
Abstract
Drug-resistant Plasmodium falciparum (Pf) infections are a major burden on the population and the healthcare system. The establishment of Pf resistance to most existing antimalarial therapies has complicated the problem, and the emergence of resistance to artemisinin derivatives is even more concerning. It is increasingly difficult to cure malaria patients due to the limited availability of effective antimalarial drugs, resulting in an urgent need for more efficacious and affordable treatments to eradicate this disease. Herein, new nucleoside analogues including morpholino-nucleoside hybrids and thio-substituted nucleoside derivatives were prepared and evaluated for in vitro and in vivo antiparasitic activity that led a few hits especially nucleoside-thiopyranoside conjugates, which are highly effective against Pf3D7 and PfRKL-9 strains in submicromolar concentration. One adenosine derivative and four pyrimidine nucleoside analogues significantly reduced the parasite burden in mouse models infected with Plasmodium berghei ANKA. Importantly, no significant hemolysis and cytotoxicity towards human cell line (RAW) was observed for the hits, suggesting their safety profile. Preliminary research suggested that these thiosugar-nucleoside conjugates could be used to accelerate the antimalarial drug development pipeline and thus deserve further investigation.
Similar content being viewed by others

Introduction
Malaria continues to be a global health concern, with 247 million infections and 625,000 deaths worldwide in 2021, mainly among children and pregnant women1. The WHO African Region is responsible for a disproportionately large amount of the worldwide malaria burden. Human malaria is caused by five species of the mosquito-borne parasitic protozoa of the genus Plasmodium, the most prevalent and deadly of which is Plasmodium falciparum (Pf), which is accountable for about 90% of malaria-related deaths2. Despite various malaria control and eradication attempts, most countries have not been able to eradicate the disease3. Drug resistance, toxicity, the lack of an effective vaccination, and low drug efficiency are all factors contributing to this4. The ongoing battle against Plasmodium drug resistance involves the exploration and development of wide range of therapeutics5. The current vaccine used for prevention of Pf malaria, RTS, S/AS016 provides only moderate protection, although a pipeline of new vaccine candidates provides optimism. Further, the effective use of the frontline chemotherapy, Artemisinin Combination Therapy (ACTs), is now threatened by the emergence of resistance7.
Due to the lack of a vaccine and the growing resistance to current drugs, it is extremely important to develop new antimalarials and, in particular, to design drug candidates with a different structure and mechanism of action than those of current therapeutics8. Because pathogenic protozoa lack the ability to synthesize purines via de novo pathway, they rely on the salvage and reutilization of preformed purines for development and proliferation. They have various enzymes that are not found in mammals and can be used as therapeutic targets9. Nucleoside and nucleotide analogues are among the most promising class of potential antimalarial drugs because they can act as inhibitors in both the de novo pathway for pyrimidine nucleotide biosynthesis and the salvage pathway for purine nucleotides of Pf10. Acyclo nucleosides11, as well as 5’-thio nucleosides12 have been reported as potent inhibitors of Pf. Saturated six-membered nitrogen heterocycles, including morpholine, are common structural elements of antiplasmodial compounds13,14.
Recently, we have developed the synthetic methods for the preparation of new types of carbohydrate-modified nucleoside analogues, which can be divided into two major groups. The first group includes morpholino-nucleoside hybrids composed of morpholino unit(s) at the 5’-end and a nucleoside or 2’-deoxyribonucleoside at the 3’-end15. The second group consists of configurationally altered, l-lyxo, d-xylo or d-arabino configured nucleoside derivatives bearing a sulfanylmethyl-linked substituent at different positions of the furanose ring, which can be prepared by photoinitiated radical addition of various thiols onto C4’, C3’ or C2’ exomethylene moiety of nucleosides16,17,18,19. Regarding the great potential of nucleoside analogues11,12 as well as morpholine-containing derivatives13,14 in antimalarial therapy, we decided to test the antiplasmodial activity of the newly developed types of nucleoside analogues. Nucleoside transporters play a role in the uptake of certain antimalarial compounds, including chloroquine. These transporters facilitate the transport of nucleosides and nucleoside-like compounds across the parasite's plasma membrane. Chloroquine has been suggested to enter Plasmodium falciparum-infected erythrocytes via nucleoside transporters, which act as entry points for the compound into the parasite. This mechanism of uptake allows chloroquine to reach its target site and exert its antimalarial effects20.
Previously prepared morpholino nucleoside hybrids (Fig. 1)15 were included in the biological studies, and a new library of thio-substituted nucleosides (Fig. 2) was also generated to investigate the structural diversity of thiol substituents and to find the group's bioactive components. In this manuscript, we present these two sets of nucleoside-based hybrid molecules that possess antiplasmodial activity based on their evaluation against chloroquine sensitive (Pf3D7) and resistant strains (PfRKL-9). Top five analogues showed inhibitory concentrations (IC50) in the submicromolar range, which were further tested against the chloroquine resistant strain PfRKL-9 and then in vivo effectiveness in mice model.

Morpholino-nucleoside hybrids15 (hit compound is highlighted in blue).

l-Lyxo-, d-xylo and d-arabino configured thiosubstituted nucleoside analogues 7–20 and nucleoside reference compounds 21–26 (hit compounds are highlighted in blue).
Results and discussion
Chemistry
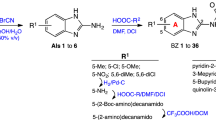
The morpholino-nucleoside hybrids 1–6 involved in antimalarial evaluation are oligonucleotide analogues composed of one or two morpholino units and a natural ribo- (1, 2, 4–6) or 2’-deoxyribonucleoside (3) linked through the nitrogen atom of the morpholine ring (Fig. 1). These hybrid derivatives were prepared from nucleoside 2’,3’-dialdehydes in a double reductive amination-cyclization reaction with 5'-aminonucleosides15. The compounds were subjected to the antimalarial tests in both protected (1–4) and free forms (5 and 6). The second set of compounds tested includes nucleoside analogues derived from adenosine (7–9), uridine (10–17 and 20), and 5-methyluridine (18 and 19) bearing a thioether-linked substituent at the 5-', 3’- or 2'-position of the furanose unit (Fig. 2). A specific feature of the thio-substituted nucleoside analogues 7–20 is that they contain a configurationally modified furanose unit instead of natural d-ribose. Using our recently established photoinduced thiol-ene coupling method21, the l-lyxo (7–9), d-xylo (10–19), and d-arabino (20) configured nucleoside analogues could be obtained with high stereoselectivity by radical mediated addition reactions between C4’-, C3’- or C2’-exomethylene nucleosides and various thiols including amino acid derivatives, 1-thiosugars or alkyl mercaptans16,17,18,19. In addition to the furanose-modified nucleoside analogues 7–20, some simple, protected or partially protected nucleoside derivatives 21–26 were included in the antimalarial studies as reference compounds. The preparation of 10,16 1918 and 21–2615,17 has previously been reported, the synthesis of 7–9, 11–18 and 20 is shown in Fig. 3 and Table 1.

Thio-click-based synthesis of 5’- and 2’-modified nucleoside analogues.
First, the adenosine-derived nucleoside analogue 7 was prepared by reacting 1-thioglucose derivative 27a with the 4’-exomethylene adenosine derivative 2817 under previously optimized thiol-ene coupling conditions using the photoinitiator 2,2-dimethoxy-2-phenylacetophenone (DPAP) and irradiation with UVA light (λmax = 365 nm) (Fig. 3). The photoinitiated thiol-ene reaction begins with the generation of a thiyl radical from the thiol by light-irradiation in the presence of the initiator, then occurs by a free-radical chain mechanism, including a propagation step and a chain transfer step, to produce an anti-Markovnikov type thioether product with full regioselectivity21,22. We used several short irradiation cycles, always adding a new dose of initiator to the reaction mixture, because this has been shown to be more effective than longer-term continuous irradiation16,22. Accordingly, alkene 28 and thiol 27a were reacted by UV irradiation for 4 × 15 min in the presence of 4 × 0.1 equiv. of DPAP. Addition reaction at − 80 °C gave the thioglucose-nucleoside conjugate 7 as a 3:1 mixture of the l-lyxo and d-ribo diastereoisomers. After chromatographic purification, a 6:1 mixture highly enriched in the l-lyxofuranosyl major product was obtained (isomeric purity of 7 was 86%).
We performed removal of protecting groups of 7 to explore their role in antimalarial ativity. Deacetylation under Zemplén conditions yielded 8, while simultanenous deprotection of the isopropylidene and triphenylmethyl groups with 90% aqueous TFA in the presence of Et3SiH provided compound 9.
To produce a 2’-modified nucleoside analogue for the antimalarial assay, the 2’-C-exomethylene-3',5'-O-silylene-acetal derivative of uridine (29) was prepared and subjected to a photoinduced hydrothiolation reaction with butyl mercaptane 27b19. The reaction proceeded with good yield and almost complete stereoselectivity providing the expected d-arabino configured 2’-C-butylsulfanylmethyl nucleoside 20 with 69% yield and an isomeric purity of 91% (20:1 d-arabino: d-ribo ratio of 20).
Next, we prepared a small library of C3’-substituted d-xylo configured nucleoside analogues by addition of thiols 27c-27f onto 3’-C-exomethylene-2’,5’-di-O-tert-butyldimethylsilyl-uridine 30 and -ribothymidine 31 (Table 1). The applied thiols included 2-mercaptomethyl naphthalene 27c, l-cysteine derivative 27d, Mesna 27e, and O-acetylated 1-thiohexopyranoses 27f-27i. The reactions were performed in various solvents selected based on the solubility of the reactants and cooling was used to achieve high diastereoselectivity. Based on our previous results, the thiol-ene couplings were carried out at low temperature (− 40/− 80 °C) in order to achieve high conversion and high stereoselectivity16,17,18,19,21. Using such conditions, the hydrothiolated products (11–18) with the expected d-xylo configuration were indeed obtained efficiently and with excellent diastereomeric excesses.
Biological evaluation
Antimalarial activity by SYBR green I based fluorescence assay
To begin with, cell-based assay was performed to evaluate the inhibitory activities of all 26 compounds (1–26) at 1 µM and 10 µM concentrations on the asynchronous culture of Pf3D7 chloroquine-sensitive strain. Growth of one intraerythrocytic cycle of Pf was monitored after treated with compounds whereas untreated parasites were considered as control. At 10 µM concentration, the majority of the molecules inhibited more than 50% of the parasites (Fig. S1). Five of the twenty-six compounds, morpholino-nucleoside hybrid 1 and thiosubstituted nucleoside analogues 7, 16, 17 and 18 were observed to have significant growth inhibition in a dose dependent manner against Pf3D7. Morpholino-nucleoside hybrids 3 and 5 also showed remarkable actvity against Pf3D7 but they have cytotoxic properties (vide infra). Other compounds, except for the above mentioned seven derivatives showed insignificant percent growth inhibition as compared to untreated control against 3D7 strain of Pf.
Half maximal inhibition concentration (IC50) of the five hit molecules (1, 7, 16–18) was measured against Pf3D7 and chloroquine resistant strain PfRKL-9 (Fig. 4). In this assay, synchronized late trophozoite parasites were treated with different concentrations of the compounds23. The percent growth inhibition of the parasites was evaluated by SYBR Green assay. The IC50 values of compounds 7, 16, 17, 18 and 1 against Pf3D7 were found to be 0.92 ± 1.8 µM, 1.78 ± 1.3 µM, 1.33 ± 1.07 µM, 1.15 ± 1.5 µM and 1.31 ± 1.1 µM, respectively. Next, all five compounds 7, 16, 17, 18 and 1 were evaluated against resistant strain PfRKL-9 and displayed IC50 as 2.1 ± 1.3 µM, 9.55 ± 1.7 µM, 1.84 ± 1.06 µM, 2.21 ± 1.3 µM and 1.83 ± 0.7 µM, respectively. The results are depicted in Fig. 4 and Table 2. The IC50 values of chloroquine against Pf3D7 and PfRKL-9 were found to be 25 nM and 125 nM, respectively. Although, compounds 3 and 5 showed remarkable growth inhibition with IC50 values of 1.19 ± 1.1 µM and 1.34 ± 2.1 µM against Pf3D7; and 4.5 ± 2.5 µM and 6.23 ± 1.5 µM against PfRKL-9 (Fig. S2) but they showed cytotoxicity towards human RBCs (Fig. S3). Due to their cytotoxic nature, these compounds were not considered for further experiments.

Estimation of in vitro growth inhibition and half maximal inhibition concentration (IC50) of hit compounds 7, 16, 17, 18 and 1 against Pf 3D7 and Pf RKL-9 strains (A–E). Graph Pad Prism 9 software was used to calculate IC50 values. Graph showing percent inhibition for the same compound. Experiment was done in triplicate and data expressed as mean values ± SD.
Structure–activity analysis
The in vitro evaluation of compounds 1–26 against the chloroquine sensitive Pf3D7 strain (Fig. S1) allows preliminary analysis of the structure-antimalarial activity relationships of the two new nucleoside chemotypes, morpholino-nucleoside hybrids (1–6) and thiosubstituted nucleoside analogues (7–20).
Five of the six morpholino-nucleoside hybrids tested showed a remarkable ~ 50% inhibitory effect against Pf3D7 strain at a concentration of 1μM (compounds 1–5, Fig. S1A), which clearly proves the high antimalarial potential of these structures.
Among the configurationally altered, thiosubstituted analogues, the thiosugar-containing derivatives were found to have excellent activity against Pf (Fig. S1). The configuration of the thiosugar affected the antimalarial activity to some extent, compounds having gluco- and galacto-configured substituents (7, 16–18) showed excellent, while the mannopyranoside conjugate 15 showed a more moderate growth inhibitory effect. However, it is important to note that even the mannopyranoside-containing nucleoside 15 showed a stronger inhibitory effect (~ 50% inhibition at 1 μM concentration) than the alkylthio-substituted compounds 10, 11 and 19 (~ 5–25% inhibition at 1 μM), suggesting that the thiopyranoside-nucleoside conjugates can have significant antimalarial potential. We have found that the place of substitution did not affect the antimalarial activity, the introduction of a suitable thiosugar in either the 5'- (7) or 3'-position (16–18) resulted in highly active derivatives.
Our study with compounds 7–9 suggests that appropriate lipophilicity is crucial for the activity of thiosubstituted nucleoside analogues. While the deacetylation (7 → 8) did not significantly affect the antimalarial activity, the removal of the lipophilic trityl groups (7 → 9) resulted in a significant decrease in growth inhibitiory effect (Fig. S1A). For the 1-thiosugar-containing hit compounds, the lipophilicity value is clogP ~ 5 (Table 2).
The antimalarial effect of purine nucleoside analogues based on the inhibition of purine metabolism pathway is well documented10,11,12, but there are hardly any results in the literature for pyrimidine-type antimalarial agents24. In this context, it is noteworthy that among the new derivatives tested in this study, several pyrimidine nucleoside analogues (1, 3 and 4 from the morpholino-nucleoside hybrids and 12, 15–18 and 20 from the thiosugar-nucleoside chemotype) showed good/excellent inhibitory effect against Pf.
Effect of compounds on human cell lines
Effect of compounds on human RBCs was checked with the help of spectrophotometry by measuring the lysis of human red blood cells (hRBCs). Compounds were tested at 0.5, 1, 5, 10 and 20 μM concentrations with 10% (v/v) RBCs suspension for 1 h and observed the percentage of RBCs lysis by compounds 7, 16, 17, 18 and 1.
No significant lysis of erythrocytes was observed at concentration of 20 µM (Fig. 5A), while compounds 3 and 5 showed toxicity (Fig. S3). To evaluate the toxic effects of the potent compounds on human RAW cells, MTT assay was carried out and no apparent toxicity effect was observed for compounds 7, 16, 17, 18 and 1 up to 500 µM concentration (Fig. 5B). Table 2 summarizes the in vitro bioassay results and clogP values of the hit compounds and the reference compound chloroquine.

(A) Effect of compounds (7, 16, 17, 18 and 1) on human RBCs. Effect of compounds on human RBCs was checked at 0.5, 1, 5, 10, 20 µM concentration. Absorbance was taken at 415 nm indicated no significant lysis when treated with compounds upto 20 µM. (B) Estimation of in vitro cytotoxicity effect of selected analogues against embryonic kidney cell lines (HEK-293) at 100 µM and 500 µM.
Mitochondrial membrane disruption potential (Δψm)
Mitochondrial membrane potential is a marker of mitochondria’s functional status. Modification in mitochondrial membrane potential leading to mitochondrial dysfunction triggers cell death25,26. Mitochondrial dysfunction was detected by using membrane permeant JC-1 dye. JC-1 is a cationic probe, due to electronegative environment inside the functionally active mitochondria with high Δψm, it aggregates in energised mitochondria and gives red color fluorescence at 590 nm, but in case of low Δψm (depolarized state), JC-1 remain in monomeric form and gives green color fluorescence at 530–10 nm. Decreased ratio of red/green fluorescence intensity of compound treated sample as compared to untreated control indicates depolarized mitochondrial membrane27,28. The damage to the parasite's mitochondria after treatment with compounds 7, 16, 17, 18 and 1 was measured according to the method reported previously28. Because of lipophilic nature of the JC-1, it is cell permeable and emits a green signal (525 nm) in the cytoplasm, but the high transmembrane potential of functional mitochondria causes it to aggregate and emit a red signal (590 nm). In contrast, as shown in Fig. 6, the parasite treated with compounds showed a considerable decrease in JC-1 red staining and an increase in diffused green mitochondrial fluorescence. The loss of mitochondrial membrane potential was demonstrated by a significant fall in the red/green ratio of the JC-1 stained counts in parasites treated with compounds. The parasites treated with IC50 concentrations of compounds 7, 16, 17, 18 and 1 exhibited reduced red/green ratio of 5.26 ± 1.13, 4.98 ± 1.77, 6.28 ± 1.55, 5.53 ± 1.40 and 5.69 ± 0.70. The untreated control parasite showed the red/green ratio of 7.04 ± 1.30 (Fig. 6A and B).

(A) Fluorescence images of parasites displaying monomeric JC-1 (green) in the cytoplasm and JC-1 aggregates (red) in mitochondria. The first row (control) exhibits untreated parasites with bright red signal at 590 nm indicating a functional mitochondrion. Subsequent rows exhibit the effects of the 7, 16, 17, 18 and 1 with bright green and faint red signals. (B) The fluorometric ratio of JC-1 (aggregates)/JC-1 (monomeric) in the parasite population after treatment with 7, 16, 17, 18 and 1 vs. untreated control. Data expressed are mean values ± SD. **P < 0.01 vs. control: Dunnett’s test. Experiments were carried out in triplicate.
In vivo antimalarial activity of compound 7 against P. berghei ANKA
The most potent compound, the 1-thioglucose-adenosine conjugate 7 with purity level of > 95% (Fig. S4) was evaluated for antiplasmodial activity in mice model. In vivo antiplasmodial efficacy of 7 was assessed in P. berghei ANKA infected BALB/c mice model. Compound dose was given intra-peritoneal injections for seven days in a row. To check the parasitemia, thin blood smears were made from P. berghei ANKA infected mice for up to seven consecutive days. At a dose of 50 mg/kg, compound 7 showed 60% inhibition against the rodent-infecting P. berghei ANKA. The percentage parasitemia of group of mice treated with 7 was observed to be 10% as compared with the control wherein percentage parasitemia was 25% on the seventh day (Fig. 7A). Compound 7 was able to effectively reduce the parasite load compared to a control group of untreated mice for seven consecutive days post infection.

Efficacy of compound 7 on P. berghei ANKA infected Balb/c mice. Untreated mice were taken as control. Infected mice was administered intra-peritoneally with 7 (50 mg/kg body weight; n = 5) for seven consecutive days. (A) Percentage parasitemia was calculated by Giemsa-stained thin blood smears from day 1 to day 7 post infection; (B). Survival of mice was observed for 21 days post-infection using Kaplan–Meier survival analysis and statistical differences in animal survival was analyzed by a log rank test.
Groups of mice were observed for mean survival rates for 21 days post-infection. Mean survival time (MST) was calculated, which showed an improvement in mean survival time of mice treated with 50 mg/kg of 7 (MST = 14 days) compared to the control (MST = 10 days). It indicates that compound 7 showed potent antiplasmodial activity against P. berghei ANKA in vivo at a dose of 50 mg/kg and significantly prolonged survival of mice compared to the control group (Fig. 7B). To make relevant inferences, statistical analyses were carried out on the in vivo data using Graphpad Prism data sheets.
Conclusion
Drug-resistant malaria has emerged as a severe threat to worldwide malaria management, necessitating the development of novel antimalarials that are effective against both drug-susceptible and drug-resistant malaria. Hybrid molecules that utilizes new entities with different pharmacophores represent a rational approach in development of novel therapeutics. The present work demonstrates that nucleoside-1-thiosugar hybrids, as synthetic products without cytotoxicity, might be suitable candidates for the development of antimalarial agents against Pf. The IC50 concentrations of the aforementioned nucleoside analogues were determined on Pf 3D7 and PfRKL-9 strains and were found to be between 0.95 µM and 2.21 µM. These analogues, when applied at IC50 concentrations, resulted in depolarization of the membrane potential of treated malaria parasites, leading to cell death. A 50 mg/kg dose of hit compound 7 administered to mice infected with the rodent parasite P. berghei ANKA showed up to a 60% reduction in parasite load, along with prolonged survival of the mice.
In addition to 1-thiosugar-nucleoside conjugates, our study also identified morpholino-nucleoside hybrids as a new chemotype of antimalarial drug candidates. Unfortunately, in the investigated morpholino-nucleoside hybrids, the strong antimalarial effect was usually accompanied by significant cytotoxicity, therefore further optimization is required for this chemotype in order to develop effective and safe antimalarial lead compounds.
Materials and methods
General
Nucleoside derivatives 1–617, 1016 and 1918, naphtylmethylmercaptane 27c29, Fmoc-cysteine derivative 27d17, 1-thiosugars 27f-i30, adenosine-4’-exomethylene 2816, uridine-2’-exomethylene 2931, uridine-3’-exomethylene 3016 and 5-methyluridine-3’-exomethylene 3116 were prepared according to literature procedures. 2,2-Dimethoxy-2-phenylacetophenone (DPAP), n-butyl mercaptane 27b, and MesNa 27e were purchased from Sigma-Aldrich Chemical Co. and used without further purification. Optical rotations were measured with a Perkin-Elmer 241 automatic polarimeter at 20 °C. Reactions were monitored by TLC (Kieselgel 60 F254, Merck) with detection by UV-light (254 nm) and immersing into sulfuric acidic ammonium molybdate solution or 5% ethanolic sulfuric acid followed by heating. Purifications were performed on silica gel 60 (Merck, 0.040–0.063 mm). The 1H NMR (360 and 400 MHz) and 13C NMR (90 and 100 MHz) spectra were recorded with Bruker DRX-360 and Bruker DRX-400 spectrometers at 25 °C. Chemical shifts are referenced to Me4Si (0.00 ppm for 1H) and to the residual solvent signals (CDCl3: 77.2, DMSO-d6: 39.5, CD3OD: 49.0 for 13C). Two-dimensional COSY and 1H − 13C HSQC experiments were used to assist NMR assignments16. The diastereomeric ratio of the new compounds were determined on the basis of their 1H NMR spectra as described earlier18,19. MALDI-TOF MS measurements were carried out with a Bruker Autoflex Speed mass spectrometer equipped with a time-of-flight (TOF) mass analyzer. In all cases 19 kV (ion source voltage 1) and 16.65 kV (ion source voltage 2) were used. For reflectron mode, 21 kV and 9.55 kV were applied as reflector voltage 1 and reflector voltage 2, respectively. A solid phase laser (355 nm, ≥ 100 μJ/pulse) operating at 500 Hz was applied to produce laser desorption and 3000 shots were summed. 2,5-Dihydroxybenzoic acid (DHB) was used as matrix and F3CCOONa as cationising agent in DMF. ESI-QTOF MS measurements were carried out on a maXis II UHR ESI-QTOF MS instrument (Bruker), in positive ionization mode32. The following parameters were applied for the electrospray ion source: capillary voltage: 3.5 kV; end plate offset: 500 V; nebulizer pressure: 0.8 bar; dry gas temperature: 200 °C and dry gas flow rate: 4.5 L/min. Constant background correction was applied for each spectrum; the background was recorded before each sample by injecting the blank sample matrix (solvent). Na-formate calibrant was injected after each sample, which enabled internal calibration during data evaluation. Mass spectra were recorded by otofControl version 4.1 (build: 3.5, Bruker) and processed by Compass DataAnalysis version 4.4 (build: 200.55.2969).
The photoinitiated reactions were carried out in a borosilicate vessel by irradiation with a low-pressure Hg-lamp (Osram Supratec UV, HTC 150–211, 150 W, 230 V, R7s) giving maximum emission at 365 nm, without any caution to exclude air or moisture16. The experimental set-up consists of the reaction vessel and the cooling medium (acetone–liquid nitrogen mixture) in a Dewar flask and an UV-lamp. Before irradiation, the entire set-up is covered by an aluminum foil tent to protect the laboratory personnel against UV light16,30. The purity of hit compounds was assessed on HPLC system (Gilson, USA) with an analytical column (C18) and a Thermo Separation Spectra SERIES UV100 detector coupled with software. The mobile phase for the analysis was prepared from acetonitrile and water (v/v) and compounds showed purity > 95%.
The study was carried out accordingly to relevant guidelines. Mice-based in vivo experiments are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Chemical synthesis
General method for photoinitiated free radical thiol-ene reaction16,30
The corresponding alkene, thiol and DPAP (0.1 eqiuv/alkene) were dissolved in the given solvent to obtain a solution with a concentration range of 0.2–0.5 M for the alkene (the solution should be as concentrated as possible). The reaction mixture was cooled to the given temperature and irradiated with UV-light for 15 min. After irradiation another 0.1 eqiuv. of DPAP was added and the irradiation continued for another 15 min. The addition of 0.1 equiv. of DPAP and the irradiation was repeated two more times. The solvent was evaporated in vacuo and the crude product was purified by flash column chromatography.
1-[2’,3’-O-isopropylidene-5’-S-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)-5’-thio-α-l-lyxofuranosyl]-N-trityl-adenine (7)
Compound 28 (200 mg, 0.376 mmol), 1-thioglucose-peracetate 27a (160 mg, 0.452 mmol, 1.2 equiv.) and DPAP (9.6 mg, 0.0376 mmol, 0.1 equiv.) dissolved in toluene (2 mL) and were reacted at − 80 °C according to the general method. The residue was purified by flash column chromatography (gradient elution hexane/acetone 85/15 → 8/2 → 6/4) to give compound an inseparable 3:1 mixture of 1-[2’,3’-O-isopropylidene-5’-S-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)-5’-thio-α-l-lyxofuranosyl]-N-trityl-adenine (7) and its 4’-epimer 2’,3’-O-isopropylidene-5’-S-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)-5’-thioadenosine (213 mg, 64%, ~ 3:1 mixture of l-lyxo: d-ribo isomers) as an amorphous solid. After a second chromatographic purification, the diastereomeric purity of 7 was increased to l-lyxo: d-ribo 6:1. Rf = 0.08 (hexane:acetone 8:2), 1H NMR (400 MHz, CDCl3) δ (ppm) 7.98, 7.83 (2xs, 2 × 1H, H-2, H-8), 7.34 (d, J = 7.1 Hz, 8H, aromatic), 7.29–7.19 (m, 13H, aromatic), 6.98 (s, 1H, NH), 5.98 (s, 1H, H-1’), 5.48 (d, J = 5.9 Hz, 1H), 5.21 (dd, J = 6.2, 3.1 Hz, 2H), 5.08 (td, J = 9.7, 4.0 Hz, 3H), 4.64–4.56 (m, 2H), 4.21 (m, 1H), 4.14 (d, J = 4.5 Hz, 1H), 4.09 (dt, J = 12.4, 3.0 Hz, 2H), 3.65 (ddd, J = 10.0, 4.3, 2.2 Hz, 2H), 3.15 (dd, J = 13.7, 7.6 Hz, 1H), 2.88 (dd, J = 13.7, 6.1 Hz, 1H), 2.05 (s, 3H), 2.03 (s, 3H), 2.02 (s, 3H), 2.00 (s, 3H), 1.99 (s, 3H),1.57 (s, 3H, i-propylidene CH3), 1.41 (s, 3H, i-propylidene CH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 170.5, 170.2, 169.4, 169.3 (4C, 4 × AcCO), 154.2 (1C, Cq adenine), 152.4 (2C, 2 × adenine CH) 148.3, 144.9 (3C, 3 × aromatic Cq), 129.0, 127.9, 127.0 (15C, 15 × aromatic CH), 121.3 (1C, Cq adenine), 113.4 (1C, i-propylidene Cq), 90.2, 85.0, 84.1, 83.6, 81.3, 76.0, 74.0, 69.9, 68.2 (9C, skeletal carbons), 71.4 (1C, N-TrtCq), 61.9 (1C, C-6’’), 28.3 (1C, C-5’), 26.4, 25.1 (2C, 2 × i-propylidene CH3), 20.7, 20.6 (4C, 4 × AcCH3). MALDI-ToF MS: m/z calcd for C46H49N5NaO12S+ [M + Na]+ 918.299, found 918.297.
1-[2’,3’-O- Isopropylidene-5’-S-(β-d-glucopyranosyl)-5’-thio-α-l-lyxofuranosyl]-N-trityl-adenine (8)
Compound 7 (150 mg, 0.166 mmol) was dissolved in dry MeOH (2.5 mL), then the pH of the solution was adjusted to approximately 10 and stirred overnight. Next day, the rection mixture was neutralized with Amberlite IR-120 (H+), filtered and evaporated under reduced pressure. The crude product was purified by flash chromatography (CHCl3/MeOH 95/5 → 9/1) to give compound 8 (119 mg, 98%, L-lyxo D-ribo ratio ~ 3:1) as a white solid. Rf = 0.36 (CH2Cl2/MeOH 9/1). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.96 (s, 2H, H-2, H-8), 7.32 (d, J = 7.6 Hz, 8H, aromatic), 7.16 (dt, J = 22.4, 7.2 Hz, 14H, aromatic), 5.99 (s, 1H, H-1’), 5.55 (br. s, 1H, OH), 5.37 (d, J = 5.6 Hz, 1H, H-2’), 5.11–5.08 (m, 1H, H-3’), 4.52 (s, 1H), 4.33 (d, J = 9.3 Hz, 1H), 3.69–3.54 (m, 3H), 3.53–3.47 (m, 1H), 3.44–3.34 (m, 1H), 3.23 (s, 2H), 3.20–3.03 (m, 2H), 3.02–2.84 (m, 2H), 1.26 (s, 6H, 2 × i-propylidene CH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 154.1, 148.4 (2C, 2 × adenine Cq), 144.9 (3C, 3 × aromatic Cq), 129.1, 128.0, 127.0 (15C, aromatic), 120.8 (1C, adenine Cq), 113.4 (1C, i-propylidene Cq), 90.0, 86.6, 85.0, 83.6, 81.1, 79.8, 77.9, 72.7, 69.3 (9C, skeletal carbons), 71.5 (1C, Trt-Cq), 61.4 (1C, C-6’’), 29.2 (1C, C-5’), 26.3, 25.0 (2C, 2 × i-propylidene CH3). MALDI-ToF MS: m/z calcd for C38H41N5NaO8S [M + Na]+ 750.2574, found 750.2568.
1-[5’-S-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl)-5’-thio-α-l-lyxofuranosyl]-adenine (9)
Compound 7 (258 mg, 0.288 mmol) was dissolved in 90% aq. TFA solution (5 mL), then Et3SiH (149 µl, 0.864 mmol, 3.0 equiv.) was added and stirred for 2 h. the reaction mixture was diluted with toluene and evaporated under educed pressure. The crude product was purified by flash chromatography (CHCl3/MeOH 97/3 → 95/5 → 9/1) to give compound 9 (92 mg, 52%, D-ribo: L-lyxo ratio ~ 1:3) as a white solid. Rf = 0.74 (CH2Cl2/MeOH 85/15), MALDI-ToF MS: m/z calcs for C24H31N5NaO12S [M + Na]+ 636.159, found 636.160.
1-[3’-Deoxy-3’-C-(2-naphtyl)methylsulfanylmethyl-2’,5’-di-O-(tert-butyldimethylsilyl)-β-D-xylofuranosyl]-uracil (11)
Compound 30 (200 mg, 0.426 mmol), naphthalen-2-ylmethanethiol 27c (148 mg, 0.853 mmol, 2.0 equiv.) and DPAP (10.6 mg, 0.043 mmol, 0.1 equiv.) were dissolved in THF (1.5 mL) and irradiated for 4 × 15 min at − 40 °C. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography (hexane/acetone 9/1) to give compound 11 (165 mg, 61%, with ~ 10:1 D-xylo:D-ribo ratio) as a colorless syrup. [α]d = + 39.2 (c = 0.12, CHCl3), Rf = 0.20 (hexane/acetone 8/2), 1H NMR (400 MHz, CDCl3) δ (ppm) 7.94 (d, J = 8.2 Hz, 1H, H-6), 7.87–7.74 (m, 4H, aromatic), 7.68 (s, 1H, aromatic), 7.50–7.45 (m, 2H, aromatic), 5.93 (d, J = 6.7 Hz, 1H, H-1’), 5.68 (dd, J = 8.1, 2.0 Hz, 1H, H-5), 4.29 (d, J = 7.4 Hz, 1H, H-4’), 4.10–4.05 (m, 1H, H-2’), 3.97 (dd, J = 11.8, 1.0 Hz, 1H, H-5’a), 3.89 (d, J = 3.0 Hz, 2H, naphthylmethylene CH2), 3.85 (dd, J = 11.8, 2.2 Hz, 1H, H-5’b), 2.72–2.59 (m, 3H, H-3’ & SCH2), 0.89 (s, 9H, t-Bu), 0.69 (s, 9H, t-Bu), 0.08 (s, 3H, SiCH3), 0.05 (s, 3H, SiCH3), − 0.19 (s, 3H, SiCH3), − 0.22 (s, 3H, SiCH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 163.4, 150.8 (2C, 2xCO), 140.5 (1C, C-6), 135.0, 133.3, 132.8 (3C, naphthalene C-4a, C-8a & C-7), 128.8, 127.7, 127.7, 127.4, 126.7, 126.3, 126.0 (7C, 7 × aromatic CH), 102.8 (1C, C-5), 87.6 (1C, C-1’), 79.3 (1C, C-4’), 77.4 (1C, C-2’), 63.7 (1C, C-5’), 46.2 (1C, C-3’), 36.9 (1C, naphthylmethylene CH2), 28.2 (1C, SCH2), 26.0, 25.4 (6C, 2 × SiC(CH3)3), 18.2, 17.6 (2C, 2 × t-BuCq), − 4.8, − 5.5, − 5.7 (4C, 4 × SiCH3). MALDI-ToF MS: m/z calcd for C33H50N2NaO5SSi2 [M + Na]+ 665.2877, found 665.2872.
1-[3’-Deoxy-3’-C-(N-Fmoc-cysteine methyl ester)thiylmethyl-2’,5’-di-O-(tert-butyldimethylsilyl)-β-D-xylofuranosyl]-uracil (12)
Compound 30 (194 mg, 0.414 mmol), N-Fmoc-cysteine-methyl-esther 27d (221 mg, 0.621 mmol, 1.5 equiv.) and DPAP (10.6 mg, 0.041 mmol, 0.1 equiv.) were dissolved in THF (2 mL) and irradiated for 4 × 15 min at − 80 °C. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography (hexane/acetone 9/1 → 85/15) to give compound 12 (219 mg, 64%, ~ 15:1 D-xylo:D-ribo ratio) as a colorless syrup. [α]d = + 43.8 (c = 0.42, CHCl3), Rf = 0.19 (hexane/acetone 8/2), 1H NMR (400 MHz, CDCl3) δ (ppm) 9.73 (s, 1H, uracil NH), 7.97 (d, J = 8.1 Hz, 1H, H-6), 7.77 (d, J = 7.5 Hz, 2H, 2xFmoc ArCH), 7.63 (d, J = 7.4 Hz, 2H, 2xFmoc ArCH), 7.41 (t, J = 7.4 Hz, 2H, 2 × Fmoc ArCH), 7.33 (t, J = 7.4 Hz, 2H, 2 × Fmoc ArCH), 5.98 (d, J = 6.6 Hz, 1H, H-1’), 5.80 (d, J = 8.0 Hz, 1H, CysNH), 5.75 (d, J = 8.1 Hz, 1H, H-5), 4.65 (dd, J = 13.1, 5.5 Hz, 1H, H-α), 4.45 (dt, J = 13.2, 6.7 Hz, 1H, Fmoc CH2a), 4.38 (dd, J = 10.5, 7.1 Hz, 1H, Fmoc CH2b), 4.32–4.26 (m, 1H, H-4’), 4.24 (d, J = 7.0 Hz, 1H, fluorene H-9), 4.15 (dd, J = 9.1, 6.7 Hz, 1H, H-2’), 3.94 (d, J = 11.7 Hz, 1H, H-5’a), 3.87 (dd, J = 11.8, 1.6 Hz, 1H, H-5’b), 3.81 (s, 3H, COOCH3), 3.10 (dd, J = 13.4, 4.9 Hz, 1H, H-βa), 2.99 (dd, J = 13.3, 5.7 Hz, 1H, H-βb), 2.80 (dd, J = 14.7, 7.7 Hz, 2H, SCH2), 2.68 (dt, J = 15.2, 9.1 Hz, 1H, H-3’), 0.96 (s, 9H, t-Bu), 0.87 (s, 9H, t-Bu), 0.15 (s, 6H, 2xSiCH3), 0.02 (s, 3H, SiCH3), − 0.10 (s, 3H, SiCH3).13C NMR (100 MHz, CDCl3) δ (ppm) 171.1 (1C, COOCH3), 163.5 (1C, C-4), 155.7 (1C, Fmoc CO), 150.8 (1C, C-2), 143.7, 141.3 (4C, 4 × Fmoc Cq), 140.5 (1C, C-6), 127.8, 127.1, 125.1, 120.0 (8C, 8 × Fmoc aromatic CH), 102.8 (1C, C-5), 87.7 (1C, C-1’), 79.1 (1C, C-4’), 77.2 (1C, C-2’), 67.3 (1C, Fmoc CH2), 63.6 (1C, C-5’), 53.6, 52.9 (2C, COOCH3 & C-α), 47.1, 46.7 (2C, fluorene C-9 & C-3’), 35.4 (1C, C-β), 30.2 (1C, SCH2), 26.0, 25.5 (6C, 2 × SiC(CH3)3), 18.2, 17.7 (2C, 2 × t-BuCq), − 4.6, − 4.73, − 5.5, − 5.7 (4C, 4xSiCH3). MALDI-ToF MS: m/z calcd for C41H59N3NaO9SSi2 [M + Na]+ 848.341, found 848.358.
1-[3’-Deoxy-3’-C-(2-sodium-sulfonatoethyl-sulfanylmethyl)-2’,5’-di-O-(tert-butyldimethylsilyl)-β-D-xylofuranosyl]-uracil (13)
Compound 30 (200 mg, 0.42 mmol), MesNa 27e (91 mg, 0.56 mmol, 1.3 equiv.) and DPAP (10.6 mg, 0.042 mmol, 0.1 equiv.) were dissolved in a mixture of THF (1.7 mL) and MeOH (0.3 mL) and irradiated for 4 × 15 min at − 80 °C. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography (CHCl3/MeOH 95/5 → 9/1 → 8/2) to give compound 13 (253 mg, 80%, ~ 10:1 D-xylo:D-ribo ratio) as a white solid. [α]d = + 45.5 (c = 0.20, CHCl3), Rf = 0.58 (CH2Cl2/MeOH 8/2), 1H NMR (400 MHz, DMSO) δ (ppm) 7.80 (d, J = 8.1 Hz, 1H, H-6), 5.78 (d, J = 6.7 Hz, 1H, H-1’), 5.70 (d, J = 8.1 Hz, 1H, H-5), 4.24 (d, J = 7.6 Hz, 1H, H-4’), 4.09 (t, J = 7.6 Hz, 1H, H-2’), 3.87 (dd, J = 11.8, 1.7 Hz, 1H, H-5’a), 3.82 (dd, J = 11.8, 1.9 Hz, 1H, H-5’b), 2.81–2.73 (m, 2H, SCH2), 2.72–2.61 (m, 5H, H-3’, CH2SO3Na, SCH2), 0.91 (s, 9H, t-Bu), 0.82 (s, 9H, t-Bu), 0.11 (s, 6H, 2 × SiCH3), 0.01 (s, 3H, SiCH3), -0.14 (s, 3H, SiCH3). 13C NMR (100 MHz, DMSO) δ (ppm) 162.8, 150.7 (2C, 2 × CO), 139.7 (1C, C-6), 102.4 (1C, C-5), 86.8 (1C, C-1’), 78.6 (1C, C-4’), 77.0 (1C, C-2’), 63.2 (1C, C-5’), 51.5 (1C, CH2SO3Na), 45.6 (1C, C-3’), 28.4, 27.3 (2C, 2 × SCH2), 25.8, 25.5 (6C, 2xSiC(CH3)3), 17.9, 17.4 (2C, 2 × t-BuCq), − 4.7, − 5.1, − 5.7, − 5.8 (4C, 4xSiCH3). MALDI-ToF MS: m/z calcd for C24H45N2Na2O8SSi2 [M + Na]+ 655.1951, found 655.1946.
1-[3’-Deoxy-3’-C-((2-acetamido-3,4,6-tri-O-acetyl-β-d-glucopyranosylthio)methyl)-2’,5’-di-O-(tert-butyldimethylsilyl)-β-d-xylofuranosyl]-5-uracil (14)
Compound 30 (100 mg, 0.21 mmol), N-acetyl-2-amino-3,4-di-O-acetyl-2-deoxy-1-thio-β-D-glucopyranose 27f (90 mg, 0.25 mmol, 1.2 equiv.) and DPAP (5.3 mg, 0.021 mmol, 0.1 equiv.) were dissolved in a mixture of toluene (1 mL) and MeOH (0.5 mL) and irradiated for 4 × 15 min at − 80 °C. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography (CH2Cl2/acetone 8/2) to give compound 14 (148 mg, 85%, ~ 12:1 D-xylo:D-ribo ratio) as a white solid. [α]d = + 17.9 (c = 1.09, CHCl3), Rf = 0.25 (hexane/acetone 7/3) 1H NMR (400 MHz, CDCl3) δ (ppm) 7.92 (d, J = 8.1 Hz, 1H, H-6), 6.41 (d, J = 9.3 Hz, 1H, NHAc), 5.87 (d, J = 6.7 Hz, 1H, H-1’), 5.66 (d, J = 8.0 Hz, 1H, H-5), 5.15 (t, J = 9.7 Hz, 1H, H-3’’), 4.94 (t, J = 9.7 Hz, 1H, H-4’’), 4.59 (d, J = 10.3 Hz, 1H, H-1’’), 4.29 (d, J = 5.8 Hz, 1H, H-4’), 4.13–3.97 (m, 4H, H-6’’ab, H-2’ and H-2’’), 3.83 (dd, J = 21.4, 11.4 Hz, 2H, H-5’ab), 3.67 (dt, J = 8.5, 4.7 Hz, 1H, H-5’’), 2.89 (d, J = 7.4 Hz, 1H, SCH2a), 2.85–2.74 (m, 2H, SCH2b & H-3’), 2.07 (s, 3H, AcCH3), 1.97 (s, 6H, 2 × AcCH3), 1.87 (s, 3H, AcCH3), 0.88 (s, 9H, t-Bu), 0.79 (s, 9H, t-Bu), 0.07 (s, 6H, 2 × SiCH3), − 0.04 (s, 3H, SiCH3), − 0.17 (s, 3H, SiCH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 171.1, 170.7, 170.2, 169.3 (4C, 4xAcCO), 163.5, 150.8 (2C, 2 × uracil CO), 140.6 (1C, C-6), 102.7 (1C, C-5), 87.3 (1C, C-1’), 86.4 (1C, C-1″), 79.1 (1C, C-4’), 77.4 (1C, C-2’), 76.0 (1C, C-5″), 73.6 (1C, C-3″), 68.4 (1C, C-4″), 63.8 (1C, C-5’), 62.6 (1C, C-6″), 53.0 (1C, C-2″), 47.4 (1C, C-3’), 29.0 (1C, SCH2), 25.9, 25.5 (6C, 2 × SiC(CH3)3), 23.1, 20.7, 20.6, 20.5 (4C, 4xAcCH3), 18.1, 17.6 (2C, 2 × t-BuCq), -4.6, -4.8, -5.5, -5.8 (4C, 4xSiCH3). MALDI-ToF MS: m/z calcd for C36H61N3NaO13SSi2 [M + Na]+ 854.3361, found 854.3356.
1-[3’-Deoxy-3’-C-(-2,3,4,6-tetra-O-acetyl-β-D-mannopyranosyl-sulfanylmethyl)-2’,5’-di-O-(tert-butyldimethylsilyl)-β-D-xylofuranosyl]-uracil (15)
Compound 30 (200 mg, 0.42 mmol), 2,3,4-tri-O-acetyl-1-thio-β-D-mannopyranose 27 g (186 mg, 0.51 mmol, 1.2 equiv.) and DPAP (10.6 mg, 0.042 mmol, 0.1 equiv.) were dissolved in a mixture of toluene (1 mL) CH2Cl2 (1 mL) and MeOH (1 mL) and irradiated for 4 × 15 min at − 80 °C. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography (hexane/acetone 8/2 → 7/3) to give compound 15 (246 mg, 69%, ~ 50:1 D-xylo:D-ribo ratio) as a white solid. [α]D = + 2.73 (c = 0.11, CHCl3), Rf = 0.32 (hexane/acetone 7/3), 1H NMR (400 MHz, CDCl3) δ (ppm) 9.83 (s, 1H, NH), 7.89 (d, J = 8.1 Hz, 1H, H-6), 5.91 (d, J = 7.0 Hz, 1H, H-1’), 5.67 (dd, J = 8.1, 1.6 Hz, 1H, H-5), 5.52 (d, J = 3.4 Hz, 1H), 5.27 (s, 1H), 5.13 (d, J = 9.9 Hz, 1H, H-4’’), 5.05 (dd, J = 10.0, 3.4 Hz, 1H), 4.69 (s, 1H, H-1″), 4.38 (d, J = 7.4 Hz, 1H, H-4’), 4.16–4.11 (m, 2H, H-6″ab), 4.06 (dd, J = 8.9, 7.4 Hz, 1H, H-2’), 3.86 (q, J = 11.6 Hz, 2H, H-5’ab), 3.76–3.67 (m, 1H, H-5’’), 3.00 (d, J = 10.0 Hz, 1H), 2.82 (d, J = 12.1 Hz, 1H, SCH2a), 2.79–2.70 (m, 2H, SCH2b & H-3’), 2.15 (s, 3H, AcCH3), 2.11 (s, 3H, AcCH3), 2.01 (s, 3H, AcCH3), 1.93 (s, 3H, AcCH3), 0.89 (s, 9H, t-Bu), 0.82 (s, 9H, t-Bu), 0.08 (s, 6H, 2xSiCH3), − 0.05 (s, 3H, SiCH3), − 0.17 (s, 3H, SiCH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 170.8, 170.0, 170.0, 169.6 (4C, 4 × AcCO), 163.3, 150.9 (2C, 2 × uracil CO), 140.2 (1C, C-6), 103.0 (1C, C-5), 87.0 (1C, C-1’), 84.6 (1C, C-1’’), 78.7 (1C, C-4’), 77.1 (1C, C-2’), 76.7 (1C, C-5’’), 71.6, 70.5, 65.5 (1C, C-4’’), 63.9 (1C, C-5’), 62.9 (1C, C-6’’), 53.5, 47.3 (1C, C-3’), 30.4 (1C, SCH2), 25.9, 25.5 (6C, 2 × SiC(CH3)3), 20.6, 20.6, 20.6, 20.5 (4C, 4 × AcCH3), 18.1, 17.7 (2C, 2 × t-BuCq), − 4.6, − 4.8, − 5.5, − 5.7 (4C, 4xSiCH3). MALDI-ToF MS: m/z calcd for C36H60N2NaO14SSi2 [M + Na]+ 855.3201, found 855.3194.
1-[3’-Deoxy-3’-C-(-2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-sulfanylmethyl)-2’,5’-di-O-(tert-butyldimethylsilyl)-β-D-xylofuranosyl]-uracil (16)
Compound 30 (200 mg, 0.42 mmol), 2,3,4-tri-O-acetyl-1-thio-β-D-galactopyranose 27 h (186 mg, 0.51 mmol, 1.2 equiv.) and DPAP (10.6 mg, 0.042 mmol, 0.1 equiv.) were dissolved in a mixture of toluene (1 mL) CH2Cl2 (1 mL) and MeOH (1 mL) and irradiated for 4 × 15 min at − 80 °C. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography (hexane/acetone 8/2 → 7/3) to give compound 16 (305 mg, 86%, ~ 18:1 D-xylo:D-ribo ratio) as a white foam. [α]d = + 32.9 (c = 0.17, CHCl3), Rf = 0.32 (hexane/acetone 7/3) 1H NMR (400 MHz, CDCl3) δ (ppm) 7.89 (d, J = 8.2 Hz, 1H, H-6), 5.89 (d, J = 6.8 Hz, 1H, H-1’), 5.66 (dd, J = 8.1, 1.9 Hz, 1H, H-5), 5.38 (d, J = 3.2 Hz, 1H, H-4’’), 5.13 (t, J = 9.9 Hz, 1H, H-2’’), 5.02 (dd, J = 10.0, 3.3 Hz, 1H, H-3’’), 4.44 (d, J = 9.9 Hz, 1H, H-1’’), 4.28 (d, J = 8.0 Hz, 1H, H-4’), 4.15–3.98 (m, 3H, H-2’ & H-6’’ab), 3.87 (ddd, J = 29.2, 12.3, 3.9 Hz, 3H, H-5’ab & H-5’’), 2.93 (dd, J = 12.4, 4.1 Hz, 1H, SCH2a), 2.82 (t, J = 11.9 Hz, 1H, SCH2b), 2.72 (ddd, J = 21.3, 11.1, 4.5 Hz, 1H, H-3’), 2.08 (s, 3H, AcCH3), 2.05 (s, 3H, AcCH3), 2.01 (s, 3H, AcCH3), 1.92 (s, 3H, AcCH3), 0.89 (s, 9H, t-Bu), 0.81 (s, 9H, t-Bu), 0.07 (s, 6H, 2xSiCH3), − 0.03 (s, 3H, SiCH3), − 0.17 (s, 3H, SiCH3). 13C NMR (100 MHz, CDCl3) δ (ppm)170.5, 170.0, 169.9, 169.4 (4C, 4 × AcCO), 163.3, 150.8 (2C, 2 × uracil CO), 140.3 (1C, C-6), 102.8 (1C, C-5), 87.2 (1C, C-1’), 85.3 (1C, C-1’’), 78.9 (1C, C-4’), 77.3 (1C, C-2’), 74.8 (1C, C-5’’), 71.6 (1C, C-3’’), 67.4 (1C, C-4’’), 66.8 (1C, C-2’’), 63.7 (1C, C-5’), 62.0 (1C, C-6’’), 47.1 (1C, C-3’), 28.5 (1C, SCH2), 25.9, 25.5 (6C, 2 × SiC(CH3)3), 20.7, 20.6, 20.5, 20.5 (4C, 4 × AcCH3), 18.1, 17.6 (2C, 2 × t-BuCq), − 4.6, − 4.8, − 5.5, − 5.8 (4C, 4 × SiCH3). ESI-ToF–MS: m/z calcd for C36H60N2NaO14SSi2 [M + Na]+ 855.3201, found 855.3196.
1-[3’-Deoxy-3’-C-(-2,3,4-tri-O-acetyl-β-D-xylopyranosyl-sulfanylmethyl)-2’,5’-di-O-(tert-butyldimethylsilyl)-β-D-xylofuranosyl]-uracil (17)
Compound 30 (200 mg, 0.42 mmol), 2,3,4-tri-O-acetyl-1-thio-β-D-xylopyranose 27i (151 mg, 0.51 mmol, 1.2 equiv.) and DPAP (10.6 mg, 0.042 mmol, 0.1 equiv.) were dissolved in a mixture of toluene (1 mL) and CH2Cl2 (1 mL) and irradiated for 4 × 15 min at − 80 °C. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography (hexane/acetone 9/1 → 8/2) to give compound 17 (286 mg, 93%, ~ 12:1 D-xylo:D-ribo ratio) as an amorphous solid. [α]D = + 4.4 (c = 0.25 CHCl3), Rf = 0.29 (hexane/acetone 8/2), 1H NMR (400 MHz, CDCl3) δ (ppm) 7.86 (d, J = 8.1 Hz, 1H, H-6), 5.84 (d, J = 6.2 Hz, 1H, H-1’), 5.64 (d, J = 8.1 Hz, 1H, H-5), 5.13 (t, J = 8.6 Hz, 1H, H-3’), 4.90–4.80 (m, 2H, H-2’’ & H-4’’), 4.43 (d, J = 8.8 Hz, 1H, H-1’’), 4.23 (d, J = 7.9 Hz, 1H, H-4’), 4.13 (dd, J = 11.5, 5.0 Hz, 1H, H-5’’a), 4.05 (dd, J = 8.3, 6.8 Hz, 1H, H-2’), 3.82 (s, 2H, H-5’ab), 3.38–3.28 (m, 1H, H-5’’b), 2.82 (s, 1H, SCH2a), 2.81 (s, 1H, SCH2b), 2.63–2.54 (m, 1H, H-3’), 1.97 (s, 9H, 3 × AcCH3), 0.88 (s, 9H, t-Bu), 0.78 (s, 9H, t-Bu), 0.06 (s, 6H, 2 × SiCH3), − 0.04 (s, 3H, SiCH3), − 0.17 (s, 3H, SiCH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 169.8, 169.6, 169.1 (3C, 3 × AcCO), 163.4, 150.8 (2C, 2 × uracil CO), 140.2 (1C, C-6), 102.6 (1C, C-5), 87.7 (1C, C-1’), 84.2 (1C, C-1’’), 79.1 (1C, C-4’), 77.4 (1C, C-2’), 72.0 (1C, C-3’’), 69.4, 68.4 (2C, C-2’’ & C-4’’), 65.5 (1C, C-5’’), 63.3 (1C, C-5’), 47.2 (1C, C-3’), 27.2 (1C, SCH2), 25.8, 25.4 (6C, 2 × SiC(CH3)3), 20.5 (3C, 3xAcCH3), 18.1, 17.5 (2C, 2 × t-BuCq), − 4.8, − 4.9, − 5.7, − 5.8 (4C, 4 × SiCH3). MALDI-ToF MS: m/z calcd for C33H56N2NaO12SSi2 [M + Na]+ 783.299, found 783.296.
1-[3’-Deoxy-3’-C-((2-acetamido-3,4,6-tri-O-acetyl-β-d-glucopyranosylthio)methyl)-2’,5’-di-O-(tert-butyldimethylsilyl)-β-d-xylofuranosyl]-5-methyluracil (18)
Compound 31 (100 mg, 0.21 mmol), N-acetyl-2-amino-3,4-di-O-acetyl-2-deoxy-1-thio-β-D-glucopyranose 27f (90 mg, 0.25 mmol, 1.2 equiv.) and DPAP (5.3 mg, 0.021 mmol, 0.1 equiv.) were dissolved in a mixture of toluene (1 mL) and MeOH (0.5 mL) and irradiated for 3 × 15 min at − 80 °C. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography (CH2Cl2/acetone 8/2) to give compound 18 (134 mg, 77%, ~ 25:1 D-xylo:D-ribo ratio) as a white solid. [α]D = + 14.0 (c = 0.10, CHCl3), Rf = 0.28 (CH2Cl2/acetone 9/1), 1H NMR (400 MHz, CDCl3) δ (ppm) 7.53 (d, J = 0.9 Hz, 1H, H-6), 5.95 (d, J = 9.3 Hz, 1H, NHAc), 5.89 (d, J = 7.0 Hz, 1H, H-1’), 5.19 (t, J = 9.8 Hz, 1H, H-3’’), 5.01 (t, J = 9.7 Hz, 1H, H-4’’), 4.61 (d, J = 10.4 Hz, 1H, H-1’’), 4.33 (d, J = 8.2 Hz, 1H, H-4’), 4.16 (d, J = 2.5 Hz, 1HH-6’’a), 4.13 (d, J = 3.8 Hz, 1H, H-6’’b), 4.12–4.08 (m, 1H, H-2’), 4.05 (d, J = 9.6 Hz, 1H, H-2’’), 3.92 (dd, J = 11.9, 1.1 Hz, 1H, H-5’a), 3.86 (dd, J = 11.8, 1.8 Hz, 1H, H-5’b), 3.71 (ddd, J = 9.8, 5.5, 2.7 Hz, 1H, H-5’’), 2.94 (d, J = 1.6 Hz, 1H, SCH2a), 2.92 (s, 1H, SCH2b), 2.82 (dt, J = 17.7, 9.0 Hz, 1H, H-3’), 2.14 (s, 3H, OAcCH3), 2.04 (s, 6H, 2 × OAcCH3), 1.95, 1.94 (2 s, 2 × 3H, NHAcCH3 & thymine CH3), 0.96 (s, 9H, t-Bu), 0.85 (s, 9H, t-Bu), 0.15 (s, 3H, SiCH3), 0.14 (s, 3H, SiCH3), 0.01 (s, 3H, SiCH3), − 0.13 (s, 3H, SiCH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 171.3, 171.0, 170.2, 169.5 (4C, 4 × AcCO), 163.9, 151.0 (2C, C-2, C-4), 135.7 (1C, C-6), 111.4 (1C, C-5), 87.2 (1C, C-1’), 86.7 (1C, C-1’’), 78.7 (1C, C-4’), 76.8 (1C, C-2’), 76.2 (1C, C-5’’), 73.6 (1C, C-3’’), 68.4 (1C, C-4’’), 63.8 (1C, C5’), 62.7 (1C, C-6’’), 53.3 (1C, C-2’’), 47.4 (1C, C-3’), 29.3 (1C, SCH2), 26.2, 25.7 (6C, 2xSiC(CH3)3), 23.3 (1C, NH AcCH3), 20.9, 20.8, 20.7 (3C, 3xOAcCH3), 18.4, 17.8 (2C, 2 × t-BuCq), 12.4 (1C, thymine CH3), − 4.4, − 4.6, − 5.2, − 5.3 (4C, 4 × SiCH3), MALDI-ToF MS: m/z calcd for C37H63N3NaO13SSi2 [M + Na]+ 868.3518, found 868.3525.
2’-Deoxy-2’-C-butylsulfanylmethyl-3’,5’-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-D-arabinofuranosyl-uracil (20)
Compound 29 (200 mg, 0.41 mmol), n-BuSH 27b (360 µL, 3.3 mmol, 8.0 equiv.) and DPAP (10.6 mg, 0.041 mmol, 0.1 equiv.) were dissolved in THF (1 mL) and irradiated for 4 × 15 min at 0 °C. The reaction mixture was concentrated under reduced pressure. The crude product was purified by flash column chromatography (hexane/acetone 9/1) to give compound 2019 (164 mg, 69% with ~ 20:1 D-arabino:D-ribo ratio) as a colourless syrup. [α]d = + 16.2 (c = 0.26, CHCl3), Rf = 0.32 (hexane/acetone 8/2), 1H NMR (400 MHz, CDCl3) δ (ppm) 9.77 (s, 1H, NH), 7.62 (d, J = 8.1 Hz, 1H, H-6), 6.23 (d, J = 4.4 Hz, 1H, H-1’), 5.69 (dd, J = 8.1, 1.6 Hz, 1H, H-5), 4.35 (t, J = 8.8 Hz, 1H, H-3’), 4.11 (dd, J = 13.3, 1.8 Hz, 1H, H-5’a), 4.01 (dd, J = 13.1, 2.7 Hz, 1H, H-5’b), 3.75 (d, J = 8.1 Hz, 1H, H-4’), 2.89–2.81 (m, 2H, H-2’ & SCH2a), 2.57–2.50 (m, 1H, SCH2b), 2.41 (td, J = 7.2, 2.0 Hz, 2H, SCH2CH2CH2CH3), 1.47 (dt, J = 14.6, 7.4 Hz, 2H, CH2CH2CH2CH3), 1.35–1.29 (m, 2H, CH2CH2CH2CH3), 1.06 (dt, J = 11.3, 4.8 Hz, 28H, 8 × i-PrCH3 & 4 × i-PrCH), 0.86 (t, J = 7.3 Hz, 3H, CH2CH2CH2CH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 163.9, 150.8 (2C, 2xCO), 102.0 (1C, C-5), 84.2 (2C, C-1’, C-4’), 60.4 (1C, C-5’), 49.3 (1C, C-2’), 33.2 (1C, SCH2CH2CH2CH3), 31.4 (1C, SCH2CH2CH2CH3), 29.0 (1C, SCH2), 21.9 (1C, CH2CH2CH2CH3), 17.5, 17.4, 17.3, 17.2, 17.1, 17.1 (8C, 8 × i-PrCH3), 14.1, 13.7, 13.0, 12.8, 12.6 (5C, 4 × i-PrCH & CH2CH2CH2CH3). MALDI-ToF MS: m/z calcd for C26H48KN2O6SSi2 [M + K]+ 611.262, found 611.317.
Parasite culture
Pf3D7 and PfRKL-9 were cultured by following procedure reported by Trager and Jensen33. In brief, culture media used for parasite culture supplemented with the complete RPMI 1640, 0.2% NaHCO3, 0.5% AlbuMax II, 0.1 mM hypoxanthine, 25 mg/mL gentamicin at 37 °C in human O+ RBCs and maintained in mixed gas environment (5% O2, 5% CO2 and 90% N2). In order to examine the morphological changes in the parasite, a blood film must be prepared. A thin blood film was air-dried, fixed with methanol, and allowed to dry before staining. Blood films were stained with a 7.5% Giemsa solution (pH of 7.2) for 15 min. Light microscopy analysis was carried out to evaluate the inhibitory effect of compounds at different concentrations against Pf3D7 and PfRKL-9.
SYBR green I based fluorescence assay
SYBR green I based fluorescence assay was performed to measure the growth inhibition effect of the 26 compounds and further dose dependent assay against Pf3D7, and PfRKL-9 (Indian field strain, source- NIMR, New Delhi, India) was carried out at different concentrations34. In brief, Pf3D7 and PfRKL-9 parasitized erythrocytes (trophozoite stage with 1% parasitemia) were diluted with noninfected erythrocytes at a constant 2% hematocrit in culture medium. Parasitized erythrocytes were treated with different concentrations of compounds ranging from 1 µM to 10 µM, whereas DMSO worked as a control. Triplicate wells of parasitized erythrocytes treated with compounds were seeded in 96 well microtiter plate following incubation at 37 °C for one cycle of parasite growth. Growth inhibition of parasites by compounds was calculated by fluorescence intensity taken at 485 nm excitation and 530 nm emission spectrum. Growth inhibition (%) was calculated as follows: % Inhibition = [1 − % Parasitemia (Treatment)/% Parasitemia (Control)] *100. Data were expressed as mean ± SD.
Cytotoxicity effect of compounds on normal human cells
Cytotoxicity against human embryonic kidney cell line (ATCC) was determined using MTT dye35. In order to measuring the cytotoxicity of the compounds we used MTT dye to detect live/dead cells. RAW cells were stored at 37 °C, 5% CO2 in 75 cm3 sterile culture flasks (Corning) in complete Dulbecco's Modified Eagle Medium (DMEM) culture medium (a) supplemented with 5% Fetal Bovine Serum (FBS), gentamicin (40 mg/mL), with changing the medium thrice a week. The monolayers were washed, counted, diluted in complete medium, distributed in 96-well microtiter plates (5000 cells/well), followed by incubation for another 24 h at 37 °C. The test compounds and DMSO were added in triplicate. After a 48 h incubation at 37 °C, the supernatant was removed and 100 µL of MTT solution in complete DMEM was added to each well, followed by 2 h incubation at 37 °C. The culture plates were read in a spectrophotometer (Tecan infinite M200, nanoquant, UK) with a 490 nm filter, and the cytotoxic concentrations were determined based on a dose response curve.
Hemolytic activity
Human RBCs were from Rotary Blood Bank, Tughlakabad Institutional Area, New Delhi, India. Effect of compounds on RBCs was checked by hemolytic assay36. In brief, RBCs suspension with 10% (v/v) washed with 1 × Phosphate-buffered saline (PBS) (pH 7.4) and again resuspended in 1 × PBS. RBCs suspension was incubated with compounds 7, 16, 17, 18 and 1 at different concentrations (0.5, 1, 5, 10 and 20 μM) for 1 h at 37 °C. Samples were centrifuged and supernatant was taken for absorbance at 415 nm. Triton X-100 with 1% (v/v) was used as a positive control. Experiment was done in triplicate.
Mitochondrial membrane potential
Mitochondrial membrane potential of the malaria parasites was determined by using MitoProbe™ JC-1 assay as described previously24. In brief, trophozoite stage parasites (10–12% parasitemia) were treated with IC50 concentration of compounds 7, 16, 17, 18 and 1 for 3 h at 37 °C in the CO2 incubator. Samples were washed with 1 × PBS by centrifugation at 500 × g for 2 min at RT. Freshly prepared JC-1 dye was added to each sample with working concentration of 5 µg/mL. Incubation was done for 30 min at 37 °C in a CO2 incubator. Cells were washed twice with 1 × PBS. Spectrofluorometer was used to observe mitochondrial membrane potential by measuring fluoroscence intensities from the parasite lysate at wavelength of excitation (520 nm) and emission (590 nm) respectively. In vivo cell imaging was done with the help of Olympus fluoview 3000 confocal microscopy. Live cell imaging was observed under a 100X objective (oil immersion) and band pass filter at 355–460 nm. Experiments were repeated thrice and only representative figures are shown (Fig. 6).
In vivo antiplasmodial activity assay
Antiplasmodial activity of hit compound 7 was investigated in mice model with the help of protocol described previously37. Briefly, intra-peritoneal injection of 1 × 107 Plasmodium berghei ANKA infected erythrocytes diluted with sterile 1 × PBS was injected into mice. In each group five mice were taken. Thin smears were made from the tail blood of mice to check the infection. Post infection, P. berghei infected mice administered intra-peritoneally with compound 7 with the dose of 50 mg/kg body weight, daily for seven consecutive days post infection. Mice treated with 1xPBS were kept as vehicle control. Thin blood smears were made from day 1 to day 7 to count the parasitemia and mice were observed for 21 days for calculating mean survival time. Data were calculated and presented as percentage increase in the parasitemia. All animal experiments were carried out in accordance with the standard approved procedures36. The study was approved by the Institutional Animal Ethics Committee (IAEC) of Jawaharlal Nehru University, New Delhi, India; and the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India.
Statistical analysis
The data was analyzed as one-way analysis of variance (ANOVA) to determine significance between the mean values obtained for control and after treatment with compounds. Dunnett’s test was used to compare the treatment and control and statistical significance was set at P ≤ 0.01.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author (A.B.) on reasonable request.
References
WHO, World malaria report; 2022. https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022 (Accessed 10 March 2023).
Sato, S. Plasmodium—A brief introduction to the parasites causing human malaria and their basic biology. J. Phys. Anthropol. 40(1), 1. https://doi.org/10.1186/s40101-020-00251-9 (2021).
Hall, B. F. & Fauci, A. S. Malaria control, elimination, and eradication: The role of the evolving biomedical research agenda. J. Infect. Dis. 200(11), 1639–1643 (2009).
Thu, A. M., Phyo, A. P., Landier, J., Parker, D. M. & Nosten, F. H. Combating multidrug-resistant Plasmodium falciparum malaria. FEBS J. 284(16), 2569–2578 (2017).
Bushman, M., Antia, R., Udhayakumar, V. & de Roode, J. C. Within-host competition can delay evolution of drug resistance in malaria. PLoS Biol. 16(8), e2005712 (2018).
Tran, T. M., Portugal, S., Draper, S. J. & Crompton, P. D. Malaria vaccines: Moving forward after encouraging first steps. Curr. Trop. Med. Rep. 2(1), 1–3 (2015).
Siddiqui, F. A., Liang, X. & Cui, L. Plasmodium falciparum resistance to ACTs: Emergence, mechanisms, and outlook. Int. J. Parasitol.-Drug 16, 102–118 (2021).
Kumar, S., Bhardwaj, T. R., Prasad, D. N. & Singh, R. K. Drug targets for resistant malaria: Historic to future perspectives. Biomed. Pharmacother. 104, 8–27 (2018).
Zheng, Z. et al. Novel nucleoside-based antimalarial compounds. Bioorg. Med. Chem. Lett. 26(12), 2861–2865 (2016).
Cheviet, T., Lefebvre-Tournier, I., Wein, S. & Peyrottes, S. Plasmodium purine metabolism and its inhibition by nucleoside and nucleotide analogues. J. Med. Chem. 62(18), 8365–8391 (2019).
Cheviet, T. et al. β-Hydroxy- and β-aminophosphonate acyclonucleosides as potent inhibitors of plasmodium falciparum growth. J. Med. Chem. 63(15), 8069–8087 (2020).
Tran, H.-A. et al. Synthesis and activity of nucleoside-based antiprotozoan compounds. Bioorg. Med. Chem. 25(7), 2091–2104 (2017).
Tse, E. G. et al. Nonclassical phenyl bioisosteres as effective replacements in a series of novel open-source antimalarials. J. Med. Chem. 63(20), 11585–11601 (2020).
O’Neill, P. M. et al. Synthesis and profiling of benzylmorpholine 1,2,4,5-tetraoxane analogue N205: Towards tetraoxane scaffolds with potential for single dose cure of malaria. Bioorg. Med. Chem. 26(11), 2996–3005 (2018).
Debreczeni, N. et al. Tightly linked morpholino-nucleoside chimeras: New, compact cationic oligonucleotide analogues. Org. Biomol. Chem. 19, 8711–8721. https://doi.org/10.1039/D1OB01174J (2021).
Bege, M. et al. A low-temperature, photoinduced thiol–ene click reaction: A mild and efficient method for the synthesis of sugar-modified nucleosides. Org. Biomol. Chem. 15(43), 9226–9233 (2017).
Bege, M. et al. Synthesis and oligomerization of cysteinyl nucleosides. Org. Biomol. Chem. 18(40), 8161–8178 (2020).
Bege, M. et al. Synthesis and cytostatic effect of 3’-deoxy-3’-C-sulfanylmethyl nucleoside derivatives with d-xylo configuration. Molecules 24(11), 2173 (2019).
Bege, M. et al. Synthesis and anticancer and antiviral activities of C-2′ -branched arabinonucleosides. Int. J. Mol. Sci. 23, 12566 (2022).
Wang, C. et al. Structural basis of the substrate recognition and inhibition mechanism of Plasmodium falciparum nucleoside transporter PfENT1. Nat. Commun. 14(1), 1727 (2023).
Borbás, A. Photoinitiated thiol-ene reactions of enoses: A powerful tool for stereoselective synthesis of glycomimetics with challenging glycosidic linkages. Chem. Eur. J. 26(28), 6090–6101 (2020).
Lázár, L., Csávás, M., Herczeg, M., Herczegh, P. & Borbás, A. Synthesis of S-linked glycoconjugates and S-disaccharides by Thiol-Ene coupling reaction of enoses. Org. Lett. 14, 4650–4653 (2012).
Sharma, N. et al. Novel antiplasmodial compounds leveraged with multistage potency against the parasite plasmodium falciparum: In vitro and in vivo evaluations and pharmacokinetic studies. J. Med. Chem. 64(12), 8666–8683 (2021).
Belen Cassera, M. B., Zhang, Y., Hazleton, K. Z. & Schramm, V. L. Purine and pyrimidine pathways as targets in plasmodium falciparum. Curr. Top. Med. Chem. 11(16), 2103–2115 (2011).
Gottlieb, E., Armour, S. M., Harris, M. H. & Thompson, C. B. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 10(6), 709–717 (2003).
Roy, A. et al. Mitochondria-dependent reactive oxygen species-mediated programmed cell death induced by 3,3′-diindolylmethane through inhibition of F0F1-ATP synthase in unicellular protozoan parasite leishmania donovani. Mol. Pharmacol. 74(5), 1292–1307 (2008).
Xie, H. et al. LDH-A Inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol. Cancer Ther. 8(3), 626–635 (2009).
Wang, J. et al. Artemisinin directly targets malarial mitochondria through its specific mitochondrial activation. PLoS ONE 5(3), e9582 (2010).
Guo, J.-R., Huang, H.-Y., Yan, Y.-L. & Liang, C.-F. Selective S-Deacetylation of functionalized thioacetates catalyzed by Dy(OTf)3. Asian J. Org. Chem. 7, 179–188 (2018).
Kelemen, V. et al. Stereoselective thioconjugation by photoinduced thiol-ene coupling reactions of hexo-and pentopyranosyl D-and L-glycals at low temperature—Reactivity and stereoselectivity study. Chem. Eur. J. 25, 14477–14477 (2019).
Lin, T.-S. et al. Synthesis and anticancer and antiviral activities of various 2’- and 3’-methylidene-substituted nucleoside analogues and crystal structure of 2’-deoxy-2’-methylidenecytidine hydrochloride. J. Med. Chem. 34, 2607–2615 (1991).
Szűcs, Z. et al. Synthesis of an amphiphilic vancomycin aglycone derivative inspired by polymyxins: Overcoming glycopeptide resistance in Gram-positive and Gram-negative bacteria in synergy with teicoplanin in vitro. Sci. Rep. 12, 20921 (2022).
Trager, W. & Jensen, J. B. Human malaria parasites in continuous culture. Science 193(4254), 673–675 (1976).
Makler, M. T. & Hinrichs, D. J. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Amer. J. Trop. Med. Hyg. 48(2), 205–210 (1993).
Woerdenbag, H. J. et al. Cytotoxicity of artemisinin-related endoperoxides to Ehrlich ascites tumor cells. J. Nat. Prod. 56(6), 849–856 (1993).
Evans, B. C. et al. Ex vivo red blood cell hemolysis assay for the evaluation of PH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs. J. Vis. Exp. 73, e50166 (2013).
Ounjaijean, S., Kotepui, M. & Somsak, V. Antimalarial activity of tinospora baenzigeri against Plasmodium berghei- infected mice. J. Trop. Med. 2019, 5464519 (2019).
Acknowledgements
This research was funded by the National Research, Development and Innovation Office of Hungary (K 132870 to AB). SS acknowledges Drug and Pharmaceuticals Research Programme (DPRP) (Project No. P/569/2016-1/TDT) and DST-SERB (Project No. CRG/2019/002231). VS is a recipient of SERB-NPDF (PDF/2017/001525) from Department of Science and Technology (DST), Government of India, New Delhi. NS acknowledges Council for Scientific and Industrial Research (CSIR) for Senior Research Fellowship. We are grateful to Advanced Instrumentation Research Facility (AIRF), Jawaharlal Nehru University for Live cell imaging. We acknowledge National Institute of Malaria Research for providing PfRKL-9 chloroquine resistant line.
Funding
Open access funding provided by University of Debrecen.
Author information
Authors and Affiliations
Contributions
A.B., S.S., P.H. and B.R. developed the concepts and designed the experiments. M.B., V.S., N.S., N.D., I.B. and P. conducted the experiments and analyzed the data under the supervision of A.B., P.H., S.S. and B.R. M.B., V.S., P., S.S., B.R. and A.B., wrote the initial draft of the manuscript. A.B. and B.R. reviewed and edited the manuscript. A.B., S.S. and B.R. was responsible for funding acquisition. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bege, M., Singh, V., Sharma, N. et al. In vitro and in vivo antiplasmodial evaluation of sugar-modified nucleoside analogues. Sci Rep 13, 12228 (2023). https://doi.org/10.1038/s41598-023-39541-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-39541-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
