
Abstract
We have used FRET-based biosensors in live cells, in a robust high-throughput screening (HTS) platform, to identify small-molecules that alter the structure and activity of the cardiac sarco/endoplasmic reticulum calcium ATPase (SERCA2a). Our primary aim is to discover drug-like small-molecule activators that improve SERCA’s function for the treatment of heart failure. We have previously demonstrated the use of an intramolecular FRET biosensor, based on human SERCA2a, by screening two different small validation libraries using novel microplate readers that detect the fluorescence lifetime or emission spectrum with high speed, precision, and resolution. Here we report results from FRET-HTS of 50,000 compounds using the same biosensor, with hit compounds functionally evaluated using assays for Ca2+-ATPase activity and Ca2+-transport. We focused on 18 hit compounds, from which we identified eight structurally unique scaffolds and four scaffold classes as SERCA modulators, approximately half of which are activators and half are inhibitors. Five of these compounds were identified as promising SERCA activators, one of which activates Ca2+-transport even more than Ca2+-ATPase activity thus improving SERCA efficiency. While both activators and inhibitors have therapeutic potential, the activators establish the basis for future testing in heart disease models and lead development, toward pharmaceutical therapy for heart failure.
Similar content being viewed by others
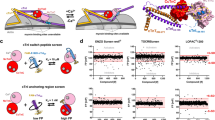


Introduction
Sarco/endoplasmic reticulum calcium ATPase (SERCA), integral to the sarcoplasmic reticulum (SR, muscle) or endoplasmic reticulum (ER, non-muscle) membrane in most mammalian cells, uses Ca2+-dependent hydrolysis of ATP to fuel active transport (uptake) of cytosolic Ca2+ into the SR or ER. The activity of SERCA1a (skeletal isoform) or SERCA2a (cardiac isoform) is essential for muscle relaxation (diastole), restoring SR Ca2+ following its release via Ca2+ channels (ryanodine receptors, RyR) for muscle contraction (systole). Decreased SERCA activity and excessive RyR leak results in failure to maintain the high gradient of [Ca2+] between the cytoplasm (sub-μM) and the SR (mM) during diastole and are associated with heart failure (HF) in humans and animals1. Decreased SERCA activity is related to multiple factors, including reduced SERCA gene expression, increased post-translational modifications, and altered interaction with regulatory proteins1. Overall, decreased SERCA activity and increased Ca2+-leak can lead to a pathophysiological state of the cardiac myocyte2 (HF, cardiac hypertrophy, diabetic hypertrophy), skeletal myofiber (Brody’s disease and myotonic dystrophy)3, or non-muscle cells (Darier’s disease, diabetes, Alzheimer’s disease)4. Altered SERCA interactions with regulatory proteins (regulins), e.g., phospholamban (PLB), have been linked to HF5. Of the seven known regulins6, the dwarf open reading frame (DWORF) peptide is the only one known to activate SERCA, both by direct activation7,8 and by competing with PLB binding9,10, preventing HF in a mouse model of dilated cardiomyopathy11.
Current therapeutic measures for HF include beta-blockers, angiotensin-converting enzyme (ACE) inhibitors, and angiotensin-receptor blockers (ARB). However, these do not directly target proteins responsible for dysfunctional Ca2+ cycling. Discovery of small molecules that target specific transporters, and their interaction with modulatory proteins, is needed to exert improved control of Ca2+ homeostasis for positive therapeutic outcomes. Here, we seek primarily SERCA2a activators to alleviate HF and related arrhythmias, a goal supported by numerous reports12,13, although some controversy remains14. Compounds that uncouple ATPase and transport activities of SERCA are of interest as enhancers of metabolism or thermogenesis, to reduce obesity or hypothermia15,16. SERCA inhibitors are proposed for treatment of cancer or malaria17,18.
SERCA2a is a large transmembrane protein, with the phosphorylation (P) and nucleotide-binding (N) domains forming the catalytic site, influenced by the actuator (A) domain (Fig. 1A). Large (5–10 nm) relative movements of these domains, detected in living cells by an intramolecular FRET biosensor (Fig. 1A)20,21, are coupled to Ca2+ transport. The interaction of small molecules with SERCA can induce structural changes, detectable by this biosensor, that correlate with function, making this a powerful tool for high throughput screening (HTS) discovery of SERCA-binding compounds20,22.

Strategy of this study. (A) FRET biosensor, human two-color SERCA2a (2CS), showing SERCA domains: nucleotide binding (N), phosphorylation (P), actuator (A) and transmembrane (TM). GFP is fused to N, RFP to A. ΔFRET (change in fluorescence resonance energy transfer), calculated from ΔFLT (change in fluorescence lifetime), is used to detect SERCA structural changes induced by compounds. The representative structural states show domain movements based on crystal structures from different nucleotide-bound SERCAs that indicate possible extents of FRET between GFP and RFP in the absence and presence of a compound (small molecule) (⎔): FRET (E2 + Tg (1IWO)), Less FRET (E1 + Ca2+, 1SU4), and More FRET (E1 + Ca2+ + ADP + AlF, 2ZBD)19. Here we focus on hits that decrease FRET (increase FLT). (B) Screening funnel describing the 5-step process in this study, involving measurements of FLT-FRET and SERCA function, with SERCA in live mammalian cells (HEK293) and in isolated pig cardiac SR membranes, respectively: (1) FLT changes caused by test compounds were measured using the SERCA-specific FRET biosensor 2CS (two-color SERCA), to identify 2960 initial hit compounds. False hits were ruled out as compounds that (2) are fluorescent or affect the donor directly, decreasing the hit compounds to 295, (3) decrease FLT (increase FRET), reducing the hit compounds to 160, and (4) affect FRET in a null-biosensor, in which donor and acceptor are separated by a non-functional flexible peptide, reducing the hit compounds to 91, of which 18 were selected for step 5 (see Results under “Removal of null-biosensor effectors”). (5) Compound concentration-dependence of FRET and SERCA function was measured to further prioritize hit compounds for future lead development. Experimental details are provided in Methods.
In previous early-stage drug discovery campaigns, we focused on the SERCA regulator, PLB, via intermolecular FRET biosensor designs23,24. We have also validated intramolecular FRET biosensor constructs of SERCA20,22,25, to detect binding of compounds directly to SERCA. For this, we engineered a “two-color” SERCA (2CS, Fig. 1A) construct with eGFP and tagRFP fluorescent proteins fused to the cytoplasmic N- and A-domains of SERCA, to detect relative motions of these domains during the enzymatic cycle responsible for Ca2+-transport22,25,26. Measuring FRET within 2CS, stably expressed in a mammalian cell line (HEK293), we previously validated this biosensor using the NCC (727 compounds)22 and LOPAC (1280 compounds)25 libraries. The next logical step, in the present study, is to use 2CS in HTS of a 50,000-compound DIVERSet library, a diverse collection of drug-like small molecules that has yielded effective hit compounds in other drug discovery projects27,28,29. HTS was enabled by the FLT-PR (fluorescence lifetime plate reader), which scans a 1536-well plate with unprecedented precision and speed, determining FLT with ~ 0.3% CV (30 times better precision than conventional intensity detection) in 2.5 min23,27,28, making possible a high-precision 50,000-compound screen in 2 days. To remove false positives, a spectral unmixing plate reader (SUPR) was used to provide complementary spectral measurement of compound-induced FRET changes26.
Although other biosensors are under development (e.g., using orange and maroon fluorescent proteins)22,23,26, the GFP/RFP 2CS biosensor has been thoroughly validated for screening the DIVERSet compound library (DIVERSet-CL) using HEK293 cells. To validate selected hit compounds and prioritize those with lead potential, we acquired concentration response curves (CRCs) using FRET and functional assays (Ca2+-ATPase activity and Ca2+-uptake). We hypothesized that the combination of improved fluorescence technology and screening a larger library of compounds would yield a larger and more diverse collection of hit compounds that improve cardiac SERCA function, thus increasing the potential for discovering lead compounds for new heart failure therapeutics.
Results
FLT-FRET HTS of 50K DIVERSet-CL
The 2CS FRET biosensor20,22,25 was incubated with compounds or DMSO (control) for FLT acquisition in the FLT-PR. FLT measurements had a median coefficient of variation (CV) of 0.4% across all plates (Fig. 2A). Plate-by-plate CV varied by < 1% (Fig. 2A).

FLT HTS of the DIVERSet library of 50,000-compounds, using the approach described in Fig. 1A to identify initial hit compounds. (A) Screening precision was determined by computing %CV for each plate using DMSO control wells, with a median value of 0.4% across 40 plates. (B) The change in lifetime (Δτ) was computed to find potential hits (red) with hit threshold set at rZ-score = ± 3, resulting in 2960 initial hits for triage with the SUPR instrument. DMSO controls (dark blue) and compounds not affecting 2CS (light blue) are grouped in the plot to illustrate plate boundaries. (C) The histogram of compounds not affecting 2CS (light blue, 1 ps bin width) shows normal distribution, similar to that of DMSO controls (no compound), as shown by a fit of the populations to Gaussian distributions. The horizontal lines in (B,C) illustrate the approximate cutoffs used, though actual cutoffs were determined on a plate-by-plate basis.
Compounds that significantly altered the structure of 2CS were determined from their change in lifetime (Δτ) vs DMSO controls (2CS plus DMSO), and Δτ was compared to the normal statistical fluctuation of the biosensor by computing the robust (r)Z-score (see Methods). FLT changes induced by potential hit compounds (Fig. 2B, red) are distinct from the normal distributions of DMSO controls (Fig. 2C, dark blue) and of compounds not affecting SERCA2a (Fig. 2C, light blue). A hit threshold was set at rZ-score = ± 3, resulting in 2960 initial hit compounds (Fig. 2B, step 1), which is ~ 10 × more initial hit compounds than we previously identified with an ATPase-based HTS assay30 using a similar threshold. Optically interfering compounds were removed (Fig. 1B, step 2) using a spectral unmixing plate reader to obtain spectra for 2CS that were analyzed relative to the donor-only 1CS sample22 (SUPR, see Methods). 295 compounds remained.
We cannot predict the direction of ΔFLT for activator vs. inhibitor. However, FLT increasers were preferred because: (a) more FLT decreasers were found to fail these tests23,25,26,31, (b) increasers offer greater reproducibility23,25,26, and (c) most previously identified SERCA modulators have been shown to be FLT increasers20,22,25. Therefore, we prioritized 160 FLT increasers (termed “hit compounds” (Fig. 1B, step 3) for retesting with a null-biosensor, to remove false positives.
Removal of null-biosensor effectors
160 hit compounds were retested (Fig. 1B, step 4) using 2CS (Fig. 3A and C; Supplementary Fig. S1) and a null-biosensor (Fig. 3B and D), GFP and RFP connected by a 32-residue unstructured flexible linker peptide (G32R)25, to rule out compounds that directly bind to the fluorescent proteins and alter FLT. ΔFLT (Δτ) from the FLT-PR and Δ(G/R) (change in the ratio of mole fractions of donor [green, G] and acceptor [red, R] in the emission spectrum as determined from a linear combination of component spectra of donor-only and acceptor-only (1CS) samples22) from the SUPR were determined for each compound. As these are two complementary measures of FRET (FRET decreases both), a strong correlation was observed in 2CS for compounds that induced a structural change much greater than observed in the null-biosensor (Fig. 3C and D) (see Methods under “FRET-HTS instrumentation and data analysis”).

Removal of null-biosensor effectors (false positives). The 160 hit compounds that passed steps 1–3 of the screening funnel in Fig. 1B were then selected and dispensed into 1536-well plates containing 10 and 30 μM [compound] (n = 3 wells for each concentration), using the same compound stock solutions as in the HTS phase (step 1 in Fig. 1B). These repeat readings establish the reproducibility needed to select a set of hit compounds for re-purchase as solids, for subsequent studies discussed below. Data are shown from the 30 μM wells for 2CS (A,C) and null-biosensor (B,D). (A) Distribution of significant ΔFLT for the 2CS biosensor. (B) The 160 hit compounds were counter-screened using a null-biosensor. Only five compounds displayed Δτ > 50 ps, indicating that our method for eliminating fluorescent compounds removes nearly all false positives. These five compounds were also excluded from further consideration (Supplementary Fig. S1A). (C) and (D) Plots of ΔFLT vs. Δ(G/R) show excellent, reproducible correlation between the two measurements for the 2CS biosensor (C), but distinct from those observed for the null-biosensor (D), indicating that the hit compounds induce a structural change in the 2CS biosensor.
Hit compounds that produced Δτ ≥ 70 ps (91 compounds, Fig. 1B, step 4; Fig. 3A, Supplementary Fig. S1A), but excluding any that exceeded a 50 ps response in the null-biosensor, were targeted for further functional testing. After determining compound availability for repurchase, we selected 18 compounds with a representative range of ΔFLT (Supplementary Fig. S1B) for CRC testing. None of these compounds were in the PAINS (Pan-Assay INterference compoundS) category32, nor were they redox agents or metal chelators.
Validation of hit compounds using FRET CRC
To further evaluate the 18 hit compounds, we determined ΔFLT for [compound] = 0.78–100 μM (Fig. 1B, step 5) in live HEK cells. All 18 hit compounds (Table 1) decreased FRET (increased FLT) of 2CS relative to 1CS (donor-only biosensor), suggesting a structural change (Fig. 1A) in the cytosolic headpiece of SERCA2a. Compounds 1 and 4 showed a significant decrease in FRET at the lower [compound] with no further effect at higher [compound]; testing was not done at even higher [compound] where non-specific effects are likely to dominate. The remaining 16 compounds decreased FRET efficiency (E) with measurable FRET-EC50 (Fig. 4 and 7C, Fig. 5 and 6B, and Table 1).

A representative activator enhances both Ca2+-ATPase activity and Ca2+-uptake at [Ca2+]MAX (pCa = 5.4) and [Ca2+]MID (pCa = 6.2). (A) Table showing the three categories (C) of activators (N, Compounds 1–9) and their effects on the Ca2+ATPase activity and Ca2+-uptake to activate, uncouple, or inhibit function. Underlined compounds are represented in Figs. 4, 5, and 6. (B) Chemical structure of DS26022409 (Compound 7). (C) CRC of normalized FRET E in live HEK cells shows decreasing FRET in response to increasing [compound]. (D) CRC of Ca2+-ATPase activity of SERCA2a in pCSR vesicles show activation at [Ca2+]MAX (black) and at [Ca2+]MID (red). (E) CRC of Ca2+-uptake shows activation at both [Ca2+]MAX and [Ca2+]MID (black and red, respectively). ΔFMAX, ΔFMID, C10, and EC50 are defined and reported in Table 1 for panels C, D, and E. Data are presented as mean ± SEM, n = 3, *p < 0.05.

A representative activator that decreases FRET and uncouples Ca2+-ATPase activity from Ca2+-uptake activity. (A) Chemical structure of DS26418355 (Compound 8). (B) CRC of normalized FRET E in 2CS biosensor in live HEK cells shows decreasing FRET response with increasing [compound]. (C) CRC shows Ca2+-ATPase activation in pCSR vesicles at both [Ca2+]MAX (black) and [Ca2+]MID (red). (D) Activation was less for Ca2+-uptake of SERCA2a in pCSR vesicles. ΔFMAX, ΔFMID, C10, and EC50 are defined and reported in Table 1 for panels B, C, and D. Data is presented as mean ± SEM, n = 3, *p < 0.05.

A representative activator that decreases FRET, increases Ca2+-ATPase activity, and has divergent effects (inhibitory) on Ca2+-uptake activity. (A) Chemical structure of Compound DS12165787 (Compound 6). (B) CRC of normalized FRET E shows decreased FRET in live HEK cells with a half maximal effect (EC50) at 7.1 ± 0.2 μM. (C) CRC shows Ca2+-ATPase activity increase of SERCA2a in pCSR vesicles at [Ca2+]MAX (black, pCa 5.4) and [Ca2+]MID (red, pCa 6.2) (D) CRC of Ca2+-uptake of SERCA2a in pCSR vesicles, showing inhibition for [Ca2+]MAX (black) and activation for [Ca2+]MID (red). The grey data point was omitted from fitting, to account for hormesis40. ΔFMAX, ΔFMID, C10, and EC50 are defined and reported in Table 1 for panels B, C, and D. Data is presented as mean ± SEM, n = 3, *p < 0.05.
Functional CRC of hit compounds
To assess the impact of hit compounds on SERCA2a function, we used an absorbance-based Ca2+-ATPase activity assay and a fluorescence-based Ca2+-uptake assay, using pig cardiac SR (pCSR) vesicles enriched for SERCA2a23 (Fig. 1B, step 5). These activities were measured at [Ca2+]MAX (saturating, pCa 5.4), [Ca2+]MID (subsaturating, midpoint, pCa 6.2), and [Ca2+]BAS (basal, pCa 8.0). For the Ca2+-ATPase activities, the values at [Ca2+]BAS were < 10% of those at [Ca2+]MAX and < 25% of those at [Ca2+]MID, whereas for Ca2+-uptake rates, values at [Ca2+]BAS were negligible. Functional data acquired at [Ca2+]MAX and [Ca2+]MID were corrected by subtracting the appropriate basal rate at pCa 8.0 and the % effect due to the compound was fitted to the Hill’s function (Fig. 4–7) to yield values in Table 1. These functional results (Table 1) at [Ca2+]MAX and [Ca2+]MID were adequate to identify activators (Compounds 1–9) and inhibitors (Compounds 10–18) (Table 1 and Fig. S2), based on the functional potency (1/EC50, where EC50 is the compound concentration at 50% of the maximum effect) and functional efficacy (ΔFMAX or ΔFMID; change in the maximal compound effect on function F). Under ideal conditions, a maximum coupling ratio (CR) of 2 Ca2+ transported per molecule of ATP hydrolyzed has been reported30,33,34,35. CR defines the efficiency of Ca2+ uptake by SERCA, which was determined from the ratio of the measured maximal Ca2+-uptake rate (VU) at [Ca2+]MAX, to the maximal Ca2+-ATPase activity (VA) at [Ca2+]MAX (VU/VA) (Table 1 and under SERCA2a Activators). CR was also used to classify and prioritize compounds. Activators were compounds that increased Ca2+-ATPase activities and/or Ca2+-uptake (at one or both [Ca2+]) (Table 1, Fig. 4–6) and were grouped in three categories: (1) increases both Ca2+-ATPase activity and Ca2+-uptake to increase CR (Compound 7), (2) increases Ca2+-ATPase activity more than Ca2+-uptake to decrease CR (Compounds 2, 4, 8, and 9), and (3) increases Ca2+-ATPase activity but inhibits (induces divergent effects on) Ca2+-uptake (Compounds 1, 3, 5, and 6). We define “divergent” to indicate that the compound induces opposing effects at two different [Ca2+] (an increase at one [Ca2+] and a decrease at the other) in one assay.

A representative inhibitor (Compound 12) that strongly decreases FRET and inhibits both Ca2+-ATPase activities and Ca2+-transport. (A) Table showing the four subcategories (C) of inhibitors (N, Compounds 11–18) and their effects on the Ca2+ATPase activity and Ca2+-uptake as strong (S), moderate (M), or mild. (B) Chemical structure of DS27464324 (Compound 12). (C) CRC of normalized FRET E shows decreasing FRET in response to [Compound 12] in 2CS biosensor in live HEK cells. (D) CRC shows inhibition of Ca2+-ATPase activity in pCSR under both [Ca2+]MAX (black) and [Ca2+]MID (red). (E) CRC shows inhibition of Ca2+-uptake in pCSR under [Ca2+]MAX (black) and [Ca2+]MID (red). ΔFMAX, ΔFMID, C10, and EC50 are defined and reported in Table 1 for panels C, D, and E. Data are presented as mean ± SEM, n = 3, *p < 0.05.
Inhibitors induced strong (≥ 68%), moderate (34 to 67%), and mild (≤ 33%) inhibition of SERCA2a and were defined in four subcategories: 1) strong inhibition of Ca2+-ATPase activities and Ca2+-uptake rates (Compounds 11, 12, 13), 2) moderate inhibition of Ca2+-ATPase activities and strong inhibition of Ca2+ -uptake (Compounds 15 and 17), 3) mild inhibition of Ca2+-ATPase activities and moderate-to-strong inhibition of Ca2+-uptake (Compounds 14 and 16), and 4) mild inhibition on Ca2+-ATPase activities and mild-to-moderate inhibition on Ca2+-uptake (Compounds 10 and 18) (Table 1, Fig. 7, and under SERCA2a Inhibitors).
Classification of compounds by physicochemical characteristics
The 18 hit compounds were subjected to cheminformatic analysis, to determine whether any shared common chemical scaffolds. Compounds with a Tanimoto coefficient and maximum common substructure (MCS)36 scores above 0.4 were binned as clusters, while those with scores below 0.4 were classified as singletons. The analysis yielded diverse scaffolds36,37 of hit compounds (Supplementary Fig. S2 and Supplementary Table S1).
Four clusters of compounds (A–D in Table 1) were found, and the remaining eight were unique (singletons) (E–L in Table 1 and Supplementary Fig. S2 and Supplementary Table S1). Compounds in cluster A (1, 2, and 3) have a common 5-(aryloxymethyl)oxazole-3-carboxamide)38; those in cluster B (4 and 5) share a N-heteroaryl-N-alkylpiperazine. Cluster C (9 and 10) is defined by an amide linkage and cluster D (11, 12, and 13) by a piperidine scaffold. Clusters E-L (6, 7, 8, 14, 15, 16, 17, and 18) contain a singleton with no common scaffold with any other hit compound in this study. All hit compounds have physicochemical properties39 that are conducive of favorable drug disposition in vivo, including low molecular weight (< 500), low cLogP (calculated partition coefficient for lipophilicity < 5), low non-H rotatable bonds that describe the molecular flexibility (< 10), low probability of hydrogen bond formation (total number of hydrogen bond acceptors and donors less than 8), and low total polar surface area (tPSA < 140 Å) (Supplementary Table S1).
SERCA2a activators
Activators (Compounds 1–9) induced varying effects on SERCA2a function (Fig. 4A). In the first category, Compound 7 (Fig. 4B) decreased FRET of 2CS in live cells with FRET-EC50 = 0.3 μM, suggesting stabilization of an open conformation of SERCA2a. It accelerated Ca2+-ATPase activity at both [Ca2+] to induce ΔFMAX = 14% and ΔFMID = 7% (Fig. 4D and Table 1). Compound 7 induced the highest increases in Ca2+-uptake of all compounds at both [Ca2+] – ΔFMAX (24%) and ΔFMID (19%) (Fig. 4E), which were greater than the increases in the Ca2+-ATPase activity (Fig. 4D). CR increased to 0.74 compared to control (0.66, Table 1), indicating increased efficiency of SERCA2a, as needed to increase rate of relaxation and improve contractile function in the heart. Saturation of both CRCs was not achieved at the highest [compound] measured, so the functional EC50 was not determined. Instead, we determined C10, the [compound] that increases function by 10%. C10 was 25 μM for Ca2+-ATPase activity at [Ca2+]MAX and was not determined at [Ca2+]MID because Compound 7 did not induce changes higher than 10%. At [Ca2+]MAX and [Ca2+]MID, C10 was 14 μM and 21 μM for Ca2+-uptake, respectively (Table 1). This lead compound will be prioritized for future optimization by medicinal chemistry, to lower its FRET-EC50 and determine functional-EC50.
In the second category of activators (Compounds 2, 4, 8, and 9), Compound 8 (singleton G) (Fig. 5A) decreased FRET (FRET-EC50 = 4.9 μM, Table 1, Fig. 5B) and increased Ca2+-ATPase activities (ΔFMAX = 49%, the largest increase observed in this study, and ΔFMID = 31%) (Table 1 and Fig. 5C). For Ca2+-uptake, the effects were smaller (10% for ΔFMAX and 7% for ΔFMID, Fig. 5D), decreasing CR from 0.66 to 0.4 (Table 1). In cardiomyocytes, this should result in a small increase in SR Ca2+ uptake, with a greater increase in ATP hydrolysis, resulting in futile cycling of ATP. EC50 values for Ca2+-ATPase activity were not significantly different at [Ca2+]MAX and [Ca2+]MID (8.3 and 10 μM, respectively), and were ~ 2 × greater than the FRET-EC50 (4.9 μM). C10 was 4.3 M ([Ca2+]MAX) and 7.8 μM ([Ca2+]MID), indicating significant ATPase activation at low dosage. C10 for uptake at [Ca2+]MAX was 25 μM and was not determined at [Ca2+]MID because Compound 8 did not induce changes higher than 10%.
Compound 2 (cluster A) induced similar Ca2+-ATPase activation (ΔFMAX = 16%, ΔFMID = 7%) as Compound 7, with slightly smaller increases in Ca2+-uptake (Table 1), decreasing CR from 0.66 to 0.53. Compound 4 (cluster B) increased Ca2+-ATPase activities at both [Ca2+]MAX and [Ca2+]MID by ~ 10%, and increased Ca2+-uptake at both [Ca2+]MAX (0.5%) and [Ca2+]MID (11%). Compound 9 (cluster C) increased ΔFMAX (21%) and ΔFMID (10%) for Ca2+-ATPase activity, though with smaller increases for Ca2+-uptake (3–5%). Thus, Compounds 4 and 9 decreased CR similarly (Table 1), indicating that SERCA2a transport efficiency was diminished slightly. Compounds 2, 4, 8, and 9 increased Ca2+ uptake (0.5–10%). Thus these four compounds join Compound 7 as promising future lead compounds (Fig. 1B).
In the third category, Compounds 1 and 3 (cluster A), 5 (cluster B), and 6 (singleton E) increased Ca2+-ATPase activities at both [Ca2+]MAX and [Ca2+]MID, but induced divergent (inhibitory) effects on Ca2+-uptake at [Ca2+]MAX, decreasing CR (0.39—0.48). In cardiomyocytes, this decrease in CR would likely result in a decrease in SR Ca2+-uptake, while increasing ATP hydrolysis. Compound 6 (Fig. 6A) decreased FRET with FRET-EC50 = 7.1 μM (Fig. 6B, Table 1), while moderately increasing Ca2+-ATPase activity (ΔFMAX = 25% and ΔFMID = 30%) (Fig. 6C), with functional EC50 = 11 μM at both [Ca2+]. It induced divergent effects on Ca2+-uptake, decreasing ΔFMAX by 21% and increasing ΔFMID by 17% (Table 1 and Fig. 6D), but had inhibitory effects at high [compound], typical of hormesis40 (biphasic dose response), which can disrupt Ca2+ homeostasis41. C10 was similar at both [Ca2+] (9.7 μM and 8.7 μM), but was significantly different for Ca2+-uptake (15 μM at [Ca2+]MAX, 4 μM at [Ca2+]MID), decreasing CR to 0.48.
Compound 5 increased Ca2+-ATPase activity moderately at [Ca2+]MAX (25%) and [Ca2+]MID (35%). Ca2+-uptake was inhibited slightly at [Ca2+]MAX (2%), but activated at [Ca2+]MID (20%) (Table 1). Compounds 1, and 3 induced low activating effects at [Ca2+]MID for Ca2+-uptake (2% and 6%), but they inhibited Ca2+-uptake at [Ca2+]MAX (10% and 20%) (Table 1). Compounds 1 and 5 induced similar decreases in the CR (to 0.49 and 0.47), while Compound 3 induced a slightly smaller CR of 0.39 (Table 1). These effects are similar to those of unphosphorylated PLB in cardiac SR42.
Compounds 2, 7, 8, and 9 induced Ca2+-dependent activation of the Ca2+-ATPase activity over the range of [compound] studied. Compounds 1, 4, and 6 showed similar activation of Ca2+-ATPase activity at the two [Ca2+] measured, while Compounds 3 and 5 showed higher activation at [Ca2+]MID compared to [Ca2+]MAX.
SERCA2a Inhibitors
Although our primary goal is to find SERCA2a activators for treatment of HF, it has been proposed that SERCA2a inhibitors or uncouplers could be effective for treating several diseases, such as cancer and malaria17,18. Compounds 10–18 decreased Ca2+-ATPase activities and Ca2+-transport at [Ca2+]MAX and [Ca2+]MID to varying extents; strong (S), moderate (M), or mild and are in four subcategories (Fig. 7A). Compared with FRET-EC50 of the activators (0.3–7 μM), most of the inhibitors (Compounds 10–18) showed weaker affinity, with FRET-EC50 values in the range of 5–32 μM, but the maximum inhibitory effects (efficacies) of the inhibitors tended to be greater (Table 1).
(1) Compounds 11, 12, and 13 (cluster D) showed similar inhibition of both SERCA2a functions, except that, compared with the Ca2+-ATPase activity, Ca2+-uptake inhibition at [Ca2+]MID required slightly higher [compound], as shown by the right-shift of the red curve (Fig. 7D). Compound 12 (cluster D, Fig. 7B) strongly inhibited both Ca2+-ATPase activity and Ca2+-uptake (Fig. 7D, E) to levels similar to the classic SERCA inhibitor thapsigargin (Tg), although Tg acts with much greater affinity (EC50 ≈ 7.5 nM23) than Compound 12 (EC50 = 3.8 μM, Fig. 7C, Table 1). (2) Compounds 15 (singleton I) and 17 (singleton K) induced moderate inhibition of both activities, decreasing ΔFMAX and ΔFMID by ~ 50% for Ca2+-ATPase activity and slightly more for Ca2+-uptake (70–95%) (Table 1). (3) Compounds 14 (singleton H) and 16 (singleton J) induced mild inhibition of Ca2+-ATPase activity, but a moderate-to-strong inhibition of the Ca2+-uptake (Fig. 7A, Table 1). (4) Compounds 10 (cluster C) and 18 (singleton L) induced mild inhibition of Ca2+-ATPase and mild-to-moderate inhibition of Ca2+-uptake (Fig. 7A, Table 1). All inhibitors decreased CR, the efficiency of Ca2+-transport.
Discussion
We have identified new compounds based on an increase in ΔFLT within a human cardiac 2CS biosensor expressed in live mammalian cells at low [Ca2+] (the normal condition in the cytoplasm of HEK cells). This decrease in FRET implies that the actuator (A) and nucleotide-binding domains (N) of SERCA2a moved apart, supporting an open configuration at low [Ca2+] in HEK cells, possibly priming SERCA in a more open state to bind Ca2+. Previous studies with a different SERCA2a biosensor indicated that the addition of Ca2+ induces a decrease in FRET21. Our functional assays at high and mid [Ca2+] show activating, uncoupling, and inhibiting effects that may correlate with structural changes. Further future elucidation of the compounds’ effects on SERCA2a conformational states will require detailed analysis of FLT-detected FRET and transient kinetics data. We identified three categories of activators that (1) increase both Ca2+-ATPase activity and Ca2+-transport to increase CR (Compound 7, Fig. 4) (2) increase Ca2+-ATPase activity and Ca2+-transport to decrease CR (Compounds 2, 4, 8, and 9, Fig. 5), and (3) increase Ca2+-ATPase activity but inhibits Ca2+-transport to decrease CR (Compounds 1, 3, 5, and 6) (Fig. 6). We identified four subcategories of inhibitors based on the extent of decrease in Ca2+ATPase activity and Ca2+-transport for SERCA2a (Table 1, Fig. 7A).
Most FRET-EC50 values were smaller (higher potency) for activators (Compounds 1–9; 0.3-7 μM) than for inhibitors (Compounds 10–18; (3-32 μM) (Table 1). However, the functional C10 and EC50 values were smaller, indicating greater potency, for inhibitors than for activators. Potencies observed by FRET and function are not precisely correlated, probably because the assays were performed on different types of samples (live cells vs. purified proteins), low nM [Ca2+] in live HEK cells43 vs. μM [Ca2+] in the pCSR in vitro assays), which measure different properties (structure vs function). Functional CRC assays showed that inhibitors tend to induce larger changes (indicating higher efficacy) than activators, in both Ca2+-ATPase activity and Ca2+-uptake. Most inhibitors induced a larger change in Ca2+-uptake than in Ca2+-ATPase activity, decreasing CR.
Most of the activators reduced CR, inducing larger changes in Ca2+-ATPase activity than in Ca2+-uptake. A notable exception is Compound 7, which increases Ca2+-transport even more than it increases Ca2+-ATPase activity, increasing CR. This compound will be a high priority as a lead compound for future efforts in medicinal chemistry and assays of physiological function. Compounds 2, 4, 8, and 9 will have only slightly lower priority.
Ten compounds were binned into four clusters (A–D); eight were singletons (E–L) (Table 1). Many compounds showed similar functional traits, suggesting that ligand-sensing sites in SERCA2a are recognized by a range of scaffolds, or that these sites are close to each other, providing potentially powerful tools in the design of future compounds44,45,46. Only Compound 7 (singleton F) induced higher activation in Ca2+ transport than in the Ca2+-ATPase activity. Compounds 2 (cluster A), 4 (cluster B), 8 (singleton G), and 9 (cluster C) induced similar effects of moderate activation of Ca2+-ATPase activity, with smaller activation of Ca2+-transport. Compounds in activator clusters A (Compounds 1 and 3) and B (Compound 5) along with Compound 6 from singleton E, showed similar functional effects: moderate ATPase activation with mild inhibition of Ca2+-transport at [Ca2+]MAX. Inhibitor compounds in clusters D (11, 12, and 13), C (10), and H–L (14–18) induced a range of effects at both [Ca2+], which will be useful in designing derivatives for structure activity relationship (SAR) analysis.
There was negligible overlap in hit compounds identified in our previous FRET-HTS of the DIVERSet-CL targeting tumor necrosis factor receptor 127. There was 81% overlap in the fluorescent compounds detected (and thus rejected) in these two HTS studies, indicating that our FRET-HTS methodologies are effective and versatile26,29. In our previous study of the DIVERSet-CL, using the SERCA2a Ca2+-ATPase activity as the primary HTS assay (ATPase-HTS), we discovered 19 activators30. While no identical activators were found in that ATPase-HTS study30 and in the current FRET-HTS study, there were several compounds with similar scaffolds that showed similar functional results. These compounds share the oxadiazol scaffold and activated the Ca2+-ATPase activity but inhibited Ca2+-uptake. Another common scaffold is the amide group; six compounds30 identified with an amide induced a smaller increase in the Ca2+-transport than in the Ca2+-ATPase activity, reducing CR similar to Compound 9 in this study. It is not surprising that the two studies did not identify the same compounds, because (a) the FRET-HTS assay was performed with human cardiac 2CS in live HEK cells in low [Ca2+], while the ATPase-HTS assay was done in purified SR from rabbit skeletal muscle (SERCA1a) under high [Ca2+]30, (b) the FRET assay is much more precise than the functional assay, (c) the relationship between SERCA structure and function is complex, and (d) the binding sites on SERCA2a for these compounds are unknown. As discussed above, a ligand-binding site may recognize several different scaffolds44,45,46. It is also possible that a compound binds to PLB or competes with PLB for binding to SERCA, thus increasing SERCA2a activity, as was shown for the activator, istaroxime47,48. These observations highlight the value of complementary HTS assays for the same target.
Activation of Ca2+-transport by SERCA2a is needed when cardiac relaxation is impaired, as in diastolic dysfunction1 or diabetic cardiomyopathy49. SERCA2a activation is a promising strategy, in combination with current drugs such as β-blockers and ACE inhibitors50. Activation of SERCA also has therapeutic potential for Alzheimer’s disease51 or Duchenne muscular dystrophy (DMD)52. Until recently, very few compounds were known to stimulate SERCA2a: CDN1163 (stimulates Ca2+ transport)24,53, CP-154526 (increases the apparent Ca2+ affinity of SERC2a)54, Ro 41–0960 (increases SERCA2a maximal activity in high Ca2+)54, and istaroxime (stimulates SERCA2a activity)55. However, our recent ATPase-HTS assay identified ~ 19 new activators of SERCA30, and we identified nine in the present study. A SERCA activator from our previous work (CDN1163) shows promise as a therapeutic agent for Alzheimer’s disease51 and for DMD52. Of all these SERCA2a activators, only istaroxime has been in phase IIb clinical trials for treatment of heart failure55,56. However, because of its unsuitability for human usage56, istaroxime must be modified47,48.
Compounds 1, 3, 5, and 6 induced small effects on the Ca2+-ATPase activity (~ 10–25% increase) and induced a negative effect on the Ca2+-transport (Fig. 6C and D), thus decreasing the CR, which is likely to increase heat output15,16,57. These effects are similar to that of SLN on SERCA1a (skeletal muscle), where SLN reduces Ca2+-transport without affecting the Ca2+-ATPase activity (SERCA1a uncoupling), thus reducing CR15. Uncoupling of SERCA1a leads to higher usage of ATP, which enhances non-shivering thermogenesis (NST)15. Another contributor to NST is Ca2+ leak from SR through resting RyR channels, stimulating SERCA to re-sequester Ca2+ into SR, thus using more ATP and generating heat58, which has been suggested as a potential therapeutic strategy for reducing obesity15,57.
Six decades of research for SERCA inhibitors as oncology therapeutics have yielded hundreds of SERCA inhibitors with varying potencies and efficacies17. Similarly, our discovery of new SERCA inhibitors with a range of potencies and efficacies is likely to be advantageous for non-cardiac applications17,18.
Here we successfully used the 2CS biosensor to identify novel small-molecule effectors of SERCA with diverse chemical scaffolds, resulting in an array of activator and inhibitor hit compounds. Most importantly, based on the amplitude of the functional effects on SERCA2a, we discovered a potential lead compound (Compound 7) that activates Ca2+-uptake more than the Ca2+-ATPase activity, increasing the CR, so this will be a high priority for future efforts in medicinal chemistry and assays of physiological function, along with four other promising SERCA2a activators. The innovative technology included two novel plate-readers – the FLT instrument used in the primary screen, and a spectral instrument – that were used to remove compounds with interfering fluorescence signals, allowing us to focus on valid SERCA activators and inhibitors. It is possible that some of the eliminated fluorescent compounds have potential as SERCA2a effectors, which could be evaluated in future work using our ATPase activity assay30. In future studies, we will evaluate these hit compounds in more functional detail, including the full range of [Ca2+], SERCA isoforms, Na+/Ca2+ exchanger, RyR, and L-type Ca2+-channels. Medicinal chemistry will be done to elucidate SAR and to design analogs with greater potency and specificity27,59, justifying studies in intact muscles and animals. Finally, we have shown that our primary screening technology can perform precise HTS on several thousand compounds per hour, making this approach capable of application on an industrial scale, screening millions of compounds.
Methods
Molecular biology
A two-color intramolecular human SERCA2a (2CS) biosensor, based on human cardiac SERCA2a fused to green fluorescent protein (eGFP) and red fluorescent protein (tagRFP) was developed to detect structural changes that are related to the functional changes of SERCA20,22,25. Briefly, tagRFP was genetically fused to the N-terminus of SERCA (A-domain) and eGFP was inserted as an intrasequence tag before residue 509 in the nucleotide-binding domain (N-domain)60,61. Donor-only and acceptor-only (1CS) biosensors were created in a similar manner as the 2CS biosensor but with the construct containing either only eGFP or only tag-RFP, respectively. The fluorescent proteins fused to SERCA in 2CS and 1CS do not significantly affect SERCA activity, in membranes purified from HEK cells23,25. A null-biosensor construct consisting of eGFP and tagRFP connected by a 32-residue unstructured flexible linker peptide (G32R) was created as described previously23,25. All constructs were cloned into expression vectors containing the genes for antibiotic resistance to G418, puromycin, or blasticidin.
Cell culture
Stable cell lines were generated using either human embryonic kidney (HEK) HEK293 (ATCC, Manassas, VA) or HEK293-6E (National Research Council, Canada) cells25. Briefly, cells were transiently transfected with 2CS, 1CS, or G32R null-biosensor plasmids using Lipofecatime 3000 or 293fectin (Thermo Fisher Scientific). Flow cytometry was used to select and enrich for the population of cells expressing respective biosensors. Stable HEK293 cell lines were maintained in phenol red-free DMEM media (Gibco, Waltham, MA) supplemented with 2 mM GlutaMAX (Gibco, Waltham, MA), 10% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA), 1 IU/mL penicillin/streptomycin (Gibco, Waltham, MA), and 250 μg/mL G418 (Fisher Scientific). Stable HEK293-6E cell lines were maintained in F17 media (Sigma Aldrich) supplemented with Kolliphor p188 (Sigma Aldrich, St. Louis, MO), 200 nM/mL GlutaMAX, and either 1 μg/mL puromycin (Invitrogen, Carlsbad, CA) or 2 μg/mL blasticidin (Goldbio) as a selection antibiotic. All cell lines were grown at 37 °C with 5% CO2.
Compound handling
A 50,000 DIVERSet-CL was purchased from ChemBridge Corporation (San Diego, CA) at a 10 mM stock concentration for each compound. All compounds met the high quality standard of 100% identification by NMR and/or LC–MS and have a minimum purity of 85% and their identity verified using LC–MS/ELSD as confirmed by the ChemBridge Corporation. For the FLT HTS initial screens, the compound library was reformatted into 384 well Echo compatible plates using the Biomek FX (Beckman Coulter, Miami, FL) and 5 nL of either compound (columns 3–22 and 27–46) or DMSO (columns 1–2, 23–26, and 47–48) was dispensed into forty 1536 well black polystyrene assay plates (Greiner, Kremsmünste, Austria) using an Echo 550 liquid dispenser (Beckman Coulter) to yield a final assay screening concentration of 10 μM. The low autofluorescence and low interwell cross-talk of these plates made them advantageous for FLT measurements. Plates were heat sealed with a PlateLoc Thermal Microplate Sealer (Agilent, Santa Clara, CA) and stored at − 20 °C prior to use. The same methods were applied for subsequent FLT retesting of the hit compounds identified in the FLT screen (Fig. 1B, steps 1–3), except that the [compound] was tested at 10 μM and 30 μM in triplicate, where the latter gave more reproducible results.
FRET CRC assay plates (0.78–100 μM compound range) with at least ten different compound concentrations were made by adding the appropriate volume of compound or DMSO into black 384 well plates (Greiner Bio-One) using a Mosquito HV (SPTLabTech, United Kingdom). Subsequent Ca2+-ATPase activity and Ca2+-transport CRC assay plates (0–50 μM compound range) with repurchased compounds were made in a similar manner using with the Echo 550 (Beckman Coulter) using either 384 well transparent plates (Greiner Bio-One) or black-walled plates with transparent bottoms (Greiner Bio-One), respectively.
HTS sample preparation and FRET measurements
On each day of screening, cells were harvested, washed three times with PBS, and centrifuged at 300 g for 5 min. Cells were filtered using a 70 µm cell strainer and diluted to 1–2 × 106 cells/mL. Cell concentration and viability were assessed using the Cell countess (Invitrogen) and trypan blue assay. During assays, cells were constantly and gently stirred using a magnetic stir bar at room temperature, keeping the cells in suspension and evenly distributed to avoid clumping. HEK 293-cells expressing 2CS were dispensed at 5 μL or 50 μL per well into assay plates (dispensed into 40 assay plates, each containing 1536 wells) pre-plated with compounds (from a DIVERset 50,000 compound library) using a Multdrop Combi liquid dispenser (Thermo Fisher Scientific, Pittsburg, PA) and sealed until needed. Because the kinetics of membrane permeability, diffusion, and/or binding of the compound to live cells may be compound-dependent, we tested two incubation times, 20 min and 120 min, for the FRET CRC. FRET EC50 values determined from both incubations were similar, but the 120 min incubation yielded a more reproducible and sigmoidal curve. Plates containing eight-point concentration curves of three tool compounds (known SERCA effectors thapsigargin, BHQ, and CPA)22 were used as positive controls for biosensor function and performance prior to running the full-scale FRET-HTS assay.
The primary FLT-FRET-HTS assay was performed over two days with a custom HTS fluorescence lifetime plate reader (FLT-PR) with donor emission detected at 517 nm (Fig. 1B, step 1). Initial hits from this FLT screen were selected (using rZ-score, discussed below), then fluorescent compounds were removed using the Similarity Index (SI) calculated from the spectral measurement acquired with the SUPR (SI, discussed below) (Fig. 1B, step 2). Both instruments were provided by Photonic Pharma LLC (Minneapolis, MN)23.
The same methods were applied for subsequent FRET retesting (Fig. 1B, step 4) of the reproducible hit compounds identified in Fig. 1B, steps 1–3, except that the compounds were tested at 10 μM and 30 μM [compound]. 160 hit compounds were selected from the library master plates and reloaded onto new assay plates for retesting with 2CS and a null-biosensor, using FLT-PR (ΔFLT) and SUPR (Δ(G/R)). This step was designed to remove compounds that bind directly to the fluorescent protein or produce other artifacts in the FLT reading that do not involve FRET. Then 18 hit compounds, representing a range of ΔFLT, were selected and purchased from ChemBridge to determine CRC from FRET, Ca2+-ATPase activity, and Ca2+-transport assays using at least ten different concentrations by repeatedly scanning the 1536-well plates.
FRET-HTS instrumentation and data analysis
A detailed description of the high-throughput fluorescence lifetime plate reader (FLT-PR) and spectral unmixing plate reader (SUPR), manufactured by Fluorescence Innovations Inc and provided by Photonic Pharma, LLC was described previously23,26. Briefly, for lifetime measurement with the FLT-PR, the observed donor-fluorescence waveform, I(t) was fit by a convolution of the measured instrument response function (IRF) (Eq. 1a) and a single-exponential decay F(t) to obtain the lifetime (τ) of the donor fluorophore22,26,62 in the absence (τD) and presence (τDA) of the acceptor as described in Eq. (1b):
In experiments with a donor-only control, FRET efficiency (E) was determined as the fractional decrease of donor FLT in the absence and in the presence of acceptor as in Eq. (2):
E was determined in the presence and absence of compound and normalized relative to E of the DMSO control. For spectral detection of FRET with SUPR, the observed fluorescence emission spectrum F(λ) was fit by least-squares minimization of a linear combination of component spectra for donor (G for green), acceptor (R for red), cellular autofluorescence (C) and water Raman (W), as described previously22. The change in ratio of the mole fractions of the G and R component spectra between compound and DMSO control (Δ(G/R)) provides a direct indication of a change in FRET due to biosensor structural changes, independent of the lifetime measurements. Together, these complementary metrics provide an effective method for eliminating false positives arising in either method.
HTS data analysis
FLT-PR data was used as the primary metric for flagging potential hit compounds. After fitting waveforms with a single exponential decay to quantify donor lifetime, the change in fluorescence lifetime due to compound (Δτ) was computed by performing a moving median subtraction in the order the plate was scanned, with a window size of 24 wells, rather than subtracting DMSO controls. The reasons for this are twofold: 1) plate gradients are often observed due to heating of the digitizer during acquisition and 2) performing Δτ computations with DMSO controls can sometimes result in artifacts as a half of the DMSO wells are on the edge of plates, which occasionally exhibit artifacts due to processes needed for the preparation of the compound library being tested. As most compounds are likely to be non-hits, and therefore DMSO like, computation of a moving median is an effective alternative to solving both gradient issues and edge-effect distortion of the primary metric for hit selection, Δτ.
Previous validation in 1536-well plates indicated that the Z' parameter – a measure of HTS assay quality that factors in the signal window and the variance of positive and negative controls (i.e., thapsigargin and DMSO vehicle)63 – yielded a value of 0.6225. A value of 0.5 ≤ Z' < 1 indicates excellent assay quality, ready for large-scale HTS63 as we recently showed for an ATP-based HTS assay30. Data from the control (tool) compounds was not needed for assessment of assay quality in the full-scale HTS in this study, instead we used the coefficient of variance (CV), computed using wells containing only DMSO, to assess the quality of the plates, in order to separate within-plate and between-day variability, as in our previous ATP-based HTS study30.
In our previous ATPase-based HTS study, we defined a hit compound as one that changes the ATPase signal by 4SD relative to the DMSO controls30. Given the increased precision (< 1% CV, Fig. 2A) afforded by the FLT measurement and the availability of complementary HTS measurements using the spectral plate reader on the same plate for further triage, we set our hit threshold to an rZ-score of ± 3, on a plate-by-plate basis, in order to include a broader range of initial hit compounds. The rZ-score was used (instead of the standard Z-score), where the median (M) and median absolute deviation (MAD) are used in place of the mean and standard deviation (Eq. 3), to best capture the most hits, as the standard Z-score is more susceptible to strong outliers23.
To remove interfering fluorescent compounds, the similarity index (SI)22 was computed by comparing a region (500–540 nm) of the donor-only spectrum (I(a)) for each well to that of the plate-wide average DMSO spectrum (I(b)) in the same wavelength band as described in Eq. 426. Compounds that reproducibly exceeded an SI rZ-score of 5 (corresponding to an SI of 2 × 10–4) were deemed likely fluorescent compounds and were removed from consideration.
Spectral (SUPR) data was processed similarly to FLT-PR data, with the Δ(G/R) metric being computed by applying the same moving median filter on the initial measurement of the ratio of donor (G) to acceptor (R) mole fractions (G/R)21,25. The hit threshold was also set using an rZ-score of ± 3. While the FLT-PR data and SUPR data showed strong correlation, the FLT-PR data exhibited some clear outliers, presumably due to compounds directly modifying the donor lifetime. To eliminate these likely interfering compounds, correlation was enforced by eliminating compounds that exceed an rZ-score of ± 3 from the median value of the ratio of Δτ over the Δ(G/R) metric.
Cardiac SR preparation
Cardiac SR vesicles were isolated from fresh porcine left ventricular tissue using differential centrifugation of the homogenized tissue as previously described20. The SR vesicles were flash-frozen and stored at − 80 °C until needed. The SERCA concentration in the ER preparations purified from HEK cell homogenate is at least 10 times less than in purified porcine cardiac SR20, but there was sufficient expression of the fluorescent SERCA2a biosensor in HEK cells to detect FRET with high precision by FLT22,23,26.
Effects of hit compounds on the Ca2+-ATPase activity of SERCA
Functional assays were performed using porcine cardiac SR (pCSR) vesicles20. An enzyme-coupled, NADH-linked ATPase assay was used to measure SERCA ATPase activity in 384-well microplates. Each well contained 50 mM MOPS (pH 7.0), 100 mM KCl, 1 mM EGTA, 0.2 mM NADH, 1 mM phosphoenol pyruvate, 10 IU/mL of pyruvate kinase, 10 IU/mL of lactate dehydrogenase, 7 µM of the calcium ionophore A23187 (Sigma), and CaCl2 was added to set free [Ca2+] to three different concentrations23. Ca2+-ATPase activity was measured at three [Ca2+]: [Ca2+]MAX (saturating, pCa 5.4), [Ca2+]MID (subsaturating, midpoint, pCa 6.2), and [Ca2+]BAS (basal non-activating, pCa 8.0). 10 μg/mL of SR vesicle, calcium, compound (0.048 to 50 μM), and assay mix were incubated for 20 min at room temperature before measurement of functional assays with each of the 18 hit compounds, because a shorter incubation time than the FRET live-cell assays achieved optimal responses. The assay was started upon the addition of MgATP, at a final concentration of 5 mM (total volume to 80 μL), and absorbance was read at 340 nm in a SpectraMax Plus384 microplate spectrophotometer (Molecular Devices, Sunnyvale, CA).
Effects of hit compounds on the Ca2+-transport activity of SERCA
Ca2+-transport assays were performed with similar porcine SR samples as in the Ca2+-ATPase assays described above. The compound effect on the Ca2+-transport activity of SERCA2a was determined using an oxalate-supported assay in which the change in fluorescence in a Ca2+-sensitive dye, Fluor-4, was determined as previously described23. A buffered solution containing 50 mM MOPS (pH 7.0), 100 mM KCl, 30 mg/mL sucrose, 1 mM EGTA, 10 mM potassium oxalate, 2 M Fluo-4, 30 µg/mL porcine cardiac SR vesicles, CaCl2 calculated to reach the free [Ca2+] (pCa 8.0, 6.2, and 5.4), and compound (0.048 to 50 μM) was dispensed into 384-well black walled, transparent bottomed plates (Greiner Bio-One) containing the tested small molecule and incubated at 22℃ for 20 min while covered and protected from light. To start the reaction, MgATP was added to a final concentration of 5 mM, and the decrease in 485-nm excited fluorescence of Fluo-4 was monitored at 520 nm for 15 min using a FLIPR Tetra (Molecular Devices, San Jose, CA).
Data analysis of FRET CRC assays of hit compounds
FRET efficiency (E) (Eq. 2) was determined as the fractional decrease of donor lifetime (τD = 2.5 ± 0.01 ns for 1CS, donor only) in the presence of acceptor (2CS, τDA = 2.33 ± 0.001 ns) due to FRET. E was plotted as “FRET Effect (E/EDMSO)” vs [compound], fitted to the Hill’s function for determination of FRET-EC50 27,31. This normalization of E corrects for variation of controls done on different days.
Data analysis of Ca2+-ATPase and Ca2+-transport activities from CRC assays
SERCA2a activity, F (rate of ATP hydrolysis or Ca2+ uptake), was measured at varying pCa and varying compound concentration. F measured at [Ca2+]MAX (saturating, pCa 5.4) or [Ca2+]MID (subsaturating, midpoint, pCa 6.2) was corrected by subtracting the basal rate at pCa 8.0) and the % effect due to compound was reported. Concentration response curves (CRC) were fitted using the Hill function to determine VMAX (the activity at saturating [compound]), and EC50, the compound concentration at 50% effect54. When the CRC did not achieve saturation, the maximal change (Δ) in activity was determined, to yield ΔFMAX and ΔFMID at the [Ca2+]MAX and [Ca2+]MID conditions, respectively (Fig. 5C, Table 1). ΔFMAX, ΔFMID, C10 (compound concentration inducing 10% effect), and EC50 are reported in Table 1.
Cheminformatic analysis of hit compounds
An online interactive program was used to perform cheminformatics analysis64 to determine whether the hit compounds had structural similarity by identifying common chemical scaffolds (core structural feature) using binning, multidimensional scaling (MDS), and compound similarity methods where the Tanimoto coefficient36 and maximum common substructure36 values were used to determine clustering (Supplementary Table S1). The physicochemical properties (for e.g. Lipinski Rule of 5) and bioactivity properties of the compounds were also used in the clustering analysis39. A cluster contained two or more compounds with similarity score > 0.4, while a unique compound with a similarity score < 0.4 was referred to as a singleton.
Statistical analysis
Analysis of two-group comparisons was done by a two-tailed unpaired Student’s t-test (*p < 0.05) using the data analysis program Microsoft Excel (Santa Rosa, CA). Data are presented as mean ± SEM calculated from a minimum of three separate experiments (n = 3).
Data availability
All the data discussed are presented within the article and Supplementary Information and are available from the corresponding authors (OR and DDT) on reasonable request.
References
Bers, D. M., Eisner, D. A. & Valdivia, H. H. Sarcoplasmic reticulum Ca2+ and heart failure: Roles of diastolic leak and Ca2+ transport. Circ. Res. 93, 487–490. https://doi.org/10.1161/01.RES.0000091871.54907.6B (2003).
Bers, D. M. Cardiac sarcoplasmic reticulum calcium leak: Basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 76, 107–127. https://doi.org/10.1146/annurev-physiol-020911-153308 (2014).
Guglielmi, V. et al. Characterization of sarcoplasmic reticulum Ca(2+) ATPase pumps in muscle of patients with myotonic dystrophy and with hypothyroid myopathy. Neuromuscul. Disord. 26, 378–385. https://doi.org/10.1016/j.nmd.2016.04.003 (2016).
Viskupicova, J. & Rezbarikova, P. Natural polyphenols as SERCA activators: Role in the endoplasmic reticulum stress-related diseases. Molecules https://doi.org/10.3390/molecules27165095 (2022).
Bers, D. M. & Despa, S. Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J. Pharmacol. Sci. 100, 315–322 (2006).
Rathod, N. et al. Nothing regular about the regulins: Distinct functional properties of SERCA transmembrane peptide regulatory subunits. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22168891 (2021).
Li, A. et al. The transmembrane peptide DWORF activates SERCA2a via dual mechanisms. J. Biol. Chem. 296, 100412. https://doi.org/10.1016/j.jbc.2021.100412 (2021).
Fisher, M. E. et al. Dwarf open reading frame (DWORF) is a direct activator of the sarcoplasmic reticulum calcium pump SERCA. Elife https://doi.org/10.7554/eLife.65545 (2021).
Rustad, M. D., Roopnarine, O., Cornea, R. L. & Thomas, D. D. Interaction of DWORF with SERCA and PLB as determined by EPR spectroscopy. Biochem. Biophys. Res. Commun. 645, 97–102. https://doi.org/10.1016/j.bbrc.2023.01.041 (2023).
Nelson, B. R. et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 351, 271–275. https://doi.org/10.1126/science.aad4076 (2016).
Makarewich, C. A. et al. The DWORF micropeptide enhances contractility and prevents heart failure in a mouse model of dilated cardiomyopathy. Elife https://doi.org/10.7554/eLife.38319 (2018).
Hadri, L. & Hajjar, R. J. Calcium cycling proteins and their association with heart failure. Clin. Pharmacol. Ther. 90, 620–624. https://doi.org/10.1038/clpt.2011.161 (2011).
Sato, D., Uchinoumi, H. & Bers, D. M. Increasing SERCA function promotes initiation of calcium sparks and breakup of calcium waves. J. Physiol. 599, 3267–3278. https://doi.org/10.1113/JP281579 (2021).
Xie, A., Liu, H., Kang, G. J., Feng, F. & Dudley, S. C. Jr. Reduced sarcoplasmic reticulum Ca(2+) pump activity is antiarrhythmic in ischemic cardiomyopathy. Heart Rhythm 19, 2107–2114. https://doi.org/10.1016/j.hrthm.2022.08.022 (2022).
Bal, N. C. & Periasamy, M. Uncoupling of sarcoendoplasmic reticulum calcium ATPase pump activity by sarcolipin as the basis for muscle non-shivering thermogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 375, 20190135. https://doi.org/10.1098/rstb.2019.0135 (2020).
Nowack, J., Giroud, S., Arnold, W. & Ruf, T. Muscle non-shivering thermogenesis and its role in the evolution of endothermy. Front. Physiol. 8, 889. https://doi.org/10.3389/fphys.2017.00889 (2017).
Michelangeli, F. & East, J. M. A diversity of SERCA Ca2+ pump inhibitors. Biochem. Soc. Trans. 39, 789–797. https://doi.org/10.1042/BST0390789 (2011).
Pagliaro, L., Marchesini, M. & Roti, G. Targeting oncogenic Notch signaling with SERCA inhibitors. J. Hematol. Oncol. 14, 8. https://doi.org/10.1186/s13045-020-01015-9 (2021).
Aguayo-Ortiz, R. & Espinoza-Fonseca, L. M. Linking biochemical and structural states of SERCA: Achievements, challenges, and new opportunities. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21114146 (2020).
Gruber, S. J. et al. Discovery of enzyme modulators via high-throughput time-resolved FRET in living cells. J. Biomol. Screen 19, 215–222. https://doi.org/10.1177/1087057113510740 (2014).
Raguimova, O. N. et al. Redistribution of SERCA calcium pump conformers during intracellular calcium signaling. J. Biol. Chem. 293, 10843–10856. https://doi.org/10.1074/jbc.RA118.002472 (2018).
Schaaf, T. M. et al. High-throughput spectral and lifetime-based FRET screening in living cells to identify small-molecule effectors of SERCA. SLAS Discov. 22, 262–273. https://doi.org/10.1177/1087057116680151 (2017).
Schaaf, T. M. et al. Live-cell cardiac-specific high-throughput screening platform for drug-like molecules that enhance Ca(2+) transport. Cells https://doi.org/10.3390/cells9051170 (2020).
Cornea, R. L. et al. High-throughput FRET assay yields allosteric SERCA activators. J. Biomol. Screen 18, 97–107. https://doi.org/10.1177/1087057112456878 (2013).
Schaaf, T. M. et al. Red-shifted FRET biosensors for high-throughput fluorescence lifetime screening. Biosensors (Basel) https://doi.org/10.3390/bios8040099 (2018).
Schaaf, T. M., Peterson, K. C., Grant, B. D., Thomas, D. D. & Gillispie, G. D. Spectral unmixing plate reader: High-throughput, high-precision FRET assays in living cells. SLAS Discov. 22, 250–261. https://doi.org/10.1177/1087057116679637 (2017).
Lo, C. H. et al. Noncompetitive inhibitors of TNFR1 probe conformational activation states. Sci. Signal https://doi.org/10.1126/scisignal.aav5637 (2019).
Lo, C. H., Schaaf, T. M., Thomas, D. D. & Sachs, J. N. Fluorescence-based TNFR1 biosensor for monitoring receptor structural and conformational dynamics and discovery of small molecule modulators. Methods Mol. Biol. 2248, 121–137. https://doi.org/10.1007/978-1-0716-1130-2_9 (2021).
Lo, C. H. et al. Discovery of small molecule inhibitors of huntingtin Exon 1 aggregation by FRET-based high-throughput screening in living cells. ACS Chem. Neurosci. 11, 2286–2295. https://doi.org/10.1021/acschemneuro.0c00226 (2020).
Bidwell, P. A. et al. A large-scale high-throughput screen for modulators of SERCA activity. Biomolecules https://doi.org/10.3390/biom12121789 (2022).
Rebbeck, R. T. et al. Novel drug discovery platform for spinocerebellar ataxia, using fluorescence technology targeting beta-III-spectrin. J. Biol. Chem. 296, 100215. https://doi.org/10.1074/jbc.RA120.015417 (2021).
Baell, J. B. & Holloway, G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740. https://doi.org/10.1021/jm901137j (2010).
Autry, J. M. et al. Sarcoplasmic reticulum from horse gluteal muscle is poised for enhanced calcium transport. Vet. Sci. https://doi.org/10.3390/vetsci8120289 (2021).
Racker, E. & Eytan, E. A coupling factor from sarcoplasmic reticulum required for the translocation of Ca2+ ions in a reconstituted Ca2+ATPase pump. J. Biol. Chem. 250, 7533–7534 (1975).
Yu, X. & Inesi, G. Variable stoichiometric efficiency of Ca2+ and Sr2+ transport by the sarcoplasmic reticulum ATPase. J. Biol. Chem. 270, 4361–4367. https://doi.org/10.1074/jbc.270.9.4361 (1995).
Willett, P. The calculation of molecular structural similarity: Principles and practice. Mol. Inform. 33, 403–413. https://doi.org/10.1002/minf.201400024 (2014).
Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U. S. FDA approved pharmaceuticals. J. Med. Chem. 57, 10257–10274. https://doi.org/10.1021/jm501100b (2014).
Shinde, S. R. et al. A systematic appraisal on catalytic synthesis of 1,3-oxazole derivatives: A mechanistic review on metal dependent synthesis. Synth. Commun. 52, 1–36. https://doi.org/10.1080/00397911.2021.1989596 (2022).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 46, 3–26. https://doi.org/10.1016/s0169-409x(00)00129-0 (2001).
Calabrese, E. J., Dhawan, G., Kapoor, R., Iavicoli, I. & Calabrese, V. HORMESIS: A fundamental concept with widespread biological and biomedical applications. Gerontology 62, 530–535. https://doi.org/10.1159/000441520 (2016).
Chambers, P. J., Juracic, E. S., Fajardo, V. A. & Tupling, A. R. Role of SERCA and sarcolipin in adaptive muscle remodeling. Am. J. Physiol. Cell Physiol. 322, C382–C394. https://doi.org/10.1152/ajpcell.00198.2021 (2022).
Kranias, E. G. & Hajjar, R. J. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 110, 1646–1660. https://doi.org/10.1161/CIRCRESAHA.111.259754 (2012).
Tong, J., McCarthy, T. V. & MacLennan, D. H. Measurement of resting cytosolic Ca2+ concentrations and Ca2+ store size in HEK-293 cells transfected with malignant hyperthermia or central core disease mutant Ca2+ release channels. J. Biol. Chem. 274, 693–702. https://doi.org/10.1074/jbc.274.2.693 (1999).
Balamurugan, R., Dekker, F. J. & Waldmann, H. Design of compound libraries based on natural product scaffolds and protein structure similarity clustering (PSSC). Mol. Biosyst. 1, 36–45. https://doi.org/10.1039/b503623b (2005).
Koch, M. A. et al. Charting biologically relevant chemical space: A structural classification of natural products (SCONP). Proc. Natl. Acad. Sci. USA 102, 17272–17277. https://doi.org/10.1073/pnas.0503647102 (2005).
Ma, B., Shatsky, M., Wolfson, H. J. & Nussinov, R. Multiple diverse ligands binding at a single protein site: A matter of pre-existing populations. Protein Sci. 11, 184–197. https://doi.org/10.1110/ps.21302 (2002).
Arici, M. et al. Istaroxime metabolite PST3093 selectively stimulates SERCA2a and reverses disease-induced changes in cardiac function. J. Pharmacol. Exp. Ther. 384, 231–244. https://doi.org/10.1124/jpet.122.001335 (2023).
Luraghi, A. et al. Highly selective SERCA2a activators: Preclinical development of a congeneric group of first-in-class drug leads against heart failure. J. Med. Chem. 65, 7324–7333. https://doi.org/10.1021/acs.jmedchem.2c00347 (2022).
Lebeche, D., Davidoff, A. J. & Hajjar, R. J. Interplay between impaired calcium regulation and insulin signaling abnormalities in diabetic cardiomyopathy. Nat. Clin. Pract. Cardiovasc. Med. 5, 715–724. https://doi.org/10.1038/ncpcardio1347 (2008).
Shareef, M. A., Anwer, L. A. & Poizat, C. Cardiac SERCA2A/B: Therapeutic targets for heart failure. Eur. J. Pharmacol. 724, 1–8. https://doi.org/10.1016/j.ejphar.2013.12.018 (2014).
Krajnak, K. & Dahl, R. A new target for Alzheimer’s disease: A small molecule SERCA activator is neuroprotective in vitro and improves memory and cognition in APP/PS1 mice. Bioorg. Med. Chem. Lett. 28, 1591–1594. https://doi.org/10.1016/j.bmcl.2018.03.052 (2018).
Nogami, K. et al. Pharmacological activation of SERCA ameliorates dystrophic phenotypes in dystrophin-deficient mdx mice. Hum. Mol. Genet. 30, 1006–1019. https://doi.org/10.1093/hmg/ddab100 (2021).
Sordi, G., Goti, A., Young, H. S., Palchetti, I. & Tadini-Buoninsegni, F. Stimulation of Ca(2+) -ATPase transport activity by a small-molecule drug. ChemMedChem 16, 3293–3299. https://doi.org/10.1002/cmdc.202100350 (2021).
Stroik, D. R. et al. Targeting protein–protein interactions for therapeutic discovery via FRET-based high-throughput screening in living cells. Sci. Rep. 8, 12560. https://doi.org/10.1038/s41598-018-29685-z (2018).
Rocchetti, M. et al. Modulation of sarcoplasmic reticulum function by PST2744 [istaroxime; (E, Z)-3-((2-aminoethoxy)imino) androstane-6,17-dione hydrochloride)] in a pressure-overload heart failure model. J. Pharmacol. Exp. Ther. 326, 957–965. https://doi.org/10.1124/jpet.108.138701 (2008).
Gheorghiade, M., Ambrosy, A. P., Ferrandi, M. & Ferrari, P. Combining SERCA2a activation and Na-K ATPase inhibition: A promising new approach to managing acute heart failure syndromes with low cardiac output. Discov. Med. 12, 141–151 (2011).
Maurya, S. K. & Periasamy, M. Sarcolipin is a novel regulator of muscle metabolism and obesity. Pharmacol. Res. 102, 270–275. https://doi.org/10.1016/j.phrs.2015.10.020 (2015).
Meizoso-Huesca, A., Pearce, L., Barclay, C. J. & Launikonis, B. S. Ca(2+) leak through ryanodine receptor 1 regulates thermogenesis in resting skeletal muscle. Proc. Natl. Acad. Sci. USA https://doi.org/10.1073/pnas.2119203119 (2022).
Nikolaienko, R. et al. New N-aryl-N-alkyl-thiophene-2-carboxamide compound enhances intracellular Ca(2+) dynamics by increasing SERCA2a Ca(2+) pumping. Biophys. J. https://doi.org/10.1016/j.bpj.2022.12.002 (2022).
Hou, Z. et al. 2-Color calcium pump reveals closure of the cytoplasmic headpiece with calcium binding. PLoS ONE 7, e40369. https://doi.org/10.1371/journal.pone.0040369 (2012).
Pallikkuth, S. et al. Phosphorylated phospholamban stabilizes a compact conformation of the cardiac calcium-ATPase. Biophys. J. 105, 1812–1821. https://doi.org/10.1016/j.bpj.2013.08.045 (2013).
Talbot, C. B. et al. Correction approach for delta function convolution model fitting of fluorescence decay data in the case of a monoexponential reference fluorophore. J. Fluoresc. 25, 1169–1182. https://doi.org/10.1007/s10895-015-1583-4 (2015).
Zhang, J. H., Chung, T. D. & Oldenburg, K. R. A Simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen 4, 67–73. https://doi.org/10.1177/108705719900400206 (1999).
Backman, T. W., Cao, Y. & Girke, T. ChemMine tools: an online service for analyzing and clustering small molecules. Nucleic Acids Res. 39, W486-491. https://doi.org/10.1093/nar/gkr320 (2011).
Acknowledgements
Prachi Bawaskar, Ben Grant, Simon J. Gruber, Evan W. Kleinboehl, Ang Li, Ji Li, Kurt Peterson, Seth L. Robia, and Tory M. Schaaf contributed to the conception of this project. Jesse E. McCaffrey, Bengt Svensson, Sarah Blakely Anderson, and J. Michael Autry provided helpful discussions. Marzena Brinkmann provided helpful advice on the chemical scaffold nomenclature. Bengt Svensson also provided technical assistance with illustrations. Fluorescence microscopy was performed at the UMN Imaging Center, flow cytometry at the UMN Lillehei Heart Institute, compound dispensing at the UMN Institute of Therapeutic Drug Discovery and Development, and spectroscopy at the UMN Biophysical Technology Center. This work was supported by NIH grants R01HL139065 (to DDT and RLC) and R37AG26160 (to DDT).
Author information
Authors and Affiliations
Contributions
D.D.T. and R.L.C. designed the research. S.L.Y., A.R.T., L.N.R., and P.A.B. prepared samples and performed experiments. S.L.Y., A.R.T., and O.R. analyzed the data. O.R. wrote the first draft of the manuscript. O.R., S.L.Y., A.R.T., R.T.R., C.C.A., R.L.C., and DDT edited and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
DDT and RLC hold equity in, and serve as executive officers for Photonic Pharma LLC (PP), which had no role in this study except for providing some instrumentation. ART and SLY are paid consultants for PP. OR is the sole proprietor of Editing Science LLC, which had no role in this study. These relationships have been reviewed and managed by the University of Minnesota. LNR, RTR, PAB, and CCA declare no conflicts of interest in regard to this manuscript.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Roopnarine, O., Yuen, S.L., Thompson, A.R. et al. Fluorescence lifetime FRET assay for live-cell high-throughput screening of the cardiac SERCA pump yields multiple classes of small-molecule allosteric modulators. Sci Rep 13, 10673 (2023). https://doi.org/10.1038/s41598-023-37704-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-37704-x
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
