
Abstract
Almost 50% of esophageal adenocarcinoma (EAC) patients progressed from Barrett’s esophagus (BE). EAC is often diagnosed at late stages and is related to dismal prognosis. However, there are still no effective methods for stratification and therapy in BE and EAC. Two public datasets (GSE26886 and GSE37200) were analyzed to identify differentially expressed genes (DEGs) between BE and EAC. Then, a series of bioinformatics analyses were performed to explore potential biomarkers associated with BE-EAC. 27 up- and 104 down-regulated genes were observed between GSE26886 and GSE37200. The GO and KEGG enrichment analysis indicated that the DEGs were highly involved in tumorigenesis. Subsequently, Weighted Gene Co-Expression Network Analysis (WGCNA) were performed to explore the potential genes related to BE-EAC, which were validated in The Cancer Genome Atlas (TCGA) database, and 5 up-regulated genes (MYO1A, ACE2, COL1A1, LGALS4, and ADRA2A) and 3 down-regulated genes (AADAC, RAB27A, and P2RY14) were found in EAC. Meanwhile, ADRA2A and AADAC could contribute to EAC pathogenesis and progression. MYO1A, ACE2, COL1A1, LGALS4, ADRA2A, AADAC, RAB27A, and P2RY14 could be potential novel diagnostic and prognostic biomarkers in BE-EAC.
Similar content being viewed by others
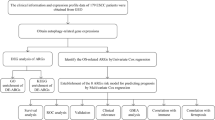


Introduction
Esophageal adenocarcinoma (EAC), the predominant subtype in the west, is one of the two main histological types of esophageal cancer, and the incidence of EAC has increased nearly six-fold over the last three decades1,2. Long-term exposure to the acid, bile, and other stomach contents causes great injury of the squamous esophageal epithelium and increases the risk of developing Barrett’s esophagus (BE) and later EAC3,4. BE is the only recognized precursor of EAC. Individuals with BE are 30–125 times more likely to develop EAC than the general population, and almost 50% of EAC patients progressed from BE5. Patients with BE must undergo regular endoscopic surveillance, as BE surveillance carries an improved prognosis6. Given the high cost of endoscopy and many patients still developing EAC during endoscopic surveillance, stratification of BE patients is indispensable. Meanwhile, EAC is often diagnosed at late stages and is related to dismal prognosis. Although tremendous progress has been made in therapy, including esophagectomy, chemotherapy, and molecular targeted drugs, the 5-year survival rate of EAC remains less than 20%7. Therefore, it is necessary to explore potential targets for diagnosis and therapy.
Several genes from genome-wide association studies have been identified as having impacts on the pathogenesis of BE to EAC. It is reported that ELF3, KLF5, GATA6, EHF, TTK, TPX2, and RAD54B are important genetic modifiers played important roles in the pathogenesis and progression of BE to EAC1,8. Spechler et al.9 noted that early CDKN2A (P16) loss or methylation and subsequent loss of P53 in non-dysplastic BE might contribute to BE-EAC progression. In addition, Dulak et al.10 indicated that SMAD4, ARID1A, PIK3CA, SPG20, TLR4, ELMO1, and DOCK2 had a significant impact on BE-EAC progression. However, there are still no effective methods for stratification and therapy in BE and EAC.
Therefore, we analyzed two public datasets to identify differentially expressed genes (DEGs) among BE and EAC. Then, Weighted Gene Co-Expression Network Analysis (WGCNA) was performed to explore the potential genes related to BE-EAC. This study aimed to screen potential genes for BE-EAC progression.
Material and methods
Data Retrieving and Processing
Data from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) were fulfilled the inclusion criteria below: ① publication date from 2010 to 2022; ② containing BE and EAC tissue samples; ③ sample size > 3 in each group. The exclusion criteria were: ① duplicated research; ② animal or cell experiments; ③ incomplete data; ④ patients with chemotherapy or radiation treatment. Then, the gene expression profiles of GSE2688611 and GSE3720012 were downloaded from GEO. Finally, 20 BE samples and 21 EAC samples in the GSE26886; and 31 BE samples and 15 EAC samples in the GSE37200 were included in this study. EAC without chemotherapy or radiation treatment data were obtained from The Cancer Genome Atlas (TCGA) database, including 70 EAC samples and 8 normal samples adjacent to EAC.
Batch effects were corrected using “limma” R packages13, and principal components analysis (PCA) was carried out. Two R packages (“GEOquery” and “limma”) were used for the analysis of DEGs. The threshold for the DEGs was set as adjusted-P value < 0.05 and |log2 fold change (FC) |≥ 1. Volcano plots and heat maps were drawn using R package “ggplot2” (https://ggplot2.tidyverse.org/) and “complexHeatmap”14. Venn diagram was performed using the jvenn tool (http://jvenn.toulouse.inra.fr/app/example.html), and the overlaps represented the intersection between the two datasets.
Gene ontology (GO) analysis and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analysis
To identify the function of DEGs, GO and KEGG analysis were performed using Metascape (metascape.org) database. GO is a commonly used bioinformatics tool that supply comprehensive information on gene function of individual genomic products based on defined features and is primarily divided into three parts, molecular function (MF), biological process (BP), and cellular component (CC). KEGG is a database resource for understanding high-level biological functions and utilities15. We determined that results were statistically significant at a level of adjusted-P < 0.05 and false discovery rate (FDR) < 0.05. Then, histograms and chord plots were generated with R package “GOplot”.
Weighted gene co-expression network analysis (WGCNA)
Considering that GSE26886 had larger sample size of EAC, it was used to detect modules highly correlated with EAC, and WGCNA was performed using R package “WGCNA”16 and carried out on all genes. The scale-free topology of the networks was assessed for various values of the β shrinkage parameter, and we chose β = 8 based on scale-free topology criterion. Finally, the dynamic tree cut algorithm was applied to the dendrogram for module identification with the mini-size of module gene numbers set as 50, and similar modules were merged following a height cutoff of 0.25. In the module-trait analysis, gene-trait significance (GS) value > 0.3 and module membership (MM) value > 0.55 were defined as a threshold17. Then, Venn diagram was performed to explore the trait-expression-related genes.
Exploration of trait-expression-related genes in TCGA database
Subsequently, the expression levels of trait-expression-related genes were estimated in TCGA database, a receiver operating characteristic (ROC) curve was performed to assess the diagnostic value of the genes by “pROC” R package, and survival analysis was also performed using “survival” and “survminer” R packages.
Statistical analysis
Statistical analysis was performed using R software (Version 4.1.0, www.r-project.org). Statistical comparisons between groups of normalized data were performed using the t-test or Mann–Whitney U-test according to the test condition, and categorical data were analyzed by the χ2 test or Fisher exact test. A difference with P < 0.05 was considered significant.
Results
Identification of DEGs in the EAC patients
The batch effects were removed (Figure S1A,B), and the PCA showed that there were obvious differences between BE and EAC (Fig. 1A,B). the DEGs between BE and EAC in GSE26886 and GSE37200 datasets were identified, respectively (Fig. 1C,F). Then, we sought for the overlapping DEGs between the two datasets, and 27 up- and 104 down-regulated genes were observed in EAC (Fig. 1G,H).

Identification of differentially expressed genes (DEGs) between BE and EAC. The principal components analysis (PCA) in GSE26886 (A) and GSE37200 (B) The volcano plot of DEGs in GSE26886 (C) and GSE37200 (D); The Heatmap of DEGs in GSE26886 (E) and GSE37200 (F); (G,H) Venn diagrams displayed the overlapping DEGs of up (G) and down (H) regulated genes between BE and EAC.
GO and KEGG pathway enrichment analysis
To explore the potential roles of DEGs between BE and EAC, GO and KEGG pathway enrichment analyses were performed. GO analysis showed that the up-regulated genes in EAC were mainly involved in biological processes (BP) associated with negative regulation of cell population proliferation, skeletal system development, blood vessel development, positive regulation of programmed cell death, and ossification (Fig. 2A). In contrast, the down-regulated genes in EAC were mainly involved in BP associated with monocarboxylic acid metabolic process, digestion, thrombin-activated receptor signaling pathway, response to zinc ion, and cellular response to fluid shear stress (Fig. 2B). These results indicated that the DEGs were highly associated with epithelial-mesenchymal transition (EMT) and nutrition. KEGG analysis indicated that the up-regulated DEGs in EAC were primarily enriched in Pertussis, IL-17 signaling pathway, cytokine-cytokine receptor interaction, ECM-receptor interaction, protein digestion and absorption, and Amoebiasis (Fig. 2C); while the down-regulated genes in EAC were enriched in chemical carcinogenesis, Amoebiasis, drug metabolism-other enzymes, steroid hormone biosynthesis, bile secretion, glycerophospholipid metabolism, and inflammatory mediator regulation of trp channels (Fig. 2D). These results demonstrated that the DEGs were highly involved in tumorigenesis.

GO and KEGG pathway enrichment analysis. GO analysis of up (A) and down (B) regulated DEGs in EAC; KEGG analysis of up (C) and down (D) regulated DEGs in EAC.
Identification of key modules by WGCNA
WGCNA analysis provides an overview of the transcriptomic organization, and the relationships between sets of genes with external, biological traits. To identify key modules related to clinical traits, WGCNA was performed by using GSE26886 dataset (Fig. 3A). The power of β = 8 (scale-free R2 = 0.90) was selected as the soft thresholding parameter to construct a scale-free network (Fig. 3B). Similar module clustering was constructed by using dynamic hybrid cutting (threshold = 0.25). A total of 25 modules were identified (Fig. 3C). The results in Fig. 3D showed that the grey module was the highest positive module correlated to EAC (R2 = 0.86, P = 2e−12) and was highly negative correlated to BE (R2 = 0.86, P = 2e−12). Figure 3E showed gene significance for BE and EAC in grey module.

WGCNA to identify trait-related modules and genes. (A) Sample dendrogram and trait heat map; (B) calculating soft-thresholding power; Left: scale-free fit indices using different soft-thresholding powers; Right: mean connectivity using different soft-thresholding powers; (C) the dendrogram clustered by Dynamic Tree Cut algorithm; (D) the heatmap profiling the correlations between module eigengenes and the clinical characteristics; (E) scatter plot of gene significance in grey module; Left: BE; Right: EAC; (F) Venn diagrams displayed the overlapping genes between trait-related genes in grey module and DEGs.
In the module-trait analysis, we intersected the trait-related genes in grey module highly associated with EAC and 131 DEGs generated from expression difference analysis, and finally extracted 27 trait-expression-related genes for the following analysis (Fig. 3F, Tables S1 and S2). These results showed that the 27 trait-expression-related genes were significantly correlated with the pathogenesis of EAC.
Exploration of trait-expression-related genes in TCGA database
Next, further validation and exploration were conducted among the 27 trait-expression-related genes in TCGA database. MYO1A, ACE2, COL1A1, LGALS4, and ADRA2A were significantly up-regulated in EAC tissue; while AADAC, RAB27A, and P2RY14 were abnormally down-expressed in EAC tissue, which indicated that these genes were repeatable in EAC (Fig. 4A). Subsequently, ROC curves were performed to estimate the diagnostic value in EAC, and the result showed that the genes mentioned above had good diagnostic properties (Fig. 4B). Later, survival analysis was performed to explore the prognostic value of the 8 genes, and the clinical data were shown in Table 1. Low- ADRA2A expression was associated with poor overall survival (OS) and disease-specific survival (DSS); while low- AADAC expression were significantly correlated with poor progress-free interval (PFI). These results illustrated that ADRA2A and AADAC could contribute to EAC pathogenesis and progression.

Further validation in TCGA database (*p < 0.05, **p < 0.01, ***p < 0.001). (A) Box plot assessing the expression of 8 genes in TCGA database; (B) ROC curve for the 8 genes; (C) Survival plots of ADRA2A and AADAC in overall survival, disease specific survival, and progress free survival.
Discussion
Currently, the pathogenesis of BE-EAC is still unclear, and the disease stratification and treatment are also limited. In the present study, we identified 27 up- and 104 down-regulated DEGs in two public datasets, and the results from GO and KEGG analysis indicated that the DEGs were highly associated with tumorigenesis. Subsequently, 27 trait-expression-related genes highly correlated with EAC were screened out by WGCNA. MYO1A, ACE2, COL1A1, LGALS4, ADRA2A, AADAC, RAB27A, and P2RY14 were also abnormally regulated in TCGA database and represented good diagnostic properties. Surprisingly, we found that ADRA2A and AADAC were correlated with EAC prognosis.
Previous studies showed that COL1A1, RAB27A, and P2RY14 were identified as the potential biomarker for esophageal squamous cell cancer (ESCC) and RAB27A associated with immune infiltration in ESCC7,18,19,20. However, there was no further experiment to verify their effects on EAC.
To the best of our knowledge, our study, for the first time, screened out 5 genes related to EAC. MYO1A is most highly expressed in the digestive tract, and it is associated with stomach adenocarcinoma and colon cancer21,22. ACE2, the receptor of COVID-19, is aberrantly expressed in many tumors23. It is reported that LGALS4, a β-galactoside binding protein, is correlated with prognosis in urothelial carcinoma of bladder and is also a tumor marker in serum immunoassay determination of colorectal carcinoma24,25. ADRA2A is thought to be involved in the progression of multiple cancer and can inhibit the activation of PI3K/Akt/mTOR pathway26. AADAC is a kind of serine hydrolase widely involved in the hydrolysis of drugs and associated with poor prognosis in stomach adenocarcinoma27,28. More future studies are needed to gain more insights into these genes.
On the one hand, our study had more strict inclusion criteria than the previous study, such as exclusion of old research, duplicated research, and patients with chemotherapy or radiation treatment, which ensured the accuracy of the results and might be enlightened for the future research or clinical guidance29. On the other hand, we explored some potential biomarkers that had not been reported in BE-EAC through multiple datasets. Nevertheless, our study also had several limitations. Firstly, further experiments were required to verify these results. Secondly, the lack of BE cases in TCGA database prevented us from comparing EAC and BE, which might impact the outcomes. However, there seemed to be no better way for us to compare the effects of the hub genes on diagnosis and prognosis.
In conclusion, MYO1A, ACE2, COL1A1, LGALS4, ADRA2A, AADAC, RAB27A, and P2RY14 could be potential novel diagnostic and prognostic biomarkers in BE-EAC. In addition, ADRA2A and AADAC could contribute to EAC progression. Although further validation is still needed, we provide useful and novel information to explore the potential candidate genes for BE-EAC prognosis and therapeutic options.
Data availability
Publicly available datasets were analyzed in this study. This data can be found here: GEO data base, accession number: GSE26886 and GSE37200.
References
Ma, S. et al. A transcriptional regulatory loop of master regulator transcription factors, PPARG, and fatty acid synthesis promotes esophageal adenocarcinoma. Cancer Res. 81, 1216–1229 (2021).
Deshpande, N. P., Riordan, S. M., Castano-Rodriguez, N., Wilkins, M. R. & Kaakoush, N. O. Signatures within the esophageal microbiome are associated with host genetics, age, and disease. Microbiome 6, 227 (2018).
Hvid-Jensen, F., Pedersen, L., Drewes, A. M., Sorensen, H. T. & Funch-Jensen, P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N. Engl. J. Med. 365, 1375–1383 (2011).
Kaakoush, N. O., Castano-Rodriguez, N., Man, S. M. & Mitchell, H. M. Is Campylobacter to esophageal adenocarcinoma as Helicobacter is to gastric adenocarcinoma?. Trends Microbiol. 23, 455–462 (2015).
Desai, T. K. et al. The incidence of oesophageal adenocarcinoma in non-dysplastic Barrett’s oesophagus: A meta-analysis. Gut 61, 970–976 (2012).
El-Serag, H. B. et al. Surveillance endoscopy is associated with improved outcomes of oesophageal adenocarcinoma detected in patients with Barrett’s oesophagus. Gut 65, 1252–1260 (2016).
Lv, J. et al. Biomarker identification and trans-regulatory network analyses in esophageal adenocarcinoma and Barrett’s esophagus. World J. Gastroenterol. 25, 233–244 (2019).
Kumar, S. et al. Integrated genomics and comprehensive validation reveal drivers of genomic evolution in esophageal adenocarcinoma. Commun. Biol. 4, 617 (2021).
Spechler, S. J., Fitzgerald, R. C., Prasad, G. A. & Wang, K. K. History, molecular mechanisms, and endoscopic treatment of Barrett’s esophagus. Gastroenterology 138, 854–869 (2010).
Dulak, A. M. et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 45, 478–486 (2013).
Wang, Q., Ma, C. & Kemmner, W. Wdr66 is a novel marker for risk stratification and involved in epithelial-mesenchymal transition of esophageal squamous cell carcinoma. BMC Cancer 13, 137 (2013).
Myers, A. L. et al. IGFBP2 modulates the chemoresistant phenotype in esophageal adenocarcinoma. Oncotarget 6, 25897–25916 (2015).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551 (2021).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Wu, C. et al. Bioinformatics analysis explores potential hub genes in nonalcoholic fatty liver disease. Front. Genet. 12, 772487 (2021).
Wang, Z. et al. Identification of potential biomarkers associated with immune infiltration in the esophageal carcinoma tumor microenvironment. Biosci. Rep. 41, 1 (2021).
Yu, F., Wu, W., Liang, M., Huang, Y. & Chen, C. Prognostic significance of Rab27A and Rab27B expression in esophageal squamous cell cancer. Cancer Manag. Res. 12, 6353–6361 (2020).
Chen, F. F., Zhang, S. R., Peng, H., Chen, Y. Z. & Cui, X. B. Integrative genomics analysis of hub genes and their relationship with prognosis and signaling pathways in esophageal squamous cell carcinoma. Mol. Med. Rep. 20, 3649–3660 (2019).
Bai, Y. et al. Development and validation of a prognostic nomogram for gastric cancer based on DNA methylation-driven differentially expressed genes. Int. J. Biol. Sci. 16, 1153–1165 (2020).
McIntosh, B. B. & Ostap, E. M. Myosin-I molecular motors at a glance. J. Cell Sci. 129, 2689–2695 (2016).
Chai, P., Yu, J., Ge, S., Jia, R. & Fan, X. Genetic alteration, RNA expression, and DNA methylation profiling of coronavirus disease 2019 (COVID-19) receptor ACE2 in malignancies: a pan-cancer analysis. J Hematol Oncol 13, 43 (2020).
Ferlizza, E. et al. Colorectal cancer screening: Assessment of CEACAM6, LGALS4, TSPAN8 and COL1A2 as blood markers in faecal immunochemical test negative subjects. J. Adv. Res. 24, 99–107 (2020).
Ding, Y., Cao, Q., Wang, C., Duan, H. & Shen, H. LGALS4 as a prognostic factor in urothelial carcinoma of bladder affects cell functions. Technol Cancer Res. Treat. 18, 1078144249 (2019).
Wang, W., Guo, X. & Dan, H. alpha2A-Adrenergic receptor inhibits the progression of cervical cancer through blocking PI3K/AKT/mTOR pathway. Onco Targets Ther. 13, 10535–10546 (2020).
Zou, Q., Lv, Y., Gan, Z., Liao, S. & Liang, Z. Identification and validation of a malignant cell subset marker-based polygenic risk score in stomach adenocarcinoma through integrated analysis of bulk and single-cell RNA sequencing data. Front. Cell. Dev. Biol. 9, 720649 (2021).
Wu, C., Wu, Z. & Tian, B. Five gene signatures were identified in the prediction of overall survival in resectable pancreatic cancer. BMC Surg. 20, 207 (2020).
Nangraj, A. S. et al. Integrated PPI- and WGCNA-retrieval of hub gene signatures shared between Barrett’s esophagus and esophageal adenocarcinoma. Front. Pharmacol. 11, 881 (2020).
Acknowledgements
The authors appreciate study investigators and staff who participated in this study.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
N.Y., H.Z., J.H., and X.X. contributed equally to this paper. N.Y., H.Z., J.H., and X.X. analyzed the study data, helped draft the manuscript, made critical revisions of the manuscript. L.L., G.Z., and M.X. assisted with data collection and the analysis. Y.L. and T.Y. supervised the research and edited the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yi, N., Zhao, H., He, J. et al. Identification of potential biomarkers in Barrett’s esophagus derived esophageal adenocarcinoma. Sci Rep 13, 2345 (2023). https://doi.org/10.1038/s41598-022-17107-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-17107-0
This article is cited by
-
Analysis of Molecular Genetic Variants of Lgals4 in Esophageal Cancer: A Preliminary Report
Biochemical Genetics (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
