
Abstract
Cardiovascular disease (CVD) is strongly associated with chronic low-grade inflammation, involving activated Toll-like receptors and their downstream cellular machinery. Moreover, CVD and other related inflammatory conditions are associated with infiltration of bacteria and viruses originating from distant body sites. Thus, in this study we aimed to map the presence of microbes in the myocardium of patients with heart disease that we previously found to display upregulated Toll-like receptor signaling. We performed metagenomics analysis of atrial cardiac tissue from patients undergoing coronary artery bypass grafting (CABG) or aortic valve replacement (AVR) and compared with atrial cardiac tissue from organ donors. A total of 119 species of bacteria and seven species of virus were detected in the cardiac tissue. RNA expression of five bacterial species were increased in the patient group of which L. kefiranofaciens correlated positively with cardiac Toll-like receptor-associated inflammation. Interaction network analysis revealed four main gene set clusters involving cell growth and proliferation, Notch signaling, G protein signaling and cell communication in association with L. kefiranofaciens RNA expression. Taken together, intracardial expression of L. kefiranofaciens RNA correlates with pro-inflammatory markers in the diseased cardiac atrium and may have an effect on specific signaling processes important for cell growth, proliferation and cell communication.
Similar content being viewed by others


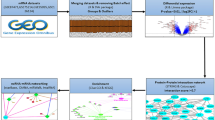
Introduction
Cardiovascular disease (CVD) is strongly associated with a chronically elevated inflammatory state, involving pro-inflammatory cytokine production following upscaled Toll-like receptor (TLR) action, triggered by various factors, such as pathogen-associated microbial patterns (PAMPs) and damage-associated molecular patterns (DAMPs), stress, hypoxia and ischemia1,2,3. Loss-of-function studies where TLRs are targeted show that a sustained inflammation is harmful to the heart and contributes to cardiac adverse remodeling4,5, which clinically is manifested as changes in size, mass and function of the heart6.
We have previously reported increased expression of TLRs 1, 3 and 7 and their mediators in the cardiac tissue of patients undergoing coronary artery bypass grafting (CABG) or aortic valve replacement (AVR)7, suggesting that triggers of viral/microbial origin might be involved in the augmented cardiac inflammatory state in these patients; TLRs 1, 3, and 7 are associated with viral infections and recognize viral single-stranded and double-stranded RNA as well as PAMPs from gram positive bacteria8,9,10.
CVD and other related pathological conditions, such as diabetes, obesity and non-alcoholic fatty liver disease, have been associated with, and shown to be modified by, bacteria and viruses, originating from distant body sites, such as the oral cavity. For example, in a large study with close to 12,000 participants, poor oral hygiene was associated with increased risk of fatal and non-fatal CVD events, including myocardial infarction, coronary artery bypass graft surgery, coronary angioplasty, stroke, heart failure and low-grade inflammation11. Altered microbial composition of the gut has also been associated with symptomatic atherosclerosis, i.e. myocardial infarction or cerebrovascular events12, and certain groups of bacteria have been shown to predict coronary artery disease13. Furthermore, the presence of bacterial DNA in human atherosclerotic plaques has been demonstrated in several reports involving different patient groups and various techniques (reviewed in ref.14). Thus, there is a large body of support for bacterial and viral expression in human tissue that is associated with low-grade inflammation and cardiovascular disease.
In this study we used metagenomics to investigate microbial gene expression in human myocardium with elevated inflammation from patients undergoing CABG or AVR and compared with normal myocardium.
Results
We performed metagenomics analysis of microbial RNA expression on right atrial tissue collected during coronary artery bypass grafting (CABG) and aortic valve replacement (AVR). A comparison was made with tissue from a control group of right atrium harvested from multi-organ donors without cardiac disease as well as commercially available right atria RNA from healthy individuals. A summary of the patient clinical data is provided in Tables 1 and 2.
A total of 119 species of bacteria and seven species of virus were detected in the cardiac tissue (Supplementary Table 1). The majority of the bacteria was classified as Firmicutes (48%), followed by Proteobacteria (23%), Bacteroidetes (4%) and Actinobacteria (4%) (Fig. 1a). Using Principal Component Analysis (PCA), an unbiased multivariate classification model, we observed a cluster separation between the control individuals and the patients with heart disease with regard to microbial gene expression (Fig. 1b). There was no clear separation between the CABG and AVR groups (Fig. 1b). A similar pattern was observed when only including microbial expression that was significantly different between the controls and patients with heart disease (Fig. 1c). Although no separation between the CABG and AVR groups was observed, there was a distinct separation within the group of individuals with heart disease (Fig. 1c). This separation could not be explained by a difference in gender, age or BMI of the individuals included (data not shown), and thus we performed bioinformatic analysis with regard to gene expression in these two groups. The result showed that the two groups differed in gene expression of genes encoding antibody light- and heavy chains (Fig. 1d) as well as various long non-coding RNAs (Fig. 1e). GSEA revealed an association of these genes with mast cell activation, phagocytosis and complement activation, particularly molecular processes involving high affinity receptor for the Fc region on immunoglobin E (FCERI) action, signaling through phosphoinositide 3-kinase (PI3K) as well as intracellular calcium signaling (Table 3 and Supplementary Fig. 1).

Cardiac microbial RNA analysis. (a) Proportion of microbial organisms and bacterial phyla detected in human atrial tissue, (b) PCA score plot showing a separation between patients with heart disease (CABG and AVR) and controls with regard to microbial RNA expression in the atrial tissue, (c) PCA score plot of microbial RNA expression significantly different between the controls and patients with heart disease in atrial tissue showing a separation of two subgroups (designated group 1 and group 2) within the patient group, (d–e) heat map of genes most significantly decreased (d) or increased (e) in group 2 versus group 1 in (c) (log2 fold change, q-value < 0.05). Heatmaps were generated using the Qlucore Omics Explorer 3.8 software (Qlucore AB, Lund, Sweden, https://qlucore.com/). n = 10 for CABG patients, 10 for AVR patients and 10 for controls.
Taken together, metagenomics analysis revealed microbial RNA expression in the heart tissue of both controls and patients with heart disease. Subsequent PCA showed an apparent separation between the groups with regard to the cardiac microbial RNA expression. Furthermore, we observed two distinct subgroups within the group with heart disease (CABG and AVR combined) that displayed genetic differences related to mast cell activation, phagocytosis and complement activation.
RNA expression from five bacterial species is significantly increased in diseased myocardium
Among the microbial expression that was significantly increased in the diseased myocardium compared to controls, the bacterial species with highest overall alignment rate percentage, ranging from 76 to 92%, were classified as Lactobacillus amylovorus, Lactobacillus backii, Lactobacillus kefiranofaciens, Lactobacillus helveticus and Lactobacillus johnsonii (Fig. 2a). The reads were evenly spread over the genomes for L. amylovorus, L. kefiranofaciens, L. helveticus and L. johnsonii, indicating a good fit to the chosen reference genome (data not shown). The reads for L. backii were more focused to a few specific regions.

Microbial RNA expression analysis in patients with heart disease compared to controls. (a) Bar graph displaying cardiac atrial RNA expression of Lactobacillus amylovorus, Lactobacillus backii, Lactobacillus kefiranofaciens, Lactobacillus helveticus and Lactobacillus johnsonii in controls, CABG patients and AVR patients. Error bars indicate SEM, *p < 0.05. (b,c) Correlation analysis between cardiac L. kefiranofaciens expression and human gene sets in patients with heart disease (CABG and AVR combined) and controls through Gene Set Enrichment Analysis (GSEA) of RNA seq-data from cardiac atrial tissue followed by correlation analysis with L. kefiranofaciens expression in the same individuals. (b), Interaction network analysis revealed four main gene set clusters that correlated with L. kefiranofaciens expression in controls: cell growth and proliferation, Notch signaling, G protein signaling and cell communication and interaction. (c), Close-up of seven gene sets that displayed opposite correlation patterns for controls compared to patients with regard to L. kefiranofaciens expression. Nodes = gene sets. Red color within the gene set nodes indicate positive correlation of those gene sets with cardiac atrial expression of L. kefiranofaciens, blue color within the gene set nodes indicate negative correlation, grey color within the gene set nodes indicate no significant correlation. The drawings were generated using the Cytoscape v. 3.9.1 software and the EnrichmentMap app (https://cytoscape.org). n = 10 for CABG patients, 10 for AVR patients and 10 for controls.
L. kefiranofaciens shows strong correlation to cardiac inflammatory markers associated with Toll-like receptor signaling
We have previously reported an increased inflammatory state in the cardiac tissue from five of the patients included in the current cohort7 and were therefore interested in investigating if there is a correlation between the expression of the assigned bacterial species and cardiac inflammatory markers associated with Toll-like receptor (TLR) signaling. Of the five bacterial species with increased RNA in the patients with cardiac disease, L. kefiranofaciens positively correlated with 31 TLR-induced inflammatory mediators (Table 4 and Supplementary Fig. 2), whereas L. amylovorus, L. helveticus, L. backii and L. johnsonii did not correlate with any of the inflammatory markers investigated (data not shown).
Plasma from the patient cohort was analyzed, using a high-throughput, multiplex immunoassay enabling analysis of 92 human cardiovascular and inflammatory biomarkers. None of the plasma CVD biomarkers from the multiplex panel correlated with the cardiac RNA expression of the observed bacterial species (Supplementary Table 2).
Taken together, out of the five bacterial species whose expression was most significantly upregulated in the patients with heart disease (CABG and AVR combined), L. kefiranofaciens had the strongest correlation with TLR-associated intracardiac inflammatory markers. Moreover, the correlation with inflammatory markers seemed confined to the cardiac tissue as no cogent correlation between intracardiac bacterial expression and CVD markers in plasma could be deduced.
L. kefiranofaciens expression is associated with transcriptional regulation by Notch, Rho activity, cytokine production and calcium signaling, and may affect these processes differently in individuals with heart disease
Based on the data showing strong correlation between L. kefiranofaciens expression and TLR-induced markers, GSEA analysis was performed to investigate possible correlation with L. kefiranofaciens expression and other signaling pathways. Interaction network analysis revealed four main gene set clusters involving cell growth and proliferation, Notch signaling, G protein signaling and cell communication and interaction in association with L. kefiranofaciens RNA expression (Fig. 2b). The majority of the nodes (gene sets) within these clusters showed a positive correlation (red color) with intracardiac L. kefiranofaciens expression in the control individuals whereas no significant correlation (grey color) was found for the patients with heart disease (CABG and AVR combined). Seven gene sets showed opposite correlation patterns for controls vs patients, where L. kefiranofaciens RNA expression in the patients was negatively correlated (blue color) with the gene sets and positively correlated in the control tissue (Fig. 2c). These gene sets and their closest neighbor gene sets included processes of transcriptional regulation by Notch, Rho GTPase activity, anti-inflammatory cytokine production and calcium signaling (Fig. 2c).
Taken together, intracardial expression of L. kefiranofaciens may have an effect on specific signaling processes important for cell growth, proliferation and cell communication of which some are differentially correlated with this bacterial species in patients with heart disease compared to controls.
Discussion
In the study presented here we sought to investigate the presence of microbial RNA in the cardiac tissue of patients undergoing CABG- or AVR- surgery and compare with tissue from individuals without heart disease.
The initial Principal Component Analysis (PCA) revealed a separation between the control individuals and the patients with heart disease with regard to microbial gene expression, without a clear separation between the CABG and AVR groups. However, there was a distinct separation within the group of individuals with heart disease that differed with respect to expression of genes encoding antibody light- and heavy chains associated with mast cell activation, phagocytosis and complement activation. It is plausible that the upregulated cardiac expression of antibodies associated with these cellular processes is impacted by specific pathogens within the cardiac tissue. However, that would need to be confirmed with more in depth mechanistic studies and is outside the scope of this report.
Dysbiosis of the intestinal or oral microbiota have long been known to be associated with increased risk of cardiovascular events, such as stroke, myocardial infarction, heart failure and atherosclerosis14,15,16. Such findings are strongly associated with an activated immune system, cytokine levels and other bacterial pro-inflammatory components, such as lipopolysaccharide (LPS) and trimethylamine oxide (TMAO), as well as increased levels of antibodies against bacterial antigens16,17,18,19,20. Besides the intestine and oral cavity, other tissues as well as the blood have been reported to harbor microbes in both healthy individuals and disease states21,22. The detection of bacteria in disparate tissue sites and blood, including bacterial DNA and RNA as well as various bacterial species, also correlate with CVD in humans23,24,25. The way that bacteria end up in distal organs and tissues following dysbiosis is thought to involve “leakage” through a compromised gut or oral epithelium where bacteria enter the blood stream and cause bacteremia. A well known example of this is endocarditis, where bacterial strains originating from the oral cavity or the gut, leak into the blood stream following inflammation and damage of the supporting tissues of the the teeth and intestinal wall, resulting in infection of the valves26. Only one of the patients in our cohort was diagnosed with endocarditis and thus endocarditis can not explain the presence of cardiac bacterial RNA in our study cohort. In addition, bacterial RNA was found also in the control group and the species found were atypical for those normally observed in cases with endocarditis.
We found RNA from 119 bacterial species and seven viral species in the cardiac tissue. RNA from five lactobacilli species was found to be increased in the diseased myocardium compared to control tissue. Additional bacterial strains or viruses might be increased in the diseased heart, but we chose to set strict cut offs and focused on the five bacterial species with the highest overall alignment rate percentage in the bioinformatics analysis.
Ziebolz et al. investigated the presence of DNA of 11 periodontal pathogens in the atrium and ventricle myocardium of 30 patients undergoing aortic valve surgery and detected bacterial DNA in both regions, with a prevalence ranging from 3 to 27% of the patients, depending on the strain investigated27. Although the study differs in several aspects from our study (93% of patients had periodontal disease, no healthy controls were included, and only 11 oral pathogens were investigated rather than a full metagenomics analysis), it supports our finding of bacterial nucleic acids in human cardiac tissue.
The bacterial species detected in our study were present also in the heart tissue of the controls. Previously reported data have suggested that the impact of infection on atherosclerosis is related to the total number of pathogens infecting an individual, referred to as pathogen burden28,29. In line with this, Koren et al. observed a correlation between the amount of bacterial DNA in atherosclerotic plaques and the leukocyte counts, suggesting that the atherosclerotic plaque bacterial load determined its inflammatory status25. In addition, Zhu et al. reported that pathogen burden significantly predicted the combined end point of myocardial infarction or death independent of C-reactive protein in 890 patients with coronary artery disease30. Furthermore, the abundance of gut microorgramisms, such Lactobacillales, has been shown to predict coronary artery disease13, and the number of Lactobacillales and the ratio of Firmicutes to Bacteroidetes increased in this patient group compared to controls31.
In light of this, we found that L. kefiranofaciens expression correlated positively with a panel of pro-inflammatory markers in the cardiac tissue in our cohort and was negatively correlated with Notch signaling, Rho GTPase activity, anti-inflammatory cytokine production and calcium signaling in the patient group (CABG and AVR combined), but positively correlated with these pathways in the control group. This suggests that L. kefiranofaciens might affect cardiac molecular signaling differently, depending on the amount of bacteria residing in the cardiac tissue.
The L. kefiranofaciens species are Gram-positive, non-motile, non-spore-forming, facultative anaerobic rod-shaped lactic acid bacteria known to produce the polysaccharide kefiran, which has been shown to exhibit antimicrobial, immunomodulating and anti-hypertensive properties32,33,34,35, and is therefore considered a probiotic. However, Lin et al. showed that direct treatment with the L. kefiranofaciens subspecies M1 induces inflammation in vitro by upregulating pro-inflammatory cytokines, such as tumor necrosis factor alpha (TNFα), and in vivo by upregulation of proinflammatory cytokines and macrophage markers in mice fed a high fat diet36. The discrepancy between different reports regarding the effects of L. kefiranofaciens are likely due to different subspecies being investigated, different celltypes/tissues studied as well as underlying pathological conditions. In support of this theory, although many lactobacilli in general are considered probiotics, this genus has been associated with contrasting outcomes when evaluated in CVD animal models, possibly due to strain-specific effects14,37. In addition, a recent survey assessing the pathogenic potential of lactobacilli, based on infection case reports, reported that serious infections caused by lactobacilli species normally considered probiotics appear to have increased in the last years. The authors concluded that pathogenic genetic traits, such as biofilm-forming capacity, should be periodically re-evaluated by genetic characterization of strains to identify non-pathogenic variants38. With regard to our study cohort, we hypothesize that a possible involvement of L. kefiranofaciens in TLR-associated inflammation would occur at a local level, within the human cardiac tissue, as we do not find a salient correlation with levels of proinflammatory markers in the plasma from the same the individuals.
Some limitations of the present study should be acknowledged. The number of individuals in our study is small and the data thus need to be confirmed in larger cohorts. Metagenomics of cardiac tissue in cohorts with other types of heart disease should be investigated as well as different locations of the human heart to put together a more extensive picture of possible intrinsic cardiac microbes in health and disease. Furthermore, it was not possible to match the individuals in the current study for gender and age, both of which could plausibly have an impact on the cardiac inflammatory state. Circulating levels of CRP and IL-6 increase with age and certain sex hormones in serum have been associated with inflammatory biomarkers39,40. Of note however, we did not find differences in circulating inflammatory markers between controls and the CABG- or AVR- patients in our previous study using the same cohort as investigated in the current study7.
In addition, it must be assumed that many other factors play a role regarding an interplay between cardiac microbes and inflammatory processes, such as environmental and genetic factors. Based on the current study, it is thus not possible to determine whether the observed correlations between bacteria and gene expressions are causal. Although it seems unlikely that for example metabolic syndrome has been an important confounding factor (included study subjects were not obese, only two individuals had type 2 diabetes and we have previously shown that this cohort does not show elevated circulating levels of pro-inflammatory markers7), this does not rule out the possibility that other confounder factors may exits. Further studies are thus needed to establish whether there is a causal relationship between bacteria and gene expressions.
In summary, we have identified RNA from five different lactobacilli species in human atrial tissue that was significantly increased in patients with heart disease. L. kefiranofaciens RNA expression was associated with cardiac TLR-induced inflammation, and negatively correlated with Notch signaling, Rho GTPase activity, anti-inflammatory cytokine production and calcium signaling in the patient group and may have an effect on specific signaling processes important for cell growth, proliferation and cell communication.
Whether an intrinsic “intracardial microbiota” exists or if dysbiosis of intracardial microbes has an impact on cardiac function and biology is an intriguing concept and our data suggest that the human heart does harbor microbes in both health and disease and that dysbiosis of these may play a role in the progression of heart disease. However, further in depth studies are necessary to confirm the data as well as elucidating underlying molecular processes, exploring possible human genetic variants of immunological mediators that might influence the susceptibility to microbial signals in relation to heart disease, as well as exploring the possibility of both beneficial and adverse ramifications of cardiac microbes in relation to cardiac function. Such new knowledge and data will frame a novel field of potential therapeutic designs for patients with heart disease, e.g. virulence blockers, antimicrobial peptides, peptidomimetics, antibodies and antisense oligonucleotides.
Methods
Human heart biopsies
Transmural biopsies were collected from the right atrium appendage just before venous cannulation for cardiopulmonary bypass in ten patients undergoing coronary artery bypass grafting and ten patients undergoing aortic valve replacement at the Sahlgrenska University Hospital, Gothenburg, Sweden. The tissue was collected in a sterile work area (during surgery), using sterile tubes, reagents and equipment. As a control group, transmural biopsies from explanted hearts were collected from the free wall of the right atrium from five multi-organ donors at the Dept. of Cardiothoracic Surgery at Sahlgrenska University Hospital were used. The donor hearts were not suitable for heart transplantation but explanted for homograft procurement in a GMP-certified facility and used in the present study after the valves were harvested. Organ donors with chronic heart failure were excluded. All tissue was immendiately put in RNA Later that had been specifically checked for contaminants using PCR, which was negative. The samples were then immediately frozen in − 80 degrees. RNA was extracted with RNAse free and sterile laboratory techniques in a laminar flow cabinet. Commercially available right atrium cardiac tissue RNA from another five healthy individuals was also added as control samples (AMS Biotechnology Europe Ltd—Abingdon, U.K.). The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in approval by the Ethical Review Board of the University of Gothenburg (reference number 560-12 and 436-15, approved on 2012-10-19 and 2015-06-25, respectively). Written informed consent was obtained from all of the included patients or, for donor hearts, next of kin, stating that their organs could be used for other medical purposes than organ transplantation.
Multiplex immunoassay analysis of plasma
Plasma EDTA samples from the same patient cohort was collected and analyzed, using Proximity Extension Assay (PEA) technology and the Proseek Multiplex CVD III kit (Olink Proteomics AB, Uppsala, Sweden), according to manufacturers instruction. Analysis of 92 human cardiovascular and inflammatory biomarkers was performed (TATAA Biocenter, Gothenburg Sweden, https://tataa.com). Linear regression calculations with bacterial RNA expression was performed using the GraphPad Prism 7.00 software (San Diego, CA). Local False Discovery Rate (local FDR) values were calculated for the obtained P-values using RStudio version 2022.02.3 with R base version 4.1.2 and the package “qvalue”, treating the analysis for each bacteria separately. P-values < 0.05 with local FDR < 0.05 were considered significant.
RNA extraction
Total RNA was isolated from heart biopsies with the RNeasy Fibrous Tissue Mini kit (Qiagen, Valencia, CA) as previously described41. The tissue was disrupted with 8 mm steal beads using a TissueLyser and treated with Proteinase K for Protease digestion (Qiagen, Valencia, CA).
Gene expression analysis of Toll-like receptor inflammatory markers
For gene expression analyses of Toll-Like Receptor (TLR)-mediated signal transduction and innate immunity, the human TLR Signaling Pathway RT2 Profiler PCR Array (PAHS-018ZA, Qiagen, Valencia, CA) was used as previously described7. The qPCR reaction was performed with an ABI 7900 HT fast real time 96 well module (Applied Biosystems, Foster City, CA). All PCR amplification was performed for 40 cycles.
Data were normalized with an automatic selection of genes from the full plate. Qiagen’s Web-based PCR Array Data Analysis Software, available at www.SABiosciences.com/pcrarraydataanalysis.php was used to automatically select an optimal set of internal reference genes for the analysis. The CT values for these genes were then geometrically averaged and used for the delta delta CT calculations. Linear regression calculations with bacterial RNA expression was performed using the GraphPad Prism 7.00 software (San Diego, CA). Local False Discovery Rate (local FDR) values were calculated for the obtained P-values using RStudio, package “qvalue”, treating the analysis for each bacteria separately. P-values < 0.05 with local FDR < 0.05 were considered significant.
RNA sequencing
RNA sequencing analysis was performed at the Genomics Core Facility at University of Gothenburg, Sweden. All samples were quality checked by the RNA integrity number (RIN) using Tapestation 2200 RNA screenTape (Agilent Technologies, Santa Clara, CA). RIN values ranged between 6.6 and 9.0 for all samples. For a detailed description of the sample preparation, see the “Supplementary materials” and “Methods” section.
Libraries were quantified and normalized with Qubit DNA HS Assay kit (Life Technologies, Carlsbad, CA) and fragment size determined by Tapestation 2200 (Agilent Technologies, Santa Clara, CA). The libraries were pooled together by using the Illumina protocol for pooling and sequenced with NovaSeq 6000 S1 (Illumina, San Diego, CA) for the read length of 2 × 100 bp.
Metagenomics analysis
The reads of each sample were classified to its best matching organism and taxonomic ID (taxid) using our in-house application Pathogen Research in Clinical Applications (PaRCA), https://github.com/ClinicalGenomicsGBG/PARCA42.
The results from PaRCA were used for Differential Expression analysis using DESeq2 where the samples were filtered prior to running DESeq2 to only contain the number of reads of the organisms that were present in at least five of the 30 samples. The normalization was performed using the DESeq2 function called variance stabilizing transformation (VST) that take into account the patient groups so that these do not contribute to the expected variance-mean trend of the experiment (setting blind to false). The results from DESeq2 were filtered for a p-adjusted value of 0.05 and a log2FoldChange ≥ 1 or log2FoldChange ≤ − 143.
Classifications were verified by screening the reads of each organism for their best matching reference, see detailed description in the supplementary materials and “Methods” section.
Multivariate analysis was carried out by principal component analysis (PCA) using SIMCA v.17.0.2 (Umetrics, Umeå, Sweden). To obtain a more normal-like distribution, data were log transformed prior to statistical analysis.
Genomics analysis – human genes
The raw data was aligned with the human GRCh38.90 reference library from the Ensembl genome browser (https://www.ensembl.org/Homo_sapiens/Info/Index), and the resulting BAM files were used for bioinformatics analysis. Transcripts with counts ≥ 10 in at least nine samples were included in the bioinformatics analysis.
Gene expression analysis and Gene Set Enrichment Analysis (GSEA)44 of the gene data presented in Fig. 1d–e were performed with Qlucore Omics Explorer 3.8 (Qlucore AB, Lund, Sweden). The Molecular Signatures Database (MSigDB) was used to obtain gene sets for GSEA, (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp). Levels of significance for differences between group means were determined with ttest or two-way ANOVA followed by Tukey’s multiple comparison tests. A false discovery rate-adjusted p-value < 0.05 was considered significant.
Gene and correlation calculations of L. kefiranofaciens (Fig. 2b,c)
All calculations were carried out using RStudio version 2022.02.3 with R base version 4.1.2, with the exception of the GSEA analysis, that in part was performed using the gene set enrichment analysis, GSEA software, and Molecular Signature Database (MSigDB)44, http://www.broad.mit.edu/gsea/.
Only genes that had at least 10 counts in at least 8 samples were retained. Normalization was performed using the DESeq2 VST function (see above). For a detailed description of the correlation calculations, see the supplementary material and “Methods” section.
Results were visulalized using the Cytoscape v. 3.9.1 software and the EnrichmentMap app (https://cytoscape.org).
Data availability
The datasets generated and/or analysed during the current study are available in the Swedish National Data Service repository, https://snd.gu.se/en, accession link https://doi.org/10.5878/e48r-gn02.
Abbreviations
- AVR:
-
Aortic valve replacement
- CABG:
-
Coronary artery bypass grafting
- CVD:
-
Cardiovascular disease
- DAMP:
-
Damage-associated molecular patterns
- GSEA:
-
Gene set enrichment analysis
- LPS:
-
Lipopolysaccharide
- PAMP:
-
Pathogen-associated microbial patterns
- PCA:
-
Principal component analysis
- TLR:
-
Toll-like receptor
- TMAO:
-
Trimethylamine oxide
References
Barr, T. L. et al. Systemic transcriptional alterations of innate and adaptive immune signaling pathways in atherosclerosis, ischemia stroke, and myocardial infarction. J Bioanal. Biomed. 7, 029–034. https://doi.org/10.4172/1948-593X.1000120 (2015).
Chao, W. Toll-like receptor signaling: A critical modulator of cell survival and ischemic injury in the heart. Am. J. Physiol. Heart Circ. Physiol. 296, H1-12. https://doi.org/10.1152/ajpheart.00995.2008 (2009).
Jiang, B. & Liao, R. The paradoxical role of inflammation in cardiac repair and regeneration. J. Cardiovasc. Transl. Res. 3, 410–416. https://doi.org/10.1007/s12265-010-9193-7 (2010).
Feng, Y. et al. Innate immune adaptor MyD88 mediates neutrophil recruitment and myocardial injury after ischemia-reperfusion in mice. Am. J. Physiol. Heart Circ. Physiol. 295, H1311–H1318. https://doi.org/10.1152/ajpheart.00119.2008 (2008).
Riad, A. et al. Toll-like receptor-4 modulates survival by induction of left ventricular remodeling after myocardial infarction in mice. J. Immunol. 180, 6954–6961 (2008).
Azevedo, P. S., Polegato, B. F., Minicucci, M. F., Paiva, S. A. & Zornoff, L. A. Cardiac remodeling: Concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq. Bras. Cardiol. 106, 62–69. https://doi.org/10.5935/abc.20160005 (2016).
Rotter Sopasakis, V. et al. Toll-like receptor-mediated inflammation markers are strongly induced in heart tissue in patients with cardiac disease under both ischemic and non-ischemic conditions. Int. J. Cardiol. 293, 238–247. https://doi.org/10.1016/j.ijcard.2019.06.033 (2019).
Miyake, K. Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin. Immunol. 19, 3–10. https://doi.org/10.1016/j.smim.2006.12.002 (2007).
Kumar, H., Kawai, T. & Akira, S. Pathogen recognition in the innate immune response. Biochem. J. 420, 1–16. https://doi.org/10.1042/BJ20090272 (2009).
Gosu, V., Basith, S., Kwon, O. P. & Choi, S. Therapeutic applications of nucleic acids and their analogues in Toll-like receptor signaling. Molecules 17, 13503–13529. https://doi.org/10.3390/molecules171113503 (2012).
de Oliveira, C., Watt, R. & Hamer, M. Toothbrushing, inflammation, and risk of cardiovascular disease: Results from Scottish Health Survey. BMJ 340, c2451. https://doi.org/10.1136/bmj.c2451 (2010).
Karlsson, F. H. et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 3, 1245. https://doi.org/10.1038/ncomms2266 (2012).
Emoto, T. et al. Characterization of gut microbiota profiles in coronary artery disease patients using data mining analysis of terminal restriction fragment length polymorphism: Gut microbiota could be a diagnostic marker of coronary artery disease. Heart Vessels 32, 39–46. https://doi.org/10.1007/s00380-016-0841-y (2017).
Jonsson, A. L. & Backhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 14, 79–87. https://doi.org/10.1038/nrcardio.2016.183 (2017).
Liccardo, D. et al. Periodontal disease: A risk factor for diabetes and cardiovascular disease. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20061414 (2019).
Carrizales-Sepulveda, E. F., Ordaz-Farias, A., Vera-Pineda, R. & Flores-Ramirez, R. Periodontal disease, systemic inflammation and the risk of cardiovascular disease. Heart Lung Circ. 27, 1327–1334. https://doi.org/10.1016/j.hlc.2018.05.102 (2018).
Jin, M., Qian, Z., Yin, J., Xu, W. & Zhou, X. The role of intestinal microbiota in cardiovascular disease. J. Cell. Mol. Med. 23, 2343–2350. https://doi.org/10.1111/jcmm.14195 (2019).
Priyamvara, A. et al. Periodontal Inflammation and the risk of cardiovascular disease. Curr. Atheroscler. Rep. 22, 28. https://doi.org/10.1007/s11883-020-00848-6 (2020).
Xu, H. et al. The gut microbiota and its interactions with cardiovascular disease. Microb. Biotechnol. 13, 637–656. https://doi.org/10.1111/1751-7915.13524 (2020).
Yoo, J. Y., Groer, M., Dutra, S. V. O., Sarkar, A. & McSkimming, D. I. Gut microbiota and immune system interactions. Microorganisms https://doi.org/10.3390/microorganisms8101587 (2020).
Moriyama, K. et al. Polymerase chain reaction detection of bacterial 16S rRNA gene in human blood. Microbiol. Immunol. 52, 375–382. https://doi.org/10.1111/j.1348-0421.2008.00048.x (2008).
Rajendhran, J., Shankar, M., Dinakaran, V., Rathinavel, A. & Gunasekaran, P. Contrasting circulating microbiome in cardiovascular disease patients and healthy individuals. Int. J. Cardiol. 168, 5118–5120. https://doi.org/10.1016/j.ijcard.2013.07.232 (2013).
Amar, J. et al. Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: The D.E.S.I.R. study. PLoS One 8, e54461. https://doi.org/10.1371/journal.pone.0054461 (2013).
Ott, S. J. et al. Detection of diverse bacterial signatures in atherosclerotic lesions of patients with coronary heart disease. Circulation 113, 929–937. https://doi.org/10.1161/CIRCULATIONAHA.105.579979 (2006).
Koren, O. et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. U. S. A. 108(Suppl 1), 4592–4598. https://doi.org/10.1073/pnas.1011383107 (2011).
Del Giudice, C. et al. Infective endocarditis: A focus on oral microbiota. Microorganisms https://doi.org/10.3390/microorganisms9061218 (2021).
Ziebolz, D. et al. Periodontal bacteria DNA findings in human cardiac tissue—Is there a link of periodontitis to heart valve disease?. Int. J. Cardiol. 251, 74–79. https://doi.org/10.1016/j.ijcard.2017.09.001 (2018).
Zhu, J. et al. Effects of total pathogen burden on coronary artery disease risk and C-reactive protein levels. Am. J. Cardiol. 85, 140–146. https://doi.org/10.1016/s0002-9149(99)00653-0 (2000).
Epstein, S. E. et al. Infection and atherosclerosis: Potential roles of pathogen burden and molecular mimicry. Arterioscler. Thromb. Vasc. Biol. 20, 1417–1420. https://doi.org/10.1161/01.atv.20.6.1417 (2000).
Zhu, J. et al. Prospective study of pathogen burden and risk of myocardial infarction or death. Circulation 103, 45–51. https://doi.org/10.1161/01.cir.103.1.45 (2001).
Emoto, T. et al. Analysis of gut microbiota in coronary artery disease patients: a possible link between gut microbiota and coronary artery disease. J. Atheroscler. Thromb. 23, 908–921. https://doi.org/10.5551/jat.32672 (2016).
Rodrigues, K. L., Caputo, L. R., Carvalho, J. C., Evangelista, J. & Schneedorf, J. M. Antimicrobial and healing activity of kefir and kefiran extract. Int. J. Antimicrob. Agents 25, 404–408. https://doi.org/10.1016/j.ijantimicag.2004.09.020 (2005).
Vinderola, G., Perdigon, G., Duarte, J., Farnworth, E. & Matar, C. Effects of the oral administration of the exopolysaccharide produced by Lactobacillus kefiranofaciens on the gut mucosal immunity. Cytokine 36, 254–260. https://doi.org/10.1016/j.cyto.2007.01.003 (2006).
Maeda, H., Zhu, X., Omura, K., Suzuki, S. & Kitamura, S. Effects of an exopolysaccharide (kefiran) on lipids, blood pressure, blood glucose, and constipation. BioFactors 22, 197–200. https://doi.org/10.1002/biof.5520220141 (2004).
Pimenta, F. S. et al. Mechanisms of action of kefir in chronic cardiovascular and metabolic diseases. Cell. Physiol. Biochem. 48, 1901–1914. https://doi.org/10.1159/000492511 (2018).
Lin, Y. C., Chen, Y. T., Li, K. Y. & Chen, M. J. Investigating the mechanistic differences of obesity-inducing lactobacillus kefiranofaciens M1 and anti-obesity Lactobacillus mali APS1 by microbolomics and metabolomics. Front. Microbiol. 11, 1454. https://doi.org/10.3389/fmicb.2020.01454 (2020).
Grazioli-Gauthier, L. et al. Lactobacillus jensenii mitral valve endocarditis: Case report, literature review and new perspectives. IDCases 27, e01401. https://doi.org/10.1016/j.idcr.2022.e01401 (2022).
Rossi, F., Amadoro, C., Gasperi, M. & Colavita, G. Lactobacilli infection case reports in the last three years and safety implications. Nutrients https://doi.org/10.3390/nu14061178 (2022).
Milan-Mattos, J. C. et al. Effects of natural aging and gender on pro-inflammatory markers. Braz. J. Med. Biol. Res. 52, e8392. https://doi.org/10.1590/1414-431X20198392 (2019).
Hatziagelaki, E. et al. Association between biomarkers of low-grade inflammation and sex hormones in women with polycystic ovary syndrome. Exp. Clin. Endocrinol. Diabetes 128, 723–730. https://doi.org/10.1055/a-0992-9114 (2020).
Sandstedt, J. et al. Markedly reduced myocardial expression of gamma-protocadherins and long non-coding RNAs in patients with heart disease. Int. J. Cardiol. 344, 149–159. https://doi.org/10.1016/j.ijcard.2021.09.046 (2021).
Olausson, J. et al. Optimization of cerebrospinal fluid microbial DNA metagenomic sequencing diagnostics. Sci. Rep. 12, 3378. https://doi.org/10.1038/s41598-022-07260-x (2022).
Love, M. I., Anders, S. & Huber, W. Analyzing RNA-seq data with DESeq2. Bioconductor 2, 1–63 (2017).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Acknowledgements
We would like to thank the Genomics and Bioinformatics Core Facility platforms at the Sahlgrenska Academy, University of Gothenburg, particularly Pernilla Ericsson and Elham Rekabdar.
Funding
Open access funding provided by University of Gothenburg. This work was supported by the Swedish Heart–Lung Foundation, the Swedish Federal Government under the LUA/ALF agreement, the Swedish Society of Medicine and the Gothenburg Society of Medicine. The funders had no role in the study design, data curation, data analysis, data interpretation, writing of the report or publication of the data.
Author information
Authors and Affiliations
Contributions
V.R.S. co-designed the study, supervised the project, performed data analysis and statistical analysis and drafted the original manuscript. L.M.H. co-designed the study, supervised the project and reviewed the manuscript. J.S. performed statistical and bioinformatics analysis and reviewed the manuscript. K.V. collected and prepared the samples and reviewed the manuscript. G.D. provided explanted heart tissue from multi-organ donors and reviewed the manuscript. A.J. provided heart tissue biopsies from patients undergoing coronary artery bypass grafting surgery and aortic valve replacement and reviewed the manuscript. All authors contributed to the article and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sandstedt, J., Vukusic, K., Dellgren, G. et al. Metagenomic sequencing of human cardiac tissue reveals Microbial RNA which correlates with Toll-like receptor-associated inflammation in patients with heart disease. Sci Rep 13, 7884 (2023). https://doi.org/10.1038/s41598-023-35157-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-35157-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
