. J. Ayurveda Integr. Med. 5(4), 251 (2014).
Pattanayak, P., Behera, P., Das, D. & Panda, S. K. Ocimum sanctum Linn. A reservoir plant for therapeutic applications: An overview. Pharmacogn. Rev. 4(7), 95 (2010).
Pandey, A. K., Singh, P. & Tripathi, N. N. Chemistry and bioactivities of essential oils of some Ocimum species: An overview. Asian Pac. J. Trop. Biomed. 4(9), 682–694 (2014).
Upadhyay, A. K. et al. Genome sequencing of herb tulsi (Ocimum tenuiflorum) unravels key genes behind its strong medicinal properties. BMC Plant Biol. 15, 1–20 (2015).
Shubhra, R. et al. Unravelling the genome of Holy basil: An “incomparable’’ “elixir of life’’ of traditional Indian medicine. BMC Genom. 16(1), 1–15 (2015).
Shubhra, R. et al. novo sequencing and comparative analysis of holy and sweet basil transcriptomes. BMC Genom. 15(1), 1–18 (2014).
Singh, V., Singh, G. & Singh, V. Tulsipin: An interologous protein interactome of Ocimum tenuiflorum. J. Proteome Res. 19(2), 884–899 (2019).
Singh, P., Kalunke, R. M. & Giri, A. P. Towards comprehension of complex chemical evolution and diversification of terpene and phenylpropanoid pathways in ocimum species. RSC Adv. 5(129), 106886–106904 (2015).
Jiang, S.-Y., Jin, J., Sarojam, R. & Ramachandran, S. A comprehensive survey on the terpene synthase gene family provides new insight into its evolutionary patterns. Genome Biol. Evol. 11(8), 2078–2098 (2019).
Lamesch, P. et al. The arabidopsis information resource (tair): Improved gene annotation and new tools. Nucleic Acids Res. 40(D1), D1202–D1210 (2012).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 7(1), 539 (2011).
Köhler, S., Bauer, S., Horn, D. & Robinson, P. N. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 82(4), 949–958 (2008).
Chen, L. et al. A computational method for the identification of new candidate carcinogenic and non-carcinogenic chemicals. Mol. BioSyst. 11(9), 2541–2550 (2015).
Pržulj, N., Corneil, D. G. & Jurisica, I. Modeling interactome: scale-free or geometric?. Bioinformatics 20(18), 3508–3515 (2004).
Pržulj, N. Biological network comparison using graphlet degree distribution. Bioinformatics 23(2), e177–e183 (2007).
Yaveroğlu, Ö. N. et al. Revealing the hidden language of complex networks. Sci. Rep. 4(1), 1–9 (2014).
Joel, C. M. & Aric, H. Efficient generation of networks with given expected degrees. In International Workshop on Algorithms and Models for the Web-Graph 115–126 (Springer, 2011).
Singh, G., Singh, V. & Singh, V. Genome-wide interologous interactome map (teagpin) of camellia sinensis. Genomics 113(1), 553–564 (2021).
Carmona-Saez, P., Chagoyen, M., Tirado, F., Carazo, J. M. & Pascual-Montano, A. Genecodis: A web-based tool for finding significant concurrent annotations in gene lists. Genome Biol. 8(1), 1–8 (2007).
Apweiler, R. et al. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 32(suppl–1), D115–D119 (2004).
Berardini, T. Z. et al. The Arabidopsis information resource: Making and mining the “gold standard’’ annotated reference plant genome. Genesis 53(8), 474–485 (2015).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215(3), 403–410 (1990).
Zhou, D., Zhou, X., Ling, Y., Zhang, Z. & Zhen, S. Agrigo: A go analysis toolkit for the agricultural community. Nucleic Acids Res. 38(suppl–2), W64–W70 (2010).
Ye, J. et al. Wego: A web tool for plotting go annotations. Nucleic Acids Res. 34(suppl–2), W293–W297 (2006).
Kanehisa, M. & Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30 (2000).
Jones, P. et al. Interproscan 5: Genome-scale protein function classification. Bioinformatics 30(9), 1236–1240 (2014).
Rastogi, S. et al. Ocimum metabolomics in response to abiotic stresses: Cold, flood, drought and salinity. PLoS ONE 14(2), e0210903 (2019).
Pandey, P., Irulappan, V., Bagavathiannan, M. V. & Senthil-Kumar, M. Impact of combined abiotic and biotic stresses on plant growth and avenues for crop improvement by exploiting physio-morphological traits. Front. Plant Sci. 8, 537 (2017).
Mizoguchi, T. et al. LHY and CCA1 are partially redundant genes required to maintain circadian rhythms in Arabidopsis. Dev. Cell 2(5), 629–641 (2002).
RobertsonMcClung, C. The genetics of plant clocks. Adv. Genet. 74, 105–139 (2011).
SheenX, L., Knowles, S. M., Andronis, C., Ong, M. S. & Tobin, E. M. Circadian clock associated1 and late elongated hypocotyl function synergistically in the circadian clock of Arabidopsis. Plant Physiol. 150(2), 834–843 (2009).
Lü, L. et al. Vital nodes identification in complex networks. Phys. Rep. 650, 1–63 (2016).
Vazquez, A., Flammini, A., Maritan, A. & Vespignani, A. Global protein function prediction from protein-protein interaction networks. Nat. Biotechnol. 21(6), 697–700 (2003).
Michael, T. P. & McClung, C. R. Enhancer trapping reveals widespread circadian clock transcriptional control in Arabidopsis. Plant Physiol. 132(2), 629–639 (2003).
Michael, T. P. et al. Network discovery pipeline elucidates conserved time-of-day-specific cis-regulatory modules. PLoS Genet. 4(2), e14 (2008).
Deepti, S. et al. Role of circadian rhythm in plant system: An update from development to stress response. Environ. Exp. Bot. 162, 256–271 (2019).
Somers, D. E., Devlin, P. F. & Kay, S. A. Phytochromes and cryptochromes in the entrainment of the Arabidopsis circadian clock. Science 282(5393), 1488–1490 (1998).
Sheerin, D. J. et al. Light-activated phytochrome a and b interact with members of the spa family to promote photomorphogenesis in Arabidopsis by reorganizing the cop1/spa complex. Plant Cell 27(1), 189–201 (2015).
Jae-Woong, Y. et al. Cop1 and elf3 control circadian function and photoperiodic flowering by regulating gi stability. Mol. Cell 32(5), 617–630 (2008).
Li, G. et al. Coordinated transcriptional regulation underlying the circadian clock in Arabidopsis. Nat. Cell Biol. 13(5), 616–622 (2011).
Han, L., Mason, M., Risseeuw, E. P., Crosby, W. L. & Somers, D. E. Formation of an SCFZTL complex is required for proper regulation of circadian timing. Plant J. 40(2), 291–301 (2004).
Jarillo, J. A. et al. An Arabidopsis circadian clock component interacts with both cry1 and phyb. Nature 410(6827), 487–490 (2001).
Kim, W.-Y. et al. Zeitlupe is a circadian photoreceptor stabilized by gigantea in blue light. Nature 449(7160), 356–360 (2007).
Ryu, J. S. et al. Phytochrome-specific type 5 phosphatase controls light signal flux by enhancing phytochrome stability and affinity for a signal transducer. Cell 120(3), 395–406 (2005).
Møller, S. G., Kim, Y.-S., Kunkel, T. & Chua, N.-H. Pp7 is a positive regulator of blue light signaling in Arabidopsis. Plant Cell 15(5), 1111–1119 (2003).
Gendron, J. M. et al. Arabidopsis circadian clock protein, toc1, is a DNA-binding transcription factor. Proc. Natl. Acad. Sci. 109(8), 3167–3172 (2012).
Pruneda-Paz, J. L., Breton, G., Para, A. & Kay, S. A. A functional genomics approach reveals CHE as a component of the Arabidopsis circadian clock. Science 323(5920), 1481–1485 (2009).
Matsushika, A., Makino, S., Kojima, M. & Mizuno, T. Circadian waves of expression of the APRR1/TOC1 family of pseudo-response regulators in Arabidopsis thaliana: Insight into the plant circadian clock. Plant Cell Physiol. 41(9), 1002–1012 (2000).
Nakamichi, N. et al. Pseudo-response regulators 9, 7, and 5 are transcriptional repressors in the Arabidopsis circadian clock. Plant Cell 22(3), 594–605 (2010).
Liu, T., Carlsson, J., Takeuchi, T., Newton, L. & Farre, E. M. Direct regulation of abiotic responses by the a rabidopsis circadian clock component PRR 7. Plant J. 76(1), 101–114 (2013).
Pokhilko, A. et al. The clock gene circuit in Arabidopsis includes a repressilator with additional feedback loops. Mol. Syst. Biol. 8(1), 574 (2012).
Xie, Q. et al. Lnk1 and lnk2 are transcriptional coactivators in the Arabidopsis circadian oscillator. Plant Cell 26(7), 2843–2857 (2014).
Farinas, B. & Mas, P. Functional implication of the MYB transcription factor rve8/lcl5 in the circadian control of histone acetylation. Plant J. 66(2), 318–329 (2011).
Clark, K. L., Larsen, P. B., Wang, X. & Chang, C. Association of the Arabidopsis ctr1 raf-like kinase with the etr1 and ers ethylene receptors. Proc. Natl. Acad. Sci. 95(9), 5401–5406 (1998).
Thain, S. C. et al. Circadian rhythms of ethylene emission in Arabidopsis. Plant Physiol. 136(3), 3751–3761 (2004).
Ueguchi-Tanaka, M. et al. Gibberellin insensitive dwarf1 encodes a soluble receptor for gibberellin. Nature 437(7059), 693–698 (2005).
Tseng, T.-S., Salomeé, P. A., McClung, C. R. & Olszewski, N. E. Spindly and gigantea interact and act in Arabidopsis thaliana pathways involved in light responses, flowering, and rhythms in cotyledon movements. Plant Cell 16(6), 1550–1563 (2004).
To, J. P. C. et al. Type-a Arabidopsis response regulators are partially redundant negative regulators of cytokinin signaling. Plant Cell 16(3), 658–671 (2004).
Nishimura, C. et al. Histidine kinase homologs that act as cytokinin receptors possess overlapping functions in the regulation of shoot and root growth in Arabidopsis. Plant Cell 16(6), 1365–1377 (2004).
Lai, A. G. et al. Circadian clock-associated 1 regulates ros homeostasis and oxidative stress responses. Proc. Natl. Acad. Sci. 109(42), 17129–17134 (2012).
Gapper, C. & Dolan, L. Control of plant development by reactive oxygen species. Plant Physiol. 141(2), 341–345 (2006).
Shi, C., Baldwin, I. T. & Jianqiang, W. Arabidopsis plants having defects in nonsense-mediated MRNA decay factors UPF1, UPF2, and UPF3 show photoperiod-dependent phenotypes in development and stress responses. J. Integr. Plant Biol. 54(2), 99–114 (2012).
Acknowledgements
VS (First author) thanks Council of Scientific and Industrial Research (CSIR), India for providing Senior Research Fellowship (JRF).
Author information
Authors and Affiliations
Contributions
V.S. (Second author) conceptualized and designed the research framework. V.S. (First author) performed the computational experiments. V.S. (First author) and V.S. (Second author) analyzed the data and interpreted results. V.S. (First author) and V.S. (Second author) wrote and finalized the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, V., Singh, V. Characterizing the circadian connectome of Ocimum tenuiflorum using an integrated network theoretic framework. Sci Rep 13, 13108 (2023). https://doi.org/10.1038/s41598-023-40212-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-40212-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
