
Abstract
In the current study, we have systematically analysed the mitochondrial DNA (mtDNA) sequence of Naegleria fowleri (N. fowleri) isolate AY27, isolated from Karachi, Pakistan. The N. fowleri isolate AY27 has a circular mtDNA (49,541 bp), which harbours 69 genes (46 protein-coding genes, 21 tRNAs and 2 rRNAs). The pan-genome analysis of N. fowleri species showed a Bpan value of 0.137048, which implies that the pan-genome is open. KEGG classified core, accessory and unique gene clusters for human disease, metabolism, environmental information processing, genetic information processing and organismal system. Similarly, COG characterization of protein showed that core and accessory genes are involved in metabolism, information storages and processing, and cellular processes and signaling. The Naegleria species (n = 6) formed a total of 47 gene clusters; 42 single-copy gene clusters and 5 orthologous gene clusters. It was noted that 100% genes of Naegleria species were present in the orthogroups. We identified 44 single nucleotide polymorphisms (SNP) in the N. fowleri isolate AY27 mtDNA using N. fowleri strain V511 as a reference. Whole mtDNA phylogenetic tree analysis showed that N. fowleri isolates AY27 is closely related to N. fowleri (Accession no. JX174181.1). The ANI (Average Nucleotide Identity) values presented a much clear grouping of the Naegleria species compared to the whole mtDNA based phylogenetic analysis. The current study gives a comprehensive understanding of mtDNA architecture as well as a comparison of Naegleria species (N. fowleri and N. gruberi species) at the mitochondrial genome sequence level.
Similar content being viewed by others

Introduction
Naegleria fowleri (N. fowleri) is a habitant of warm lakes, streams, spas, pools, domestic water reservoirs and domestic water supplies1,2,3. N. fowleri species causes primary amoebic meningoencephalitis (PAM), which is an infection of the brain that results in the complete damage of brain tissue. Last year, between May 2021 to July 2021, a total of five PAM cases of brain-eating amoeba N. fowleri meningitis fatality have been reported in the megacity Karachi, Pakistan (https://www.dawn.com/news/1634761/another-naegleria-death-reported-in-karachi).
In protozoa, the organization of mitochondrial genomes (mtDNA) and their gene structure are more diverse compared to multicellular eukaryotes. N. fowleri mtDNA code for the major and minor rRNA components, some tRNAs, 46 proteins of the respiratory chain and ribosomes4.
A single genome sequence does not reflect all of a N. fowleri genetic diversity. A large number of genomic data is required for complex analyses such as molecular evolutionary and genomics pathogenesis. Fortunately, the ongoing advancement of sequencing technologies has helped in decreasing the time and cost in sequencing. As a result, there has been an exponential increase in the genomic databases. Comparative genomics, for example, is a new scientific topic that compares the genetic content of many taxonomically related microbes5.
Several alternative approaches, for instance, average nucleotide identity (ANI), can be used to assess taxonomic relationships in the post-genomic period6.
A pan-genome is a group of orthologous and unique genes found in a group of organisms. Pan and core genome analyses are critical for studying a phylogenetic lineage's, genomic and metabolic repertoires of an organism7,8. Although the pan-genome can refer to the whole collection of genes in a particular taxon, it is primarily defined for the species, which includes dispensable genes, all core genes and strain-specific genes9,10,11. Pan-genome analysis has been used to assess microorganism genomic diversity, evolution, pathogenicity, and other traits12.
Orthologous genes are groups of genes from distinct species that arose from a single ancestor gene, and generally these genes are involved in essential functions. The study of orthologous gene clusters in various strains might be useful to explore gene organization, gene function, and genome molecular evolution13. Furthermore, accurate recognition of orthologous genes helps in functional gene annotation, comparison, and evolutionary genomics research.
Naegleria gruberi (N. gruberi) is a non-pathogenic and non-thermotolerant closely relative of N. fowleri. So, a comparative genomics-based study could reveal new information about why N. fowleri causes such severe and fatal disease. Specifically, the genetic elements that give N. fowleri pathogenetic characteristics can be explored using a comparative genomics approach.
There are few data available on mitochondrial genome features and the evolution of Naegleria species. Only six complete mitochondrial genome sequences were publicly available till 1st August 2021 in the NCBI database.
In the current study, our main objective is to extend information on the mitochondrial genome of these species. We performed the complete mtDNA sequencing of N. fowleri isolate AY27 (Accession no. MZ461463), and studied the phylogeny and diversity using a comparative genomics approach. To the best of our knowledge, this is the first data on the characterization of mtDNA of Naegleria species based on ANI value, orthologous genes and pan-genome analysis. The findings of this study provide a comprehensive genetic landscape of the Naegleria species, as well as valuable insight into the protozoan's mtDNA.
Materials and methods
Patient sample
A cerebrospinal fluid (CSF) specimen number (AY-27) of 28 years-old male PAM patient was collected from “Karachi Diagnostic Center and Molecular Biology Laboratory” (https://www.kldc.pk/), approved by the Karachi Diagnostic Center and Molecular Biology Laboratory Ethical Committee (EC Ref No. REC-NF02). All methods were carried out following ethical regulations. The patient’s informed consent was obtained for CSF sample collection.
Sample collection and identification
A direct microscopic examination was performed under a compound microscope (version n trademark) at 40 × after the CSF sample was incubated at 37 °C for 30 min. The sediments in the CSF sample were gently re-suspended in residual supernatant after centrifugation for 10 min at 250×g. Under sterile conditions 2 to 3 drops of re-suspended media was inoculated onto the Non-Nutrient Agar (NNA) plates covered with PAGE amoeba saline suspension containing E. coli ATCC25922.
The NNA cultured plates were covered using parafilm and incubated for 10 days at 42 °C14,15. Differentiation of N. fowleri from other Naegleria species based on cellular morphology is not simple. Generally, PCR-based identification is used for identification of N. fowleri from other Naegleria species16,17,18,19.
PCR product amplification was carried out in a total volume of 25 μl, containing 9.5 μl ddH2O, 0.5 μl primer (10 μM), 10 μl Green Master Mix (Promega, USA), and 5.0 μl genomic DNA isolated from CSF samples18,19,20,21. For PCR (40 cycle), the initial denaturation at 95 °C for 5 min, denaturation at 95 °C for 3 s, annealing at 53 °C for 30 s, extension at 72 °C for 30 s and the final extension at 72 °C for 5 min. The PCR product was visualized in a 2% agarose gel16.
We used two pairs of N. fowleri species-specific primers for the identification of N. fowleri; NaeglF192 (5′-GTGCTGAAACCTAGCTATTGTAACTCAGT-3′) and NaeglR344 (5′-CACTAGAAAAAGCAAACCTGAAAGG-3′)20; Nae3-F (5′-CAAACACCGTTATGACAGGG and Nae3-R TGGTTTCCCTCACCTTACG-3′)21.
Library preparation and sequencing
The genomic DNA of trophozoite was isolate using commercially available DNA extraction kit (QIAmp DNA Mini Kit, QIAGEN). The concentration of DNA was calculated using Qubit 2.0 fluorometer (Invitrogen, ThermoFisher Scientific, USA). The genomic DNA library was prepared using the NEBnext Ultra kit (Illumina, San Diego, CA) and the Illumina platform (HiSeq 4000) was used for sequencing.
Quality assessment, assembly, and gene annotation
FastQC was used to check the quality of the reads and the raw reads were pre-processed and the adapters were trimmed using Trimmomatic version 0.392. The mtDNA was assembled in two steps: first, the reads were mapped onto the reference mtDNA (Accession Number: KX580903.1) using BWA tool22 and then spades software23 was used to assemble the mapped reads. To fill the gaps, we used Geneious Prime software (https://www.geneious.com/prime/). The quality of the assembly was checked using Quast software24. The mtDNA was annotated using the Pokka annotation tools25.
The GenBank file was used for graphical representation of BLAST results for DNA vs DNA and CDS vs CDS at cgview service (http://cgview.ca/viewer) and GenomeVX service (http://wolfe.ucd.ie/GenomeVx/) was used to plot the genomic features of N. fowleri isolate AY27
Third-party sequencing data
We also used sequencing data of Naegleria species submitted to NCBI database (till 1st August 2021) by other research groups for comparative genomics. The details of Naegleria species used in the current study are given in supplementary Table S1. This included two strains isolated from the USA (KX580902.1, KX580903.1) one strain from Canada (JX174181.1), and one strain from Pakistan (OD958694.1) and N. gruberi (AF288092.1) was used as an outgroup.
Evolutionary relationships of taxa
The evolutionary distances were calculated via the maximum composite likelihood method2 and evolutionary studies were conducted using MEGA X3 software. ANI (average nucleotide identify) matrix values were calculated for Naegleria species using OTA software (https://www.ezbiocloud.net/tools/orthoani).
Evaluation of BPGA features with Naegleria species
To evaluate the PAN genome, the complete mtDNA sequences of five strains of N. fowleri including N. fowleri strain V511 (KX580902.1), N. fowleri strain V419 (KX580903.1), N. fowleri (JX174181.1), and N. fowleri isolate AY27 (MZ461463) were used for the analysis.
These sequences were annotated using Pokka software25. The gbk files were used as an input file in the BPGA pipeline26. BPGA uses USEARCH as the default clustering tool and the clustering output is used to perform pan-genome analysis. We used this pipline with 99% sequence identity as the cut-off value for pan-genome analysis. We also used BPGA pipeline26 for clusters of orthologous groups (COGs) and Kyoto Encyclopedia of Genes and Genomes (KEGG)27 pathway determination.
Orthologous clustering analysis
The OrthoVenn2 web platform28 was used for orthologous clustering analysis of the protein-coding genes among Naegleria species. Orthofiner29 was used for calculation of overall statistics about orthogroups sizes and proportion of genes assigned to orthogroups.
Nucleotide sequence accession number
The whole mtDNA sequence of N. fowleri isolates AY27 was deposited at GenBank database (Accession no. MZ461463).
Results and discussion
Isolation and identification of N. fowleri
In the CSF sample (sample id = AY-27), direct microscopy (supplementary video 1: shows the motile forms of N. fowleri trophozoites in cerebrospinal fluid movement) revealed alive motile amoebic cells with pseudopodia (Fig. 1A). The trophozoite state of amoeba was suggested by the continuous change in cell morphology and formation of pseudopods (in sample AY-27). The trophozoites were about 12 to15 μm in size. Using eruptive pseudopods, the crawling amoeba was noted to move at a rate of ~ 1 µm/s. Selective identification of Naegleria species based on cellular morphology is difficult. Therefore, the PCR method was used for the detection of Naegleria species 16,17,18,19. We used two pairs of N. fowleri species-specific primers for the detection of N. fowleri. The amplified product size of Nae3-F_Nae3-F (Nae3) and NaeglF192-F_NaeglR344-R (Naegl) were found to be 183 bp and 153 bp, respectively (Fig. 1B, Supplementary Fig. S1).

Agarose gel (2.0%) electrophoresis of the PCR products for analysis of patient CSF with primary amoebic meningoencephalitis (PAM) infection; line M: 100 bp DNA ladder, line NC: Negative control without DNA, line Nae3 (183 pb) and Naegl (153 bp): N. fowleri specific primers used for amplification of DNA from the patient CSF (A). Microscopy images: arrow indicating the N. fowleri observed in the patient CSF (B).
N. fowleri species mtDNA profile
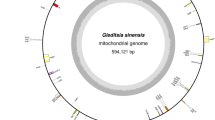
Figure 2 shows the mtDNA profile of N. fowleri. The size of mtDNA of our clinically isolated N. fowleri was found to be 49,541 bp.

The complete mitochondrial of N. fowleri with GenomeVX service visualization shows the protein-coding genes, rRNAs, tRNAs and non-coding regions.
The AT content of N. fowleri isolate AY27 is approximately 74.7%, while the GC content is 25.3%. The mtDNA of N. fowleri isolate AY27 consists of 69 genes (46 protein-coding genes, 2 rRNAs and 21 tRNAs). Among the protein-coding genes, 17 are ribosomal proteins (6 large and 11 small subunit). A total of 22 genes were found to encode various oxidative phosphorylation enzymes. Moreover, one copy of the tatC gene, which codes for the sec-independent translocase protein, one copy of the heme lyase gene, one copy of the ABC transporter subunit gene and four hypothetical proteins were also present (Supplementary Table S2).
Evolutionary relationships of taxa
This analysis involved six whole mtDNA sequences; N. gruberi (AF288092.1), N. fowleri Karachi NF001 (OD958694.1), N. fowleri strain V511 (KX580902.1), N. fowleri strain V419 (KX580903.1), N. fowleri (JX174181.1) and N. fowleri isolate AY27 (MZ461463). The phylogenetic tree analysis reveals that there were two clades (Fig. 3A). Clad I consist of N. fowleri strain V511 (KX580902.1), N. fowleri strain V419 (KX580903.1), N. fowleri (JX174181.1), and N. fowleri isolate AY27 (MZ461463). N. gruberi (AF288092.1) was also present in clade I but in a separate subclade. Clad II consist of N. fowleri Karachi NF001 (OD958694.1); It is interesting to note that N. fowleri Karachi NF001 (OD958694.1) was distinct from the rest of the three N. fowleri strains. Our N. fowleri isolates AY27 isolated from Karachi, presented higher similarities with N. fowleri (JX174181.1) compare to other strains.

Whole mtDNA sequences were aligned and the phylogenetic tree was generated using the neighbour-joining method with 1000 bootstrap replicates (A) and heatmap showing the OrthoANI (Orthologous Average Nucleotide Identity) between N. fowleri isolate AY27 (MZ461463)) and other N. fowleri isolate calculated with OAT software, and N. gruberi species (AF288092.1) used as the outgroup (B).
The average nucleotide identity (ANI) is a method for assessing genetic relatedness, therefore we calculated the ANI values for Naegleria species. Briefly, ANI value gives the average nucleotide identity of all genes shared among two genomes30. As shown in Fig. 3B, the ANI values presented a much clear grouping of the Naegleria species compared to the whole mtDNA based phylogenetic analysis. N. gruberi species present in clade I presented ANI values in the range of 76.26% to 77.72%. On the other hand, in clade II, all the N. fowleri species ANI values were in the range of 99.88% to 99.98%.
Pan and core-genome analysis of N. fowleri species
Figure 4A shows the pan-genome analyses of five N. fowleri species. The pan-genome of N. fowleri species showed a Bpan values of 0.137048 (i.e.,< 1) (Table 1), suggesting that the pan genome is still open but may be closed soon. This implies that for efficient environmental adaptations, the mtDNA is subjected to few regular evolutionary changes through gains and losses or lateral gene transfers. Thus, in the pan-genome, the number of gene families will continuously increase with the addition of new genomes to the analysis. Table 2 highlights the core, accessory, unique and exclusively absent genes. We identified 35 core genes in all five N. fowleri species. Moreover, all the five N. fowleri isolates have accessory genes and only N. fowleri strain V511 lacks both unique and exclusively absent genes.

Pan-genome analysis (A), and COG (B) and KEGG (C) major category distribution of core, accessory and unique genes.
COG and KEGG distribution
COG classified all genomic sequences into three major groups; core, accessory and unique genes (Fig. 4B). We observed that core and accessory genes are involved in metabolism, information storages and processing, and cellular processes and signaling. Unique genes are associated to cellular processes and signaling, and metabolism. The details of COG distribution are given in Supplementary Figure S2; there are more (> 60%) core, accessory and unique genes for energy production conversion. Moreover, unique genes (30%) were present for post-translational modification. Similarly, accessory genes were also related to post-translational modification, protein turnover chaperones [O] and translation, ribosomal structure biogenesis [J].
The comparative KEGG distribution/details are plotted in Fig. 4C. The KEGG percentage shows core, accessory and unique gene clusters for human disease, metabolism, environmental information processing, genetic information processing and organismal system. Accessory genes were only involved in genetic information processing. Moreover, there are categories of unique genes for environmental information processing and genetic information processing. The details of KEGG distribution are given in Supplementary Figure S3. It is interesting to noted that core, accessory and unique genes were related to neurodegenerative diseases as well as endocrine and metabolic diseases.
Core and pan phylogeny
The core (Fig. 5A) and pan phylogeny (Fig. 5B) indicates that just two groups initially arise from a single common ancestor. However, the first clad consist of four N. fowleri species and second clade consist of only one. In core and pan phylogeny, N. fowleri (JX17481.1) and N. fowleri strain v419(KX580903.1) were presented as a single separate group, respectively.

Phylogenetic analysis of core genome (A) and pan-genome (B) of N. fowleri species by BPGA pipeline.
Comparison of orthologous genes between the Naegleria species
Orthologous genes (277 genes) of Naegleria species (number of species 6) were analyzed and noted that 100% were present in the orthogroups (Table 3). The total number of orthogroups was found to be 46. The mean orthogroups size and median orthogroups size were found to be 6. Furthermore, number of single copy-orthogroups and number of orthogroups with all species present were 44 and 45, respectively. We also calculated orthogroups statistics for each Naegleria species and the details are given in Table 4.
The six Naegleria species shared 42 proteins (Fig. 6A). At the protein sequence level, the six Naegleria species form a total of 47 clusters; 42 single-copy gene clusters and 5 orthologous clusters (Fig. 6B). Three singleton genes (genes for which no orthologs could be found in any of the other species) were present in N. gruberi species (AF288092.1). Figure 6C shows the occurrence pattern of shared orthologous groups among five N. fowleri species. A green cell represents the presence of a cluster group in the corresponding species, and a grey bar represents the absence of a cluster group in that species. The pattern to the left shows cluster count and protein count in the shared clusters. The Gene Ontology (GO) distribution of the N. fowleri species of the 42 shared protein are given in Supplementary Table S3.

Venn diagram showing the distribution of orthologous gene clusters across Naegleria species and the total number of orthologous gene clusters of each organism (A). The orthologous cluster and singletons gene cluster (B). The occurrence table shows the occurrence pattern of shared orthologous groups among Naegleria species (C); the pattern towards the left shows the cluster count (the number of clusters shared between species) and protein count (number of protein members in the shared clusters).
Comparison of Naegleria species whole mtDNA
A comparative analysis of whole mitochondrial genomic DNA of Naegleria species was done using the cgview service. For this purpose, we used N. fowleri isolate AY27 (MZ461463) as a reference and the other five Naegleria species were taken as queries (Fig. 7).

Graphical representation of BLAST results using N. fowleri isolate AY27 (MZ461463) mitochondrial DNA as a reference and other Naegleria species mitochondrial DNA as a queries. From the circle inward: the first ring represents the COG (Clusters of Orthologous Groups of proteins) grouping, second, third and fourth rings consist of CDS, tRNA, rRNA and other genes; fifth ring in black color represent the reference and six to nine rings are representing five N. fowleri species and the tenth ring is for N. gruberi species.
From the circle inward: the first ring represents the COG grouping, the second, third and fourth rings consist of CDS, tRNA, rRNA and other genes; fifth rings in black color represent the reference and six to nine rings are representing five N. fowleri species and the tenth ring is for N. gruberi species. These results portray that all the N. fowleri species were more than 98% identical. As expected, when we used N. gruberi (AF288092.1) mtDNA as a reference and other five N. fowleri isolates as queries, the result showed that N. gruberi (AF288092.1) mtDNA was distinct from the N. fowleri species (Fig. 8). The result presented gaps/mismatches in the rings at several locations. However, at a few locations in the DNA, both N. fowleri species and N. gruberi species shared conserved regions.

Graphical representation of BLAST results using N. gruberi species (AF288092.1) mitochondrial DNA as a reference and other Naegleria species mitochondrial DNA as a query. From the circle inward: the first and second rings consisting of CDS, tRNA, rRNA and other genes, third ring in black color represent the reference and six to eight rings are representing five N. fowleri species.
Single nucleotide polymorphisms (SNPs) in mtDNA of N. fowleri isolate AY27 were analyzed using N. fowleri strain V511 as reference. We identified 43 SNPs in the N. fowleri isolate AY27 mtDNA (Supplementary Table S4). Most of the SNPs were present in the rRNA-large subunit ribosomal RNA gene. SNPs were also present in ATP1, ORF145, NAD4 and tRNA-Lys genes. Moreover, in the non-coding region, three SNPs were identified.
Conclusion
The N. fowleri isolate AY27 mtDNA sequence has circular DNA with 69 genes and out of which 46 are CDS. The pan-genome analysis of N. fowleri species presented an open pan-genome characteristic. The core, accessory and unique gene were linked to human disease (> 40%). Orthologous gene analysis revealed that most of the mtDNA was conserved in the Naegleria species. We identified several SNPs in the N. fowleri isolate AY27 mtDNA using N. fowleri strain V511 as a reference. The data generated in the current study will help to understand these two species at the mitochondrial level.
References
Shakoor, S. et al. Primary amebic meningoencephalitis caused by Naegleria fowleri, Karachi, Pakistan. Emerg. Infect. Dis. 17, 258–261. https://doi.org/10.3201/eid1702.100442 (2011).
De Jonckheere, J. F. The impact of man on the occurrence of the pathogenic free-living amoeboflagellate Naegleria fowleri. Future Microbiol. 7, 5–7. https://doi.org/10.2217/fmb.11.141 (2012).
Marciano-Cabral, F., MacLean, R., Mensah, A. & LaPat-Polasko, L. Identification of Naegleria fowleri in domestic water sources by nested PCR. Appl. Environ. Microbiol. 69, 5864–5869. https://doi.org/10.1128/aem.69.10.5864-5869.2003 (2003).
Odintsova, M. S. & Iurina, N. P. The mitochondrial genome of protists. Genetika 38, 773–788 (2002).
Sivashankari, S. & Shanmughavel, P. Comparative genomics—A perspective. Bioinformation 1, 376–378. https://doi.org/10.6026/97320630001376 (2007).
Kim, M., Oh, H.-S., Park, S.-C. & Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. https://doi.org/10.1099/ijs.0.059774-0 (2014).
Chun, B. H., Kim, K. H., Jeon, H. H., Lee, S. H. & Jeon, C. O. Pan-genomic and transcriptomic analyses of Leuconostoc mesenteroides provide insights into its genomic and metabolic features and roles in kimchi fermentation. Sci. Rep. 7, 11504. https://doi.org/10.1038/s41598-017-12016-z (2017).
Shin, J., Song, Y., Jeong, Y. & Cho, B. K. Analysis of the core genome and pan-genome of autotrophic acetogenic bacteria. Front. Microbiol. 7, 1531. https://doi.org/10.3389/fmicb.2016.01531 (2016).
Medini, D., Donati, C., Tettelin, H., Masignani, V. & Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594. https://doi.org/10.1016/j.gde.2005.09.006 (2005).
Vernikos, G., Medini, D., Riley, D. R. & Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154. https://doi.org/10.1016/j.mib.2014.11.016 (2015).
Bosi, E. et al. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. USA 113, E3801-3809. https://doi.org/10.1073/pnas.1523199113 (2016).
Luis Carlos, G. et al. Inside the Pan-genome—Methods and software overview. Curr. Genomics 16, 245–252. https://doi.org/10.2174/1389202916666150423002311 (2015).
Wang, Y., Coleman-Derr, D., Chen, G. & Gu, Y. Q. OrthoVenn: a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 43, W78–W84. https://doi.org/10.1093/nar/gkv487 (2015).
Ithoi, I. et al. Detection of Naegleria species in environmental samples from Peninsular Malaysia. PLoS ONE 6, e24327. https://doi.org/10.1371/journal.pone.0024327 (2011).
Barnett, N. D., Kaplan, A. M., Hopkin, R. J., Saubolle, M. A. & Rudinsky, M. F. Primary amoebic meningoencephalitis with Naegleria fowleri: Clinical review. Pediatr. Neurol. 15, 230–234. https://doi.org/10.1016/s0887-8994(96)00173-7 (1996).
Kang, H. et al. Effective PCR-based detection of Naegleria fowleri from cultured sample and PAM-developed mouse. Eur. J. Protistol. 51, 401–408. https://doi.org/10.1016/j.ejop.2015.07.003 (2015).
Wittwer, C. T. et al. The LightCycler: a microvolume multisample fluorimeter with rapid temperature control. Biotechniques 22, 176–181. https://doi.org/10.2144/97221pf02 (1997).
Fotedar, R. et al. Laboratory diagnostic techniques for Entamoeba species. Clin. Microbiol. Rev. 20, 511–532. https://doi.org/10.1128/cmr.00004-07 (2007) (table of contents).
Behera, H. S., Satpathy, G. & Tripathi, M. Isolation and genotyping of Acanthamoeba spp. from Acanthamoeba meningitis/meningoencephalitis (AME) patients in India. Parasit Vectors 9, 442–442. https://doi.org/10.1186/s13071-016-1729-5 (2016).
Qvarnstrom, Y., Visvesvara, G. S., Sriram, R. & da Silva, A. J. Multiplex real-time PCR assay for simultaneous detection of Acanthamoeba spp., Balamuthia mandrillaris, and Naegleria fowleri. J. Clin. Microbiol. 44, 3589–3595. https://doi.org/10.1128/JCM.00875-06 (2006).
Schild, M., Gianinazzi, C., Gottstein, B. & Müller, N. PCR-based diagnosis of Naegleria sp. infection in formalin-fixed and paraffin-embedded brain sections. J. Clin. Microbiol. 45, 564–567. https://doi.org/10.1128/JCM.01357-06 (2007).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. https://doi.org/10.1093/bioinformatics/btp324%JBioinformatics (2009).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. https://doi.org/10.1093/bioinformatics/btt086%JBioinformatics (2013).
Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. https://doi.org/10.1093/bioinformatics/btu153 (2014).
Chaudhari, N. M., Gupta, V. K. & Dutta, C. BPGA—An ultra-fast pan-genome analysis pipeline. Sci. Rep. 6, 24373. https://doi.org/10.1038/srep24373 (2016).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Xu, L. et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47, W52-w58. https://doi.org/10.1093/nar/gkz333 (2019).
Emms, D. M. & Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. https://doi.org/10.1186/s13059-019-1832-y (2019).
Seabolt, M. H., Konstantinidis, K. T. & Roellig, D. M. Hidden diversity within common protozoan parasites as revealed by a novel genomotyping scheme. Appl. Environ. Microbiol. 87, e02275-20. https://doi.org/10.1128/aem.02275-20 (2021).
Acknowledgements
We are very grateful to Prof. Dr. M. Iqbal Choudhary, Director International Center for Chemical and Biological Sciences (ICCBS), University of Karachi for given us dry lab facilities for bioinformatic analysis at Jamil-ur-Rahman Center for Genome Research, Dr. Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Sciences (ICCBS), University of Karachi, Pakistan. The author would like to dedicate this paper to Prof. Dr. Atta-ur-Rahman, the founding father of the Jamil-ur-Rahman Center for Genome Research, PCMD, ICCBS, University of Karachi, on the occasion of his 80th birthday.
Author information
Authors and Affiliations
Contributions
M.A. and Y.R. collected the sample. M.A., S.H.A.N. and M.K.A. help in the designing the wet lab experiments. H.M.T.M. did the genome assembly and rest of the bioinformatic analysis was performed by A.K.. A.K. wrote the paper and M.Y. did the review and editing of the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aurongzeb, M., Rashid, Y., Habib Ahmed Naqvi, S. et al. Insights into genome evolution, pan-genome, and phylogenetic implication through mitochondrial genome sequence of Naegleria fowleri species. Sci Rep 12, 13152 (2022). https://doi.org/10.1038/s41598-022-17006-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-17006-4
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}