
Abstract
Colorectal cancer (CRC) is the third most common cancer worldwide. Dysbiosis of human gut microbiota has been linked to sporadic CRC. This study aimed to compare the gut microbiota profiles of 80 Thai volunteers over 50 years of age among 25 CRC patients, 33 patients with adenomatous polyp, and 22 healthy controls. The 16S rRNA sequencing was utilized to characterize the gut microbiome in both mucosal tissue and stool samples. The results revealed that the luminal microbiota incompletely represented the intestinal bacteria at the mucus layer. The mucosal microbiota in beta diversity differed significantly among the three groups. The stepwise increase of Bacteroides and Parabacteroides according to the adenomas–carcinomas sequence was found. Moreover, linear discriminant analysis effect size showed a higher level of Erysipelatoclostridium ramosum (ER), an opportunistic pathogen in the immunocompromised host, in both sample types of CRC patients. These findings indicated that the imbalance of intestinal microorganisms might involve in CRC tumorigenesis. Additionally, absolute quantitation of bacterial burden by quantitative real–time PCR (qPCR) confirmed the increasing ER levels in both sample types of cancer cases. Using ER as a stool–based biomarker for CRC detection by qPCR could predict CRC in stool samples with a specificity of 72.7% and a sensitivity of 64.7%. These results suggested ER might be a potential noninvasive marker for CRC screening development. However, a larger sample size is required to validate this candidate biomarker in diagnosing CRC.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is the third leading cause of cancer–related deaths worldwide, with an estimated 1.9 million new cases and 935,000 deaths annually1. Approximately 60–65% of sporadic CRCs, which occur spontaneously without a family history of CRC or inherited genetic mutation, are the major proportion of CRC cases2. Adenomatous polyps or adenomas are precancerous lesions in almost all sporadic CRCs and were found in up to 50% of persons above 50 years of age undergoing colonoscopy3. Early CRC screening and removal of benign or precancerous polyps are recommended for effective prevention4. Apart from known risk factors such as age 50 or greater, unhealthy dietary habits, ethnicity, and smoking5, emerging evidence indicates that dysbiosis of intestinal bacteria is associated with the pathogenesis of sporadic CRC6. Numerous bacterial species have been studied, of which representative culturable isolates interplayed with cancer cell lines and induced disease pathogenesis in vivo7,8,9,10,11. However, there was no consensus on microbial signatures in CRC patients. The inconsistency might be caused by different technical approaches and geographical locations12.
Furthermore, microbiota in the colon mucosa (mucosal microbiota) and the feces (luminal microbiota) are different, although their patterns were shown to be partially correlated13,14. Gut microbiome profiles from both stool and colon mucosa of Asian people have been scarcely reported. In this study, we conducted a comparative analysis of the gut microbiome profile using stool and mucosal samples from Thai CRC patients, individuals with adenomas, and healthy volunteers aged 50 and above using 16S rRNA sequencing analysis. We then performed the quantitative PCR to validate the predominant bacterial signature in both sample types from Thai CRC patients.
Results
Demographic and clinical data of CRC patients, individuals with adenomas, and healthy volunteers
A total of 80 Thai participants (range; 51–85 years old) consisting of 25 CRC, 33 adenomas (AD), and 22 healthy control (HC) groups were recruited from June 2019 to December 2020. Detailed demographic distribution among the three groups is summarized in Table 1. There were no differences among CRC, AD, and HC groups according to gender, age, BMI, or underlying diseases, i.e., hypertension (HT) and dyslipidemia (DLP), except for type 2 diabetes mellitus (T2DM) (more T2DM in the CRC patients, p < 0.05). In the CRC group, most of the tumors were classified in stage T3 (60%). Other clinicopathological data were described in Supplementary Table S1.
Alpha diversity and beta–diversity metrics of the mucosal microbiota
There were no statistically significant differences in alpha–diversity indices including Chao1’s index (p = 0.104) (Fig. 1a), Shannon’s index (p = 0.877) (Fig. 1b), and Simpson’s index (p = 0.715) (Fig. 1c) among CRC, AD and HC groups. However, a significant difference in beta diversity was observed in Bray–Curtis’s distance among the three groups (p = 0.039) (Fig. 1d and e).

Microbial diversity analyses and microbial abundance profiling in mucosal tissue samples among healthy control (HC), adenoma (AD), and colorectal cancer (CRC) subjects, (a) Chao1’s index, (b) Shannon’s index, (c) Simpson’s index, (d) Principal Coordinate Analysis (PCoA) analysis based on Bray–Curtis distance in 2–Dimension, (e) PCoA analysis based on Bray–Curtis distance in 3–Dimension, (f) Taxonomic composition type at the phylum level, (g) Taxonomic composition at the genus level.
Altered gut mucosal microbiota in CRC and AD groups
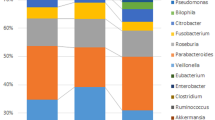
The relative taxonomic pattern of mucosal microbiota from CRC, AD, and HC groups was examined. Overall microbial compositions of the AD and the CRC groups were shifted compared to that of healthy controls. At the phylum level, Bacteroidetes was the most prevalent in the AD and the CRC groups (63% and 58%, respectively), while Proteobacteria predominated in the HC group (42%) (Fig. 1f). At the genus level, a stepwise increase of Bacteroides and Parabacteroides in the CRC group (55.2% and 3.5%) was observed when compared with the AD (50.4% and 3.4%) and the HC groups (36.0% and 1.2%). On the other hand, Escherichia–Shigella in the AD (8.8%) and the CRC (16.4%) groups decreased compared with the HC group (35%). Furthermore, there was a decrease in Faecalibacterium in the CRC patients (0.7%) compared with the AD (2.4%) and the HC groups (4%) (Fig. 1g).
Determining group–wise alteration in the bacterial abundance of AD or CRC at the genus level, the heat trees displayed only significant differences in relative abundance of the individual taxon in which the terminal nodes corresponding to the bacterial genera (Fig. 2). Three comparisons between groups were accomplished as follows: (1) HC versus (vs.) CRC; (2) AD vs CRC (3) HC vs AD. The highest difference in bacterial composition was observed between CRC patients and healthy controls. Compared to HC, the bacterial diminution in CRC was detected in the genus Faecalibacterium, Escherichia–Shigella, and within the Lachnospiraceae family. In the opposite direction, bacteria within the phylum Bacteroidetes, the genus Bacteroides (p = 0.012), Parabacteroides (p = 0.004), and Butyricimonas (p = 0.042) increased in the HC group compared to the CRC group. Moreover, genus Collinsella (p = 0.026), Erysipelatoclostridium (p = 0.0001), Ruminococacease (p = 0.002), Flavonifractor (p = 0.009), and Phocea (p = 0.029) were significantly predominated in the CRC patients. As for bacterial comparisons between AD subjects and others, only alteration of genus Flavobacterium belonging to the phylum Bacteroidetes was significantly over–represented in the AD group compared with the HC group. Interestingly, a significant rise in this genus was found in the AD group compared with the CRC group.

Heat tree analysis illustrating the taxonomic alterations in the mucosal microbiota composition according to adenoma–carcinoma sequence based on the log2 ratio median abundance. Significant alterations of taxa are displayed by name at the corresponding node. The nodes indicate the hierarchical structure of taxa. Three comparisons were conducted: colorectal cancer (CRC) (green) vs healthy control (HC) (yellow); CRC (green) vs adenoma (AD) (yellow); and AD (green) vs HC (yellow). The dominant color corresponds to a higher number of amplicon sequence variants (ASVs). The Log2 ratio is 0 (gray) when the compared groups are not significantly different.
For exploring a differential abundance of gut microbiota at the species level, the univariate analysis in MicrobiomeAnalyst was carried out with default parameters (p < 0.05). The analysis identified seven bacterial species that were significantly abundant among the three groups (Fig. 3). Five species representing Erysipelatoclostridium ramosum (p < 0.001), Bacteroides thetaiotaomicron (p = 0.002), Flavonifractor plautii (p = 0.021), Parabacteriodes merdae (p = 0.012), and Parabacteriodes distasonis (p = 0.020) were more abundant in the CRC group. While Escherichia coli–Shigella sp. (p = 0.019) and one of the unassigned taxa (p = 0.013) were significantly lower in CRC patients.

The significant difference in bacterial abundance of the individual bacterium at the species level in the mucosal tissue samples among healthy control (HC), adenoma (AD), and colorectal cancer (CRC) subjects. Data are shown as log–transformed count and p < 0.05.
Luminal microbiota partially reflected mucosal microbiota
Early detection of CRC is essential to reduce the mortality rate. Fecal biomarkers are more practical than utilizing tissue samples for CRC screening. We analyzed whether luminal microbiota could represent a proxy of mucosal microbiota in CRC. Luminal and mucosal microbiota of paired samples from 47 participants were significantly different between sample types in terms of alpha diversity and beta diversity (Supplementary Fig. S1). The distinction of microbial diversity was detected when all available samples (paired and unpaired samples) were included in the analysis (Fig. 4a–e). Bacterial comparison between sample types at the phylum level showed the luminal microbiota harbored more Bacteroidetes, Firmicutes, and Actinobacteria, whereas Proteobacteria and Fusobacterium over–represented in the mucosal microbiota (Fig. 4f). At the genus level, Escherichia–Shigella, and Fusobacterium were found more abundance in the mucosal microbiota than the luminal microbiota (Fig. 4g). However, the luminal microbiota among CRC patients, individuals with AD, and HC subjects showed no difference in overall microbiota (alpha and beta diversity) (Supplementary Fig. S2a–e). Nonetheless, the univariate analysis of fecal microbiota found significant differences in the abundance of three bacterial species among three groups (E. ramosum (p < 0.001), B. vulgatus (p = 0.020), and Eggerthella lenta (p = 0.024)) (Supplementary Fig. S2f). Thus, even though luminal and mucosal microbiota were significantly different, and luminal microbiota only incompletely reflected the intestinal microorganisms at the host mucus layer, the microbial imbalance associated with cancer was still apparent.

Microbial diversity analyses and microbial abundance profiling between feces and mucosal tissue samples, (a) Chao1’s index, (b) Shannon’s index, (c) Simpson’s index, (d) Principal Coordinate Analysis (PCoA) analysis based on Bray–Curtis distance in 2–Dimension, (e) PCoA analysis based on Bray–Curtis distance in 3–Dimension, (f) Taxonomic composition type at the phylum level, (g) Taxonomic composition at the genus level.
Identification of a novel microbial signature for CRC screening
To further confirm the taxonomic differences among CRC, AD, and HC groups, biomarker discovery using LEfSe analysis was performed with an LDA score ≥ 3.0 for both sample types. As shown in Fig. 5a, the mucosal microbiota of CRC was over–represented by B. thetaiotaomicron, P. merdae, P. distasonis, E. ramosum, and F. plautii. In addition, unspecified bacteria and Escherichia coli–Shigella sp. predominated in HC tissue. In fecal samples, E. ramosum and E. lenta were enriched in the CRC group, while B. vulgatus was predominant in the HC group. Additionally, only E. ramosum was explicitly detected in the CRC patients in fecal and tissue samples (Fig. 5b). However, this study did not find putative biomarkers that could differentiate AD patients from the HC group.

Linear discriminant analysis (LDA) effect size (LEfSe) analysis of mucosa (tissue) and lumen (feces)–associated microbiota among healthy control (HC), adenoma (AD), and colorectal cancer (CRC) groups, (a) Histogram of the LDA scores for significantly abundant species, (b) Venn diagram represented the number of unique and overlapping significantly abundant species.
Bacterial quantification in the clinical samples by absolute qPCR
To validate the result of E. ramosum (ER) in both stool and tissue samples of CRC patients, the quantification of ER in another aliquot of DNA from the identical samples used in this study was conducted by absolute qPCR. Based on the designed primers targeting the 16S rRNA gene of ER, the nucleotide sequence of the constructed plasmid used as a positive control revealed 93.1% similarity to that of E. ramosum strain D40–2 (accession number MT275475.1). We found that the copy number per gram weight (CN/g) of ER was significantly enriched in tissue samples of CRC patients compared with the AD (p = 0.027) and healthy donors (p = 0.002) (Fig. 6a). Nevertheless, there was no significant difference in the abundance of fecal ER among the three groups (Fig. 6a). Bivariate correlation analysis showed that the absolute quantity of ER measured by qPCR assay was significantly correlated with the 16S rRNA sequencing abundance (Spearman’s r = 0.48, p < 0.001) (Fig. 6b). Binary logistic regression models were generated using the ER quantity to differentiate according to three pair–wise analyses: (1) HC vs CRC; (2) AD vs CRC; (3) HC vs AD. Of these predictive models in the stool (Fig. 6c) and tissue samples (Fig. 6d), ROC analysis showed that mucosal ER provided greater discrimination in CRC detection than fecal ER. Mucosal ER could distinguish patients with CRC from HC, and CRC from individuals with AD at similar performance, giving an area under the ROC curve (AUC) of 0.789 (sensitivity (sn) = 86.7%, specificity (sp) = 65.5%), and 0.793 (sn = 86.7%, sp = 75.0%), respectively (Fig. 6d and e). Moreover, at the best cut–off value (4.44E + 05 CN/g), the fecal ER could discriminate cancer patients from HC with a sensitivity of 64.7%, and specificity of 72.7% (Fig. 6e). However, the bacterial markers of ER in both sample types indicated poorer performance in AD detection (HC vs AD) than those in cancer detection (HC vs CRC, and AD vs CRC) (Fig. 6c–e).

Absolute quantification and diagnostic performance of CRC–associated Erysipelatoclostridium ramosum in the clinical samples, (a) absolute quantity of E. ramosum of both fecal and tissue samples. The scatter plot is expressed as log10 copy number per gram weight, and the data are displayed as mean ± SD. Each dot represents one sample. (b) Spearman’s rank correlation coefficient between 16S rRNA gene sequencing and qPCR assay abundance for E. ramosum. Data was shown in log10 of reads and copy number per gram weight, respectively. (c) ROC curve for colorectal cancer (CRC) and adenoma (AD) detection in fecal samples. (d) ROC curve for CRC and AD detection in tissue samples. (e) Diagnostic performance of E. ramosum as CRC biomarker. Abbreviations: *p < 0.05; **p < 0.01.
Discussion
Accumulating evidence has indicated that alteration of intestinal microbiota composition possibly affects the initiation and progression of CRC15,16. Although adherent bacteria might be more inclined to interact with host cells than transient bacteria that are shed daily in the stool, there were a limited number of studies on gut microbiome analysis using colon tissue samples of CRC patients17,18,19. Matched non–tumor tissues from CRC patients were usually used for comparison20,21. In addition, worldwide studies, especially data from the Asia region, are rarely conducted in stool and mucosal samples to comprehensively analyze luminal and mucosal microbiota in CRC13,22,23. In this study, the 16S rRNA gene sequencing tool was utilized to compare the bacterial composition in stool and mucosal tissues among three groups of Thai participants aged 50 years or above, consisting of healthy volunteers, patients with AD as CRC precancerous lesions, and patients with CRC. The structural segregation of gut microbiota between mucosal tissues and fecal samples showed significant differences in bacterial richness, diversity, and overall microbial profile. However, fecal dysbiosis related to disease status was still observed (Supplementary Fig. S3) and partially overlapped with mucosal microbiota. These findings confirm the previous studies suggesting luminal microbiota incompletely represented the microbiota on the mucus layer13,22.
Tjalsma and his team proposed the bacterial driver–passenger model for CRC development24. Certain bacteria in the colon can initiate multistep colorectal carcinogenesis consisting of induced inflammation, increased cell proliferation, and/or genotoxin production. After epithelial DNA damage, colorectal tumorigenesis might be associated with alteration of the gut microenvironment that facilitates the outnumber of colonic commensals with either tumor–promoting or tumor–suppressive features considered as passenger bacteria. Furthermore, bacterial drivers may be replaced by passenger bacteria that could take the growth benefit of the cancerous microenvironment. Concerning the mucosal microbiota in this study, Flavobacterium, an opportunistic pathogen in immunocompromised patients25, was significantly over–represented in the mucosal tissues of the AD group but not in CRC patients. This observation is consistent with a previous report on patients with intestinal metaplasia26. This bacterium might be associated with the early or precancerous stage of CRC and act as driver bacteria but is subsequently outcompeted by passenger bacteria in CRC patients.
As for passenger bacteria, the interesting and consistent observation was the significant enrichment of Flavonifractor plautii and Parabacteroides distasonis on the mucosal tissue in Thai CRC patients, similar to the results of an Indian CRC study27. These bacterial species converted beneficial dietary flavonoids, found in a plant–based diet, e.g., green tea, wine, and cocoa, into the human intestine28. Although flavonoids are primarily composed of polyphenolic compounds with a wide spectrum of pharmacological properties, including their influential role in anti–cancer activity29, spacious degradation of flavonoids by intestinal microorganisms may lead to lower total bioavailability and lower opportunity for intact flavonoids to be absorbed in the epithelial cells30. Therefore, the elevated abundance of these bacteria may be related to the higher activity of flavonoid degradation that reduces the anti–carcinogenic effects and bioavailability of flavonoids in CRC patients. Meanwhile, the protective role of P. distasonis in tumor development and their property in the maintenance of gut barrier has been proposed in tumor–bearing mice31. Nevertheless, in this study, we cannot conclude the definite functional pathway co–involved with gut microbiota in Thai CRC subjects. Thus, further studies are required to investigate whether these bacteria play the potential role of passenger bacteria with tumor–suppressive or tumor–promoting features.
On the other hand, the significant overrepresentation of the genus Collinsella in individuals with CRC was observed and agreed with previous studies32,33. Collinsella was shown to have a role in elevating gut barrier permeability and inducing the production of proinflammatory IL–17 in patients with rheumatoid arthritis34. Another remarkable observation of mucosal microbiota, Erysipelatoclostridium ramosum was less abundant in the HC and AD groups while it was significantly elevated in the cancer tissues. This bacterium could be an opportunistic pathogen in immunocompromised hosts35, but no study has evaluated its function in CRC. Some strains of E. ramosum were shown to secrete human immunoglobin A proteases36. The IgA proteases can cleave anti–microbial IgA translocating across the colonic mucosa resulting in increased host susceptibility. These results suggest that E. ramosum might serve as one of the passenger bacteria with tumor–promoting roles that preferentially colonize in the tumor microenvironment.
Escherichia coli–Shigella sp. were more abundant in the HC tissues and generally referred to as normal flora with potential pathogenic aspects in the large intestine37. The abundance of the bacteria in a healthy population was previously reported38. However, certain E. coli strains can produce genotoxin39 and are usually considered as causing agents in the CRC–associated driver–passenger model24. The discrepancy between our study and other studies might be explained by 1) the ethnic difference in the susceptibility to colonization by Escherichia coli–Shigella sp., 2) the difference in virulence mechanisms and functional roles among strains, i.e., the genera could not be differentiated between normal flora or pathogen in our study, and 3) the possibility of currently healthy volunteers to be a high–risk group for CRC.
Although mucosal bacteria directly interact with colon tissue, using tissue samples in CRC screening is invasive. Fecal samples are more practical for the detection of CRC–related biomarkers. Interestingly, based on LEfSe analysis, E. ramosum was solely found in significant abundance in both stool and tissue samples of CRC patients. In our validation via qPCR, the absolute quantification of mucosal E. ramosum confirmed the results from 16S rRNA sequencing. In addition, the increasing tendency of fecal E. ramosum was observed in CRC patients. This bacterium could be a putative biomarker candidate in CRC screening, providing a potential specificity and sensitivity. Furthermore, previous studies investigated the utilization of fecal microbial biomarkers, such as Fusobacterium nucleatum and/or a panel of microbes, has been shown to improve the detection of CRC in independent clinical studies40,41 and systematic reviews42,43,44. Additionally, quantitation of microbial biomarkers could improve the accuracy of the fecal immunochemical test (FIT), a current non–invasive screening test, for the prediction of advanced neoplasia and CRC40,45,46. Therefore, the bacterial markers should potentially facilitate the early identification of individuals at risk and in need of close surveillance. However, the predominant abundance of E. ramosum associated with CRC in our study has not been widely reported in studies from other countries6,12. Dissimilar microbial profiles between studies could be the result of distinctions in external factors, including living environments, diet, and lifestyle, or variations in technical characteristics12. Multicentric clinical studies using the same workflow and methods are crucial to validate promising candidate biomarkers in a larger group of participants in different geographic locations. The results should reveal common and unique microbial biomarkers for the early detection of CRC in general and specific populations, respectively.
This study has some limitations that might explain different results compared with other studies. Firstly, as for disparate protocols, the selection of universal 16S rRNA gene primers is one of the factors that can cause different results of gut microbiota profiling between studies12,47. Most microbiome studies used the primers targeting V338,48, V423,49,50,51, or V3/V413,52,53 of hypervariable regions of the 16S rRNA gene. However, our preliminary study using all these primers showed low detection sensitivity in the tissue samples (data not shown). Therefore, we performed the 16S rRNA gene sequencing using V1/V2–targeted primers, consistent with previous studies18,54,55. Secondly, regarding confounders, the CRC group had significantly more patients with type 2 diabetes mellitus (T2DM) than the HC group. T2DM is associated with an increased risk of CRC56. Therefore, it could bias the CRC–associated microbiota result. However, if T2DM patients were excluded from all groups, the impact of mucosal E. ramosum still significantly increased in CRC patients compared to HC subjects (Supplementary Fig. S4). Because CRC is a multifactorial disease, this study still included the results from cases with T2DM to perform the microbiome analysis. Thirdly, we designed specific primers to amplify the 16S rRNA gene of E. ramosum for qPCR. Nevertheless, the primers should be further verified for specificity and sensitivity before used to quantify E. ramosum in clinical application.
In conclusion, this study identified the dysbiosis signature of gut microbiota according to the adenoma–adenocarcinoma sequence for the Thai population. Apart from the host factors, our results implied that the dysbiosis of colonic microorganisms accompanied by driver and passenger bacteria might be involved in CRC tumorigenesis. The microbiome analysis uncovered the putative biomarkers for CRC from mucosal and luminal microbiota. However, a larger sample size is required to further validate the candidate biomarker. Furthermore, the functional profiles of gut microbiota that interact with human hosts or studies in animal models are needed to provide new insights into their roles in CRC carcinogenesis.
Materials and methods
Volunteer recruitment, including inclusion and exclusion criteria
A total of 80 Thai volunteers consisting of 25 patients with CRC scheduled for colorectal surgery, 33 subjects with adenomatous polyp, and 22 healthy volunteers who underwent screening colonoscopy for CRC were recruited between June 2019 and December 2020 from the King Chulalongkorn Memorial Hospital (Bangkok, Thailand). All participants were selected according to the following inclusion criteria: Thai nationality, age above 50, and written informed consent. Exclusion criteria were as follows: (1) antibiotics usage within 3 months before sampling; (2) probiotics consumption in any forms within 1 week before enrollment; (3) history of inflammatory bowel disease (IBD); (4) active bowel infection within one month before participation; (5) current immunosuppressive drug usage; (6) previously intake of chemotherapy or radiotherapy; and (7) history of colonoscopy within one month before sample collection. This study was conducted with the approval of the Institutional Review Board (IRB) of the Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand (IRB approval number 182/62) according to the international guidelines for human research protection as the Helsinki Declaration, the Belmont Report, CIOMS Guidelines and International Conference on Harmonization in Good Clinical Practice guidelines. Written informed consent was obtained from each participant.
Sample collection
All participants were requested to self–collect their feces using a provided stool collection kit before polyethylene glycol (PEG) intake as a bowel preparation step. Fecal samples were immediately collected and preserved in 3 mL DNA/RNA protection reagent (New England Biolabs), transported on an icepack to the laboratory within 12 h, and stored at –80 °C before use. During surgery or colonoscopy, one tissue or biopsy from the peri–lesion site (2–3 mm from the lesion) was collected and preserved in 0.5 mL DNA/RNA protection reagent. For the HC group, randomly one biopsy from the proximal colon (PC) and another biopsy from the distal colon (DC) were collected and preserved in a 0.5 mL DNA/RNA protection reagent and stored at –80 °C before use. In the total of 146 samples, 16 mucosa tissues and 17 fecal samples were obtained from the CRC group. For 7 CRC patients, both stool and tissue samples were obtained. Twenty–six mucosa tissues and 31 fecal samples were obtained from the AD group, and 25 individuals with AD provided paired stool and tissue samples. For healthy volunteers, 34 tissue samples (17 PC and 17 DC) and 22 fecal samples were obtained, and corresponding stool and tissue samples were obtained from 17 healthy subjects.
DNA extraction
The genomic DNA of stool and tissue samples were extracted using the QIAamp PowerFecal Pro DNA kit (Qiagen) and the QIAamp Fast DNA Tissue Kit (Qiagen), respectively, following the manufacturer’s instructions. The concentration and purity of DNA were determined by NanoDrop2000 spectrophotometer (Thermo Fisher Scientific), and the integrity of DNA was checked by 1.5% (w/v) agarose gel electrophoresis. All DNA samples were stored at –20 °C until further processing.
16S rRNA gene sequencing and bioinformatics analysis
Paired–end sequencing was conducted using the Illumina MiSeq 250 bp platform (Illumina) at Génome Québec Innovation Centre (Montréal). The V1–V2 hypervariable regions of the 16S rRNA gene were targeted using the forward primers: 27bF (5′– AGRGTTTGATCMTGGCTCAG–3′) and the reverse primers: 338R (5′–TGCTGCCTCCCGTAGGAGT–3′). The raw 16S rRNA amplicon sequences were cleaned with primer and adapter trimming, then the chimeric sequences were removed using Cutadapt57. Briefly, these clean data were clustered as amplicon sequence variants (ASVs) using DADA258. Then taxonomy was annotated with the SILVA v.138.1 16S rRNA reference gene database59.
Microbiome data analysis
The microbiota diversity and abundance were analyzed using the MicrobiomeAnalyst web–based platform (https://www.microbiomeanalyst.ca/)60. Gene abundance data were analyzed by Marker Data Profiling (MDP). Data were filtered to remove features with less count of 4 and less than 20% of prevalence, as a minimum, and a low variance filter of 20%, based on the interquartile range. Alpha–diversity profiling was calculated based on the total number of ASV analyzed using the nonparametric tests. Moreover, the beta–diversity was calculated using Bray-Curtis distance and permutational multivariate analysis of variance (PERMANOVA) and visualized by principal coordinate analysis (PCoA) plot. Heat tree analysis was generated for pairwise comparisons of microbial communities. Reingold–Tilfold graph layout and Log2 fold change of relative abundance were displayed. The different taxonomy abundance among groups was also identified with classical univariate statistical comparison. Values were considered statistically significant when the p < 0.05. The robust biomarkers of CRC were identified using the linear discriminant analysis (LDA) effect size (LEfSe) approach61 with an LDA score ≥ 3.0 and a p–value of 0.05.
Quantification of Erysipelatoclostridium ramosum in the clinical samples by quantitative PCR
DNA standard curve
In this study, total fecal DNA was used as a PCR template to amplify the 16S rRNA gene of E. ramosum used for the positive control plasmid. The specific primers were designed and checked in silico specificity via Primer–BLAST62. The nucleotide sequences of the primers targeted were as follows: ERAM2–F: 5′–AGGATGGACTTATGGCGCATT–3′, ERAM2–R: 5′–TACCGTCACTCGGCTACCAT–3′. Based on T/A cloning, the gene fragment amplified using Taq DNA polymerase (Thermo Fisher Scientific) were cloned into the pGEM–T easy vector (Promega, WI, USA) according to the manufacturer’s protocol. The resulting recombinant plasmids were transformed into competent Escherichia coli DH5α (Novagen) by heat shock procedure. The DH5α cells carrying the recombinant vector were selected on Luria Bertani (LB) medium with ampicillin (100 µg/ml). The transformant colonies were picked up to check the inserted plasmid via colony PCR using specific primers. The nucleotide sequences of the constructed plasmid were confirmed by DNA sequencing using M13 primers (M13F: 5′–GTAAAACGACGGCCAGT–3′ and M13R: 5′– GCGGATAACAATTTCACACAGG–3′) (Macrogen Inc.). The plasmid was extracted from the colonies using HiYield™ Plasmid Mini Kit (RBC Bioscience). The plasmid copy number was calculated based on the length of the PCR product and the DNA concentration using the following formula: gene copy number = (amount × 6.022 × 1023)/(length × 109 × 650). The positive control plasmid was tenfold serially diluted from 2 × 107 to 2 × 102 copies/µl to be used as a standard curve to quantitate the copy number of E. ramosum per gram of each sample.
Absolute quantitative real–time PCR (absolute qPCR)
The absolute qPCR was performed using QuantStudio6 Flex Real–Time PCR systems (Applied Biosystem) and the Luna Universal qPCR Master Mix (New England Biolabs). The qPCR condition consisted of an initial denaturation at 95 °C for 5 min; 40 cycles of denaturation at 94 °C for 60 s, annealing at 60 °C for 30 s, and extension at 72 °C for 30 s; and a final extension cycle at 72 °C for 8 min. The samples, standard curve, and negative control were all simultaneously assayed in duplicate. The specificity of the PCR product was conducted by the melting curve analysis. The cycle threshold (Ct) of each sample was compared with the Ct of the standard curve to calculate the bacterial quantity. The data were normalized to the total weight of extracted samples and represented as a copy number of bacterium per gram weight.
Statistical analysis
Statistical analysis was conducted using IBM SPSS Statistics version 22 (SPSS Inc.), GraphPad Prism 8.0 (GraphPad Software Inc.), and R software version 4.1.3. The Chi–square test was used to compare categorical variables. The nonparametric Kruskal–Wallis test with Dunn’s post hoc was used to compare the differences in continuous variables among three clinical groups (i.e., HC, AD, and CRC groups). Spearman’s rank correlation coefficient was used to determine the associations between continuous variables. The independent variables related to CRC or AD diagnosis were estimated using the binary logistic regression model. The area under the receiver operating characteristic (ROC) curve (AUC) was used to evaluate the diagnostic value of potential bacterial markers in discriminating among CRC patients, AD patients, and the HC group. Youden’s index (J = Sensitivity + Specificity–1) was used to identify the best cut–off value that maximizes sensitivity and specificity in disease detection. A p < 0.05 was considered statistically significant.
Data availability
The sequence data are available in NCBI Sequence Read Archive (SRA) database with the following BioProject ID: PRJNA898111 (https://www.ncbi.nlm.nih.gov/sra/PRJNA898111) and (https://dataview.ncbi.nlm.nih.gov/object/PRJNA898111?reviewer=gv3202bucf9racuatd86do5li5, read–only format).
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 Countries. CA Cancer J. Clin. 71, 209–249. https://doi.org/10.3322/caac.21660 (2021).
Keum, N. & Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 16, 713–732. https://doi.org/10.1038/s41575-019-0189-8 (2019).
Levine, J. S. & Ahnen, D. J. Clinical practice. Adenomatous polyps of the colon. N. Engl. J. Med. 355, 2551–2557. https://doi.org/10.1056/NEJMcp063038 (2006).
Sung, J. J. et al. An updated Asia Pacific consensus recommendations on colorectal cancer screening. Gut 64, 121–132. https://doi.org/10.1136/gutjnl-2013-306503 (2015).
Thrumurthy, S. G., Thrumurthy, S. S., Gilbert, C. E., Ross, P. & Haji, A. Colorectal adenocarcinoma: Risks, prevention and diagnosis. BMJ 354, 3590. https://doi.org/10.1136/bmj.i3590 (2016).
Ternes, D. et al. Microbiome in colorectal cancer: How to get from meta–omics to mechanism?. Trends Microbiol. 28, 401–423. https://doi.org/10.1016/j.tim.2020.01.001 (2020).
Rubinstein, M. R. et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-Cadherin/β–catenin signaling via its FadA Adhesin. Cell Host Microbe 14, 195–206. https://doi.org/10.1016/j.chom.2013.07.012 (2013).
Kumar, R. et al. Streptococcus gallolyticus subsp gallolyticus promotes colorectal tumor development. PLOS Pathog. 13, 1006440. https://doi.org/10.1371/journal.ppat.1006440 (2017).
Cuevas–Ramos, G. et al. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. 107, 11537–11542. https://doi.org/10.1073/pnas.1001261107 (2010).
Tsoi, H. et al. Peptostreptococcus anaerobius induces intracellular cholesterol biosynthesis in colon cells to induce proliferation and causes dysplasia in mice. Gastroenterology 152, 1419-1433.e1415. https://doi.org/10.1053/j.gastro.2017.01.009 (2017).
He, Z. et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut 68, 289. https://doi.org/10.1136/gutjnl-2018-317200 (2019).
Osman, M. A., Neoh, H. M., Ab Mutalib, N. S., Chin, S. F. & Jamal, R. 16S rRNA gene sequencing for deciphering the colorectal cancer gut microbiome: Current protocols and workflows. Front. Microbiol. https://doi.org/10.3389/fmicb.2018.00767 (2018).
Flemer, B. et al. Tumour–associated and non–tumour–associated microbiota in colorectal cancer. Gut 66, 633–643. https://doi.org/10.1136/gutjnl-2015-309595 (2017).
Tap, J. et al. Identification of an intestinal microbiota signature associated with severity of irritable bowel syndrome. Gastroenterology 152, 111-123.e118. https://doi.org/10.1053/j.gastro.2016.09.049 (2017).
Rajagopala, S. V. et al. The human microbiome and cancer. Cancer Prev. Res. 10, 226–234. https://doi.org/10.1158/1940-6207.Capr-16-0249 (2017).
García–Castillo, V., Sanhueza, E., McNerney, E., Onate, S. A. & García, A. Microbiota dysbiosis: A new piece in the understanding of the carcinogenesis puzzle. J. Med. Microbiol. 65, 1347–1362. https://doi.org/10.1099/jmm.0.000371 (2016).
Geng, J. et al. Co–occurrence of driver and passenger bacteria in human colorectal cancer. Gut Pathog. 6, 26. https://doi.org/10.1186/1757-4749-6-26 (2014).
Mira–Pascual, L. et al. Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. J. Gastroenterol. 50, 167–179. https://doi.org/10.1007/s00535-014-0963-x (2015).
Nakatsu, G. et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun. 6, 8727. https://doi.org/10.1038/ncomms9727 (2015).
Kostic, A. D. et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor–immune microenvironment. Cell Host Microbe 14, 207–215. https://doi.org/10.1016/j.chom.2013.07.007 (2013).
Castellarin, M. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22, 299–306. https://doi.org/10.1101/gr.126516.111 (2012).
Chen, W., Liu, F., Ling, Z., Tong, X. & Xiang, C. Human intestinal lumen and mucosa–associated microbiota in patients with colorectal cancer. PLoS ONE 7, e39743. https://doi.org/10.1371/journal.pone.0039743 (2012).
Zeller, G. et al. Potential of fecal microbiota for early–stage detection of colorectal cancer. Mol. Syst. Biol. 10, 766. https://doi.org/10.15252/msb.20145645 (2014).
Tjalsma, H., Boleij, A., Marchesi, J. R. & Dutilh, B. E. A bacterial driver–passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 10, 575–582. https://doi.org/10.1038/nrmicro2819 (2012).
Manfredi, R. et al. Flavobacterium spp. organisms as opportunistic bacterial pathogens during advanced HIV disease. J. Infect. 39, 146–152. https://doi.org/10.1016/s0163-4453(99)90007-5 (1999).
Gong, J., Li, L., Zuo, X. & Li, Y. Change of the duodenal mucosa–associated microbiota is related to intestinal metaplasia. BMC Microbiol. 19, 275. https://doi.org/10.1186/s12866-019-1666-5 (2019).
Gupta, A. et al. Association of Flavonifractor plautii, a flavonoid–degrading bacterium, with the gut microbiome of colorectal cancer patients in India. mSystems 4, 00438–01419. https://doi.org/10.1128/mSystems.00438-19 (2019).
Braune, A. & Blaut, M. Bacterial species involved in the conversion of dietary flavonoids in the human gut. Gut Microbes 7, 216–234. https://doi.org/10.1080/19490976.2016.1158395 (2016).
Batra, P. & Sharma, A. K. Anti–cancer potential of flavonoids: Recent trends and future perspectives. 3 Biotech 3, 439–459. https://doi.org/10.1007/s13205-013-0117-5 (2013).
Simons, A. L., Renouf, M., Hendrich, S. & Murphy, P. A. Human gut microbial degradation of flavonoids: Structure−function relationships. J. Agric. Food Chem. 53, 4258–4263. https://doi.org/10.1021/jf0500177 (2005).
Nosho, K. et al. Association of Fusobacterium nucleatum with immunity and molecular alterations in colorectal cancer. World J. Gastroenterol. 22, 557–566. https://doi.org/10.3748/wjg.v22.i2.557 (2016).
Marchesi, J. R. et al. Towards the human colorectal cancer microbiome. PLoS ONE 6, e20447. https://doi.org/10.1371/journal.pone.0020447 (2011).
Thomas, A. M. et al. Tissue-associated bacterial alterations in rectal carcinoma patients revealed by 16S rRNA community profiling. Front. Cell. Infect. Microbiol. https://doi.org/10.3389/fcimb.2016.00179 (2016).
Chen, J. et al. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 8, 43. https://doi.org/10.1186/s13073-016-0299-7 (2016).
Milosavljevic, M. N. et al. Antimicrobial treatment of Erysipelatoclostridium ramosum invasive infections: A systematic review. Rev. Inst. Med. Trop. Sao Paulo 63, 30. https://doi.org/10.1590/s1678-9946202163030 (2021).
Kosowska, K. et al. The Clostridium ramosum IgA proteinase represents a novel type of metalloendopeptidase*. J. Biol. Chem. 277, 11987–11994. https://doi.org/10.1074/jbc.M110883200 (2002).
Kaper, J. B., Nataro, J. P. & Mobley, H. L. T. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2, 123–140. https://doi.org/10.1038/nrmicro818 (2004).
Gao, Z., Guo, B., Gao, R., Zhu, Q. & Qin, H. Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 6, 20. https://doi.org/10.3389/fmicb.2015.00020 (2015).
Nougayrède, J. P. et al. Escherichia coli induces DNA double–strand breaks in eukaryotic cells. Science 313, 848–851. https://doi.org/10.1126/science.1127059 (2006).
Eklof, V. et al. Cancer–associated fecal microbial markers in colorectal cancer detection. Int. J. Cancer 141, 2528–2536. https://doi.org/10.1002/ijc.31011 (2017).
Liang, Q. et al. Fecal bacteria act as novel biomarkers for noninvasive diagnosis of colorectal cancer. Clin. Cancer Res. 23, 2061–2070. https://doi.org/10.1158/1078-0432.Ccr-16-1599 (2017).
Ai, L. et al. Systematic evaluation of supervised classifiers for fecal microbiota–based prediction of colorectal cancer. Oncotarget 8, 9546–9556. https://doi.org/10.18632/oncotarget.14488 (2017).
Shah, M. S. et al. Leveraging sequence–based faecal microbial community survey data to identify a composite biomarker for colorectal cancer. Gut 67, 882–891. https://doi.org/10.1136/gutjnl-2016-313189 (2018).
Amitay, E. L., Krilaviciute, A. & Brenner, H. Systematic review: Gut microbiota in fecal samples and detection of colorectal neoplasms. Gut Microbes 9, 293–307. https://doi.org/10.1080/19490976.2018.1445957 (2018).
Wong, S. H. et al. Quantitation of faecal Fusobacterium improves faecal immunochemical test in detecting advanced colorectal neoplasia. Gut 66, 1441–1448. https://doi.org/10.1136/gutjnl-2016-312766 (2017).
Xie, Y. H. et al. Fecal Clostridium symbiosum for noninvasive detection of early and advanced colorectal cancer: Test and validation studies. EBioMedicine 25, 32–40. https://doi.org/10.1016/j.ebiom.2017.10.005 (2017).
Chen, Z. et al. Impact of preservation method and 16S rRNA hypervariable region on gut microbiota profiling. mSystems 4, 00271–01218. https://doi.org/10.1128/mSystems.00271-18 (2019).
Wu, N. et al. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb. Ecol. 66, 462–470. https://doi.org/10.1007/s00248-013-0245-9 (2013).
Zackular, J. P., Baxter, N. T., Chen, G. Y. & Schloss, P. D. Manipulation of the gut microbiota reveals role in colon tumorigenesis. mSphere https://doi.org/10.1128/mSphere.00001-15 (2016).
Weir, T. L. et al. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS ONE 8, e70803. https://doi.org/10.1371/journal.pone.0070803 (2013).
Baxter, N. T., Ruffin, M. T., Rogers, M. A. M. & Schloss, P. D. Microbiota–based model improves the sensitivity of fecal immunochemical test for detecting colonic lesions. Genome Med. 8, 37. https://doi.org/10.1186/s13073-016-0290-3 (2016).
Ahn, J. et al. Human gut microbiome and risk for colorectal cancer. J. Natl. Cancer Inst. 105, 1907–1911. https://doi.org/10.1093/jnci/djt300 (2013).
Sobhani, I. et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS ONE 6, 16393. https://doi.org/10.1371/journal.pone.0016393 (2011).
Allali, I. et al. Gut microbiome compositional and functional differences between tumor and non–tumor adjacent tissues from cohorts from the US and Spain. Gut Microbes 6, 161–172. https://doi.org/10.1080/19490976.2015.1039223 (2015).
Geng, J., Fan, H., Tang, X., Zhai, H. & Zhang, Z. Diversified pattern of the human colorectal cancer microbiome. Gut Pathog. 5, 2. https://doi.org/10.1186/1757-4749-5-2 (2013).
Yang, Y. X., Hennessy, S. & Lewis, J. D. Type 2 diabetes mellitus and the risk of colorectal cancer. Clin. Gastroenterol. Hepatol. 3, 587–594. https://doi.org/10.1016/s1542-3565(05)00152-7 (2005).
Martin, M. Cutadapt removes adapter sequences from high–throughput sequencing reads. EMBnet J. 17(3), 2011. https://doi.org/10.14806/ej.17.1.200 (2011).
Callahan, B. J. et al. DADA2: High–resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. https://doi.org/10.1038/nmeth.3869 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web–based tools. Nucleic Acids Res 41, D590-596. https://doi.org/10.1093/nar/gks1219 (2013).
Chong, J., Liu, P., Zhou, G. & Xia, J. Using microbiome analyst for comprehensive statistical, functional, and meta–analysis of microbiome data. Nat. Protoc. 15, 799–821. https://doi.org/10.1038/s41596-019-0264-1 (2020).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. https://doi.org/10.1186/gb-2011-12-6-r60 (2011).
Ye, J. et al. Primer–BLAST: A tool to design target–specific primers for polymerase chain reaction. BMC Bioinform. 13, 134. https://doi.org/10.1186/1471-2105-13-134 (2012).
Acknowledgements
We sincerely thank all participants who were willing to volunteer in sample collections, and we would like to extend our sincere gratitude to all collaborations successfully processed at King Chulalongkorn Memorial Hospital, Bangkok, Thailand, including Geriatric Excellence Center, Excellence Center for GI Endoscopy, Excellence Center for Colorectal Cancer, and Center of Excellence in Hepatitis and Liver Cancer. The Scholarship from the Graduate School, Chulalongkorn University to commemorate the 72nd anniversary of his Majesty King Bhumibol Adulyadej is gratefully acknowledged.
Funding
This project is funded by the National Research Council of Thailand and Graduate School, Chulalongkorn University, Thailand.
Author information
Authors and Affiliations
Contributions
K.Pa. designed the project. N.I., T.P., S.M., K.Pa., A.P., and K.Pr. were involved in the collection of clinical samples. N.I. and T.T. conducted the experiments. N.I., N.C., and T.J. performed the microbiome analysis. N.I., N.C., T.T., and K.Pa. drafted the manuscript. All authors revised and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Iadsee, N., Chuaypen, N., Techawiwattanaboon, T. et al. Identification of a novel gut microbiota signature associated with colorectal cancer in Thai population. Sci Rep 13, 6702 (2023). https://doi.org/10.1038/s41598-023-33794-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-33794-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
