
Abstract
Balamuthia mandrillaris is a pathogenic protozoan that causes a rare but almost always fatal infection of the central nervous system and, in some cases, cutaneous lesions. Currently, the genomic data for this free-living amoeba include the description of several complete mitochondrial genomes. In contrast, two complete genomes with draft quality are available in GenBank, but none of these have a functional annotation. In the present study, the complete genome of B. mandrillaris isolated from a freshwater artificial lagoon was sequenced and assembled, obtaining an assembled genome with better assembly quality parameter values than the currently available genomes. Afterward, the genome mentioned earlier, along with strains V039 and 2046, were subjected to functional annotation. Finally, comparative genomics analysis was performed, and it was found that homologous genes in the core genome potentially involved in the virulence of Acanthamoeba spp. and Trypanosoma cruzi. Moreover, eleven of fifteen genes were identified in the three strains described as potential target genes to develop new treatment approaches for B. mandrillaris infections. These results describe proteins in this protozoan's complete genome and help prioritize which target genes could be used to develop new treatments.
Similar content being viewed by others

Introduction
Balamuthia mandrillaris is a free-living amoeba (FLA) widely distributed in the environment of the warmer countries1. It is the causal agent of a chronic infection called Balamuthia amoebic encephalitis (BAE), and in some cases, skin lesions precede infection of the central nervous system (CNS)2. Additionally, it has been reported that this infection affects immunocompetent and immunocompromised people, and currently, more than 200 cases have been reported worldwide with a mortality rate > 90%, with most of these cases occurring in the United States and South America3. This high mortality rate is primarily due to the difficulty of obtaining an early diagnosis (when the disease may be manageable), coupled with the lack of specific drugs for B. mandrillaris infections; the current treatment consists of a combination of antimicrobials selected mostly empirically, resulting in few cases of survival at present4. For this reason, it is necessary to implement techniques that involve omic sciences in the study of this FLA to identify known protein domains for advancing to functional annotation and provide tools for the knowledge of the pathogenomics of this protozoan5.
Currently, the genomic information of this microorganism is scarce, and only the mitochondrial genomes of different isolates have been annotated, with lengths ranging from 39.8 to 42.8 Kb, 2 ribosomal RNAs (rRNAs), 13 to 18 transfer RNAs (tRNAs), and 33 to 38 protein-coding sequences4, 6. In a recent study, the B. mandrillaris transcriptome was analyzed, approximately 40% of the predicted proteins were functionally annotated, and 15 target genes for new treatment approaches for B. mandrillaris infections were identified7. However, there is no complete annotated genome of this FLA in GenBank, and only two draft quality genomes are available for strains 2046 and V039, which vary in size from 44 to 68 Mb, respectively8, 9.
Regarding other microorganisms of medical relevance, studies that combine functional annotation and comparative genomics have been reported to identify genes related to antibiotic resistance, virulence factors, transcriptional regulators, motility, and others10,11,12,13. Pangenome analysis of FLA revealed unique genes in pathogenic Acanthamoeba and Naegleria fowleri species. For Acanthamoeba, genes involved in virulence were reported as metalloproteases, laminin-binding proteins, and heat shock proteins14. For Naegleria fowleri, genes related to autophagy, cytoskeletal and membrane dynamics, motility, secretory products, response to stress, and posttranslational modifications were identified15.
The scarcity of genomic information has hampered the development of new compounds against B. mandrillaris. Therefore, combining functional annotation and comparative genomics of this pathogenic protozoan could help understand the genomic biology and identify conserved genes among different strains7, 16, 17. This study presents the annotation of the draft genomes of B. mandrillaris in GenBank, the genome assembly and annotation of a strain isolated in an artificial lagoon, and comparative genomics of the different strains.
Materials and methods
Maintenance of B. mandrillaris
The B. mandrillaris strain ITSON01 was isolated in 2014 from an artificial lagoon in Ciudad Obregon, Mexico18. Trophozoites were cultured axenically with Balamuthia mandrillaris ITSON medium in 75 cm2 ventilated cell culture bottles at 37 °C19. Trophozoites were harvested for DNA extraction.
DNA extraction and sequencing
Trophozoites were resuspended in phosphate-buffered saline (PBS, pH 7.4) using 6 cell culture bottles of 75 cm2 (approximately 10.8 × 106 cells). DNA extraction was performed using the Wizard SV Genomic DNA Purification System (Promega, Madison, WI) according to the manufacturer's instructions, obtaining 0.46 µg total DNA. The libraries were then sequenced at the genomic services laboratory (LABSERGEN, Irapuato, Gto) using the Illumina NextSeq platform with 150 bp paired-end reads, yielding approximately 50 million reads.
Furthermore, DNA extraction for sequencing with Oxford Nanopore Technologies (ONT) was performed by harvesting trophozoites with PBS washes as previously described, and a total of 20 cell culture bottles of 75 cm2 (approximately 36 × 106 cells) were used. Subsequently, the extraction was performed using the Wizard HMW DNA Extraction Kit (Promega, Madison, WI) according to the manufacturer's instructions, obtaining 6.89 µg total DNA. The libraries were then sequenced at the company Health GeneTech (HGT, Taoyuan, TW) with the ONT gridION platform, yielding a total of approximately 3 million reads (8.5 Gb of total bases). The length distribution of the raw long ONT reads was plotted using NanoPlot v1.41.020.
Assembly and annotation of the mitochondrial genome of B. mandrillaris strain ITSON01
The mitochondrial genome (mtDNA) assembly of B. mandrillaris was performed using short reads only (Illumina). Raw reads were filtered with default parameters for quality and minimum length using Trim Galore v0.6.421 and de novo assembled using SPAdes v3.1322. Once the assembly was obtained, to identify the mtDNA, the synteny was defined by alignment with 8 complete mtDNA (GenBank accession number: KT175738, KT175739, KT030672, KT175740, KT030671, KT030673, KT175741, and KT030670) using Mauve23. Subsequently, the aforementioned mitochondrial genome was isolated, and annotation of tRNA and protein coding genes (CDS) was performed with GeSeq24, whereas the rRNAs were identified with barrnap v0.925. Finally, the rRNAs were appended to the GeSeq output file by manual curation using Artemis26. A comparison of ITSON01 mtDNA was performed with the 8 genomes mentioned above and one more recently published genome (GenBank accession number: OM994889) using the CGview Comparison Tool (CCT)27.
Hybrid genome assembly of B. mandrillaris strain ITSON01 and genome reassembly of B. mandrillaris strain 2046
The long ONT raw reads were subjected to adapter removal with Porechop v0.2.428 and quality filtering with Filtlong v0.2.029, which eliminated reads with lengths less than 1 kb and ignored the phred quality values of the ONT reads, instead judging the quality using K-mer matches with the short Illumina reads30,31,32. The hybrid assembly was performed with default parameters using MaSuRCA v4.0.533, taking the raw short Illumina reads and filtered long ONT reads as input. The genome of B. mandrillaris strain 2046 was also reassembled using the reads available (GenBank accession numbers: SRR8980854, SRR8980855, and SRR8980856) for this strain and assembled with MaSuRCA v4.0.98.
RNA extraction and sequencing from B. mandrillaris ITSON01
RNA extraction for poly(A) and total RNA sequencing was performed using a RNeasy Minikit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions (only extraction temperature at 4 °C and centrifugation time were modified from 15 s to 1 min). RNA integrity was determined by the 2100 Bioanalyzer System (Agilent, Santa Clara, CA). The poly(A) libraries were sequenced at LC Sciences (Houston, TX) on an Illumina NovaSeq platform with 150 bp paired-end reads, yielding approximately 40 million reads per library. Moreover, the total RNA library was sequenced on the same company and platform, with 150 bp paired-end reads and a sequencing depth of approximately 200 million reads.
Annotation of proteins and noncoding genes in B. mandrillaris genomes
The complete genomes of the three strains were subjected to gene prediction and annotation using Funannotate v1.8.1434, taking as input the genome assembly of each strain and RNA-seq reads. Pannzer235 was used when Funannotate was unable to assign a functional description with the following parameter settings: minimum query coverage 0.4 or minimum sbjct coverage 0.4 and minimum alignment length 50, obtaining the proteins and tRNA annotated36, 37. Subsequently, the rRNAs (28S, 18S, and 5S) and long noncoding RNAs (lncRNAs) were predicted using the genome assembly of each strain as input with StructRNAfinder38, obtaining the location in the assembly and structure of these nonprotein-coding RNAs as output. Once these nonprotein-coding genes were obtained, manual curation was performed using Geneious Prime v2023.0.439. Finally, gene ontology (GO) terms were extracted from the Funannotate results and visualized on WEGO 2.040.
Comparative genomics
Comparative genomics analysis was performed using annotated protein sequences of the three B. mandrillaris strains (ITSON01, CDC-V039 and 2046) with default parameters using GET_HOMOLOGUES41, 42, obtaining the sequence cluster belonging to the pan/core genome as an output file.
Results
Filtering on Illumina and ONT DNA-Seq reads
After filtering with Trim Galore, approximately 0.18% of the Illumina reads were removed, reducing from 50,109,114 to 50,020,496 reads. The low number of reads removed is due to the high quality and depth coverage of the sequences from the Illumina platforms43. In the ONT reads, after adapter trimming with Porechop and quality filtering with the Illumina reads as a reference with Filtlong, approximately 43.74% of the reads were removed, reducing from 3,095,072 to 1,741,403 reads, possibly due to the large number of reads smaller than 1 kb (Fig. 1). However, most of the total bases were retained, eliminating approximately 11% after the filtering process.

Length distribution of the raw long ONT reads.
Assembly and annotation of the mitochondrial genome of B. mandrillaris ITSON01
After assembly and annotation, mtDNA was obtained with a length of 41,385 bp, 13 tRNA, 37 CDS, and 2 rRNA subunits. The mtDNA of B. mandrillaris strain ITSON01 was compared against some mtDNA of different strains available in GenBank, showing that most have an identical percentage > 98% except for strains V451 and KM-20 (Fig. 2).

Comparison of B. mandrillaris mtDNA against mtDNA from different strains (from edge to center, the strains are V118, GAM19, RP5, 2046, OK1, SAM, V039, V451, and KM-20).
Assembly of the B. mandrillaris strain ITSON01 genome and reassembly of the B. mandrillaris strain 2046 genome
After the hybrid assembly of B. mandrillaris strain ITSON01, a genome of approximately 65 Mb was obtained with better assembly quality values, such as the number of contigs, N50, L50, and low amount of "N" in the genome, than those currently available. Instead, reassembling the genome of B. mandrillaris strain 2046 resulted in a less fragmented genome, larger genome size, and lower "N" than the current genome of this strain (Table 1)8, 9. This reassembly was used for functional and comparative genomic annotation.
Functional annotation of B. mandrillaris genomes
For the ITSON01 strain, 67% of its genes were annotated as proteins with functional descriptions, 31% as proteins without functional descriptions (hypothetical proteins), and 2% as noncoding genes (rRNA and tRNA). In the case of the V039 strain, 63% of its genes were identified as proteins with functional descriptions, 35% as hypothetical proteins, and 2% as rRNA and tRNA. Finally, for the 2046 strain, 63% of its genes were described as proteins with functional descriptions, 35% as hypothetical proteins, and 2% as tRNA.
It should be noted that in the case of the 2046 strain, complete ribosomal RNAs could not be annotated due to the high fragmentation of the genome. A detailed summary of the annotation results for each strain of B. mandrillaris is presented in Table 2. Regarding lncRNAs, in the ITSON01 strain, two were annotated as MESTIT1, one as NPPA-AS1, and one as TCL6, whereas in the V039 strain, three were annotated as CDKN2B-AS, NPPA-AS1, and Six3os1.
Regarding the length distribution of the rRNA structures, in the ITSON01 strain, the large subunit (LSU) varied from 3625 to 5239 bp, and the small subunit (SSU) varied from 2017 to 2022 bp. In the V039 strain, the LSU varied from 3487 to 3853 bp, and the SSU varied from 2010 to 2028 bp. Additionally, the length of 5S rRNA was 119 bp in both strains. Some examples of structures of such rRNA obtained with StructRNAfinder are presented below (Fig. 3).

rRNA structures obtained with StructRNAfinder (created with BioRender.com).
The GO term annotation comparison revealed a similar profile for the 3 strains, except for some smaller gene families that represented less than 0.1% of the genes (Fig. 4). This analysis also revealed that the GO terms with the highest representation in the biological process category were "cellular process" (GO: 0009987) and "metabolic process" (GO: 0008152); for the cellular component category, they were "cell" (GO: 0005623) and "cell part" (GO: 0044464); and finally, for the molecular function category, they were "catalytic activity" (GO: 0003824) and "binding" (GO: 0005488).

Level 2 GO annotations, proteins of B. mandrillaris 2046 (green), B. mandrillaris ITSON01 (yellow) and B. mandrillaris V039 (blue), percentages of genes and total number of genes are log scale (10).
Comparative genomics
The results of the comparative genomics analysis were expressed in a Venn diagram (Fig. 5), which shows the overlap between orthologous groups of the different strains of B. mandrillaris. It should be noted that the orthologous gene clusters of the core genome represent approximately 6% of the proteins of each strain. At the same time, the numbers of unique protein genes, including the paralogs of each strain, were 4123 (13.8% of the total proteins), 6357 (22% of the total proteins), and 9732 (33% of the total proteins) for the ITSON01, V039, and 2046 strains, respectively.

Venn diagram of overlap among orthologous groups in the proteomes of different strains of B. mandrillaris.
Discussion
Previous studies have described the different variations in the mitochondrial genomes of B. mandrillaris, one of which is the location of an open reading frame (ORF) endonuclease containing the sequence LAGLIDADG; in the case of the V039 strain, this is not present in the genome. In the 2046, OK1, RP-5, SAM and KM-20 strains, this sequence disrupts the cox1 gene, whereas in the V451, GAM-19, V188 and ITSON01 strains, it is inserted in the 23S ribosomal gene. Although more mitochondrial genomes are required for possible genotyping of B. mandrillaris, the contribution of the mitochondrial genome of the ITSON01 strain could help to achieve this in the future5.
The better assembly metrics of the nuclear genome observed in the ITSON01 strain compared to previous works are mainly due to the use of both short and long reads (Illumina and ONT), as well as the use of MaSuRCA software, which was designed for the assembly of large genomes and has been characterized for obtaining the best hybrid assembly quality parameters in various eukaryotic genomes44,45,46. In contrast, the genome reassembly of the 2046 strain considerably improved with respect to the original assembly due to the use of MaSuRCA; this is possibly because this program has a record of obtaining better N50 length values than SPAdes and therefore having lower fragmentation in the assembled genomes using this program47. Furthermore, the reassembly was not improved compared to the genomes of the other strains (ITSON01 and V039) because only short reads were used; it is known that the use of only short reads in eukaryotic genome assemblies results in higher fragmentation (more scaffolds)48.
A larger number of genes with annotated functional descriptions were obtained due to the use of two programs, Funannotate and Pannzer2. Funannotate uses various curated databases to perform functional annotations, such as PFAM, InterPro, MEROPS, and CAZy, and to determine gene names and descriptions using EggNOG and UniProtKb/SwissProt, the latter consisting of manually reviewed high-quality protein sequences (approximately 0.5 million sequences). Additionally, Pannzer2 uses two UniProtKb databases, SwissProt and TrEMBL, the latter consisting of computationally reviewed high-quality protein sequences (approximately 208 million sequences). Therefore, the large number of sequences consulted was of great help for the functional homology annotation of the B. mandrillaris genome49,50,51.
According to the Venn diagram, the 2046 strain has a higher number of unique proteins than the other strains, which could be because genome fragmentation can be correlated with the duplication of specific sequences within the assembled genome52. In the cloud genome of strain ITSON01, several paralogous genes coding for protease (without group classification) and Vps9 domain-containing protein were identified, the latter of which has been found in other FLAs of medical importance, such as Naegleria fowleri5. Meanwhile, within the orthologous groups of the core genome of the three strains, homologous genes potentially involved in host invasion of pathogenic Acanthamoeba species were identified, which are SH3 domain-containing protein, filamin repeat domain-containing protein, myosin II light chain 1, myosin IA, heat shock protein 20, superoxide dismutase, metacaspase, and RAP7; the first 4 genes are related to the cytoskeleton, ability to tolerate high temperatures, defense against reactive oxygen species, phagocytosis process and endosomal delivery after phagocytosis (dominates energy production and cell growth), respectively (Supplementary Table S1)14, 53. Furthermore, homology was also found for a cysteine protease called cruzipain from the pathogenic protozoan Trypanosoma cruzi, which plays important functions in this protozoan, such as evasion of the immune response, differentiation, metabolism and invasion of host cells54. Therefore, it is likely that these aforementioned genes participate in similar molecular pathways and therefore have the same molecular functions in B. mandrillaris.
In other ways, we observed that within the three strains, a total of 11 of the previously published 15 sequences responsible for encoding target proteins to development of new treatments for B. mandrillaris infections were identified, which are methionyl-tRNA synthetase, xylose isomerase, heat shock protein 90, lanosterol 14-alpha demethylase, histone deacetylase, 3-hydroxy-3 methylglutaryl coenzyme A reductase, two types of DNA topoisomerase, calcium ATPase, glucokinase, and exportin-17. Additionally, coding sequences for enzymes that facilitate destruction and migration through the host, such as metalloproteinases, phospholipase A2, and phospholipase D, were found in all 3 strains4.
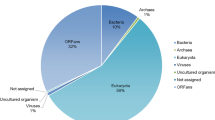
In the present study, homology was found with Acanthamoeba castellanii in the core genome of a gene encoding a serine carboxypeptidase. This type of enzyme has been described in silico as a pharmacological target in infections generated by N. fowleri because it is related to the virulence of this protozoan, as proven by genomic and transcriptomic studies. In their conclusion, they suggested that this enzyme has a ligand binding site suitable for design based on the structure of specific inhibitors, postulating it as a reliable target for treating primary amoebic meningoencephalitis (PAM) with drugs specifically aimed at blocking proliferation by inhibiting molecular function55.
Regarding the shell genome (ITSON01-V039), an extracellular protein aminopeptidase family M20/M25/M40 was determined to be homologous with the same pathogenic species of Acanthamoeba mentioned. This enzyme was shown to be involved in the Acanthamoeba pathogenesis process by pretreatment of proteins secreted by this FLA with leucine aminopeptidase inhibitor or specific antibiotic against the enzyme mentioned above, and a reduction in cell-based assay damage was observed56. In contrast to the other pathogenic species of FLA, nonpathogenic species of the genus Balamuthia have not yet been described. A new species of this genus has recently been reported (Balamuthia spinosa); however, it has not yet been described whether it is pathogenic in humans57. Therefore, differential expression analysis is still lacking to determine which proteins are involved in the pathogenesis of B. mandrillaris, but based on the evidence presented, the proteins, as mentioned earlier, could be related to the mechanisms employed by this pathogenic protozoan.
Regarding treatment development against B. mandrillaris, in a recent study, it was determined that formulations composed of azole (fluconazole and itraconazole) and 5-nitroimidazole (metronidazole) had a considerable antiparasitic effect against N. fowleri and B. mandrillaris amoebae, showing limited cytotoxic damage in human cells and reduction of host cell death caused by the pathogen58. In the present study, we found homology with bacteria and archaea (Heimdallarchaeota) in the core genome and shell (ITSON01-V039) genes coding for nitroreductases. These enzymes are important for the effectiveness of antimicrobials such as metronidazole, which requires a reduction in its nitro group to show antimicrobial effects59. Homology was also found with the FLA A. castellanii in the core genome genes coding for lanosterol 14-alpha demethylase, which has been described as a target in treatment with azoles60.
Another interesting finding to highlight is the identification of lncRNAs in the genome of B. mandrillaris. This type of noncoding RNA has been shown to be significant for its participation in development and physiological processes through its regulation of gene expression61. One of the lncRNAs shared between the ITSON01 and V039 strains was NPPA-AS1. In previous studies, an increase in this type of lncRNA was observed in HCT-8 cells infected with the pathogenic protozoan Cryptosporidium parvum, suggesting that this lncRNA, among others, could be involved in infection with this microorganism62. Regarding the other lncRNAs identified, no function related to infectious diseases is yet known. One aspect to note is that viral lncRNAs have also been attributed the ability to not induce an immune response compared to viral proteins, suggesting that viruses could use them as another strategy to invade their hosts61. Therefore, it is a desirable study area for pathogenic amoebas and other microorganisms capable of producing infections.
Conclusion
In the present study, annotation of the nuclear and mitochondrial genomes of B. mandrillaris was achieved, obtaining valuable information about possible genes involved in the pathogenicity of this protozoan through homologs with other pathogenic protozoan species. However, studies supported in functional genomics to determine genes related to the virulence of this FLA are still lacking. In addition, the comparative genomics of different strains performed in this study helped to identify the homology between strains of target genes for possible treatment against B. mandrillaris infections, which could help in prioritizing the development of treatments for those target sequences presented.
Data availability
The datasets presented in this study can be found in online repositories. The repository names and accession numbers can be found under the BioProject: PRJNA975899 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA975899?reviewer=slsojv2t5rc6q14qgbarhgnlp0).
References
Levinson, S. et al. Balamuthia mandrillaris brain infection: A rare cause of a ring-enhancing central nervous system lesion. Illustrative case. J. Neurosurg Case Lessons 3, 25 (2022).
Wu, X. et al. Diagnosing Balamuthia mandrillaris encephalitis via next-generation sequencing in a 13-year-old girl. Emerg. Microbes Infect. 9, 1379–1387 (2020).
Xu, C. et al. Subacute Balamuthia mandrillaris encephalitis in an immunocompetent patient diagnosed by next-generation sequencing. J. Int. Med. 50, 25 (2022).
Bhosale, N. K. & Parija, S. C. Balamuthia mandrillaris: An opportunistic, free-living ameba—an updated review. Trop. Parasitol. 11, 78 (2021).
Rodriguez-Anaya, L. Z., Félix-Sastré, Á. J., Lares-Villa, F., Lares-Jiménez, L. F. & Gonzalez-Galaviz, J. R. Application of the omics sciences to the study of Naegleria fowleri, Acanthamoeba spp., and Balamuthia mandrillaris: Current status and future projections. Parasite 28, 25 (2021).
Law, C. T. Y. et al. Mitochondrial genome diversity of Balamuthia mandrillaris revealed by a fatal case of granulomatous amoebic encephalitis. Front. Microbiol. 14, 25 (2023).
Phan, I. Q. et al. The transcriptome of Balamuthia mandrillaris trophozoites for structure-guided drug design. Sci. Rep. 11, 25 (2021).
Greninger, A. L. et al. Clinical metagenomic identification of Balamuthia mandrillaris encephalitis and assembly of the draft genome: The continuing case for reference genome sequencing. Genome Med. 7, 1–14 (2015).
Detering, H. et al. First draft genome sequence of Balamuthia mandrillaris, the causative agent of amoebic encephalitis. Genome Announc. 3, 25 (2015).
Kumar, R. et al. Comparative genomic analysis of rapidly evolving SARS-CoV-2 reveals mosaic pattern of phylogeographical distribution. mSystems 5, 25 (2020).
González, L. M. et al. Comparative and functional genomics of the protozoan parasite Babesia divergens highlighting the invasion and egress processes. PLoS Negl. Trop. Dis. 13, 25 (2019).
Kilian, M. & Tettelin, H. Identification of virulence-associated properties by comparative genome analysis of Streptococcus pneumoniae, S. pseudopneumoniae, S. mitis, three S. oralis subspecies, and S. infantis. MBio 10, 25 (2019).
Freschi, L. et al. The Pseudomonas aeruginosa pan-genome provides new insights on its population structure, horizontal gene transfer, and pathogenicity. Genome Biol. Evol. 11, 109–120 (2019).
Gu, X. et al. A comparative genomic approach to determine the virulence factors and horizontal gene transfer events of clinical Acanthamoeba Isolates. Microbiol. Spectr. 10, 25 (2022).
Dereeper, A. et al. Naegleria genus pangenome reveals new structural and functional insights into the versatility of these free-living amoebae. Front. Microbiol. 13, 25 (2023).
Ejigu, G. F. & Jung, J. Review on the computational genome annotation of sequences obtained by next-generation sequencing. Biology 9, 1–27 (2020).
Dong, Y., Li, C., Kim, K., Cui, L. & Liu, X. Genome annotation of disease-causing microorganisms. Brief. Bioinform. 22, 845–854 (2021).
Lares-Jiménez, L. F., Booton, G. C., Lares-Villa, F., Velázquez-Contreras, C. A. & Fuerst, P. A. Genetic analysis among environmental strains of Balamuthia mandrillaris recovered from an artificial lagoon and from soil in Sonora, Mexico. Exp. Parasitol. 145, S57–S61 (2014).
Lares-Jiménez, L. F., Gámez-Gutiérrez, R. A. & Lares-Villa, F. Novel culture medium for the axenic growth of Balamuthia mandrillaris. Diagn. Microbiol. Infect. Dis. 82, 286–288 (2015).
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669 (2018).
Krueger, F. TrimGalore. GitHub. https://github.com/FelixKrueger/TrimGalore (2023).
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A. & Korobeynikov, A. Using SPAdes de novo assembler. Curr. Protoc. Bioinform. 70, 25 (2020).
Darling, A. C. E., Mau, B., Blattner, F. R. & Perna, N. T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403 (2004).
Tillich, M. et al. GeSeq—versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, 6–11 (2017).
Seemann, T. Barrnap. GitHub. https://github.com/tseemann/barrnap (2018).
Carver, T., Harris, S. R., Berriman, M., Parkhill, J. & McQuillan, J. A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469 (2012).
Grant, J. R., Arantes, A. S. & Stothard, P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genom. 13, 25 (2012).
Wick, R. Porechop. GitHub. https://github.com/rrwick/Porechop (2018).
Wick, R. Filtlong. GitHub. https://github.com/rrwick/Filtlong (2021).
Chernikova, T. N. et al. Hydrocarbon-degrading bacteria alcanivorax and marinobacter associated with microalgae Pavlova lutheri and Nannochloropsis oculata. Front. Microbiol. 11, 25 (2020).
Asif, K. et al. Whole-genome based strain identification of fowlpox virus directly from cutaneous tissue and propagated virus. PLoS One 16, 25 (2021).
Bhandari, P. & Hill, J. E. Transport and utilization of glycogen breakdown products by Gardnerella spp. from the human vaginal microbiome. Microbiol. Spectr. 20, 20 (2023).
Zimin, A. V. et al. The MaSuRCA genome assembler. Bioinformatics 29, 2669–2677 (2013).
Palmer, J. & Stajich, J. Funannotate. GitHub. https://github.com/nextgenusfs/funannotate (2023).
Törönen, P., Medlar, A. & Holm, L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 46, W84–W88 (2018).
Morabito, C., Aiese Cigliano, R., Maréchal, E., Rébeillé, F. & Amato, A. Illumina and PacBio DNA sequencing data, de novo assembly and annotation of the genome of Aurantiochytrium limacinum strain CCAP_4062/1. Data Brief 31, 105729 (2020).
Deragon, E. et al. An oil hyper-accumulator mutant highlights peroxisomal ATP import as a regulatory step for fatty acid metabolism in Aurantiochytrium limacinum. Cells 10, 25 (2021).
Arias-Carrasco, R., Vásquez-Morán, Y., Nakaya, H. I. & Maracaja-Coutinho, V. StructRNAfinder: An automated pipeline and web server for RNA families prediction. BMC Bioinform. 19, 1–7 (2018).
Kearse, M. et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Ye, J. et al. WEGO 20: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 46, W71–W75 (2018).
Contreras-Moreira, B. & Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701 (2013).
Vinuesa, P. & Contreras-Moreira, B. Robust identification of orthologues and paralogues for microbial pan-genomics using GET_HOMOLOGUES: A case study of pIncA/C plasmids. Methods Mol. Biol. 1231, 203–232 (2015).
Hu, T., Chitnis, N., Monos, D. & Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 82, 801–811 (2021).
Jiang, J. B. et al. A hybrid de novo assembly of the sea pansy (Renilla muelleri) genome. Gigascience 8, 25 (2019).
Minei, R., Hoshina, R. & Ogura, A. D. novo assembly of middle-sized genome using MinION and Illumina sequencers. BMC Genom. 19, 25 (2018).
Tomé, L. M. R. et al. Hybrid assembly improves genome quality and completeness of Trametes villosa CCMB561 and reveals a huge potential for lignocellulose breakdown. J. Fungi 8, 25 (2022).
Sohn, J. I. & Nam, J. W. The present and future of de novo whole-genome assembly. Brief Bioinform. 19, 23–40 (2018).
Rayamajhi, N., Cheng, C. H. C. & Catchen, J. M. Evaluating illumina-, nanopore-, and PacBio-based genome assembly strategies with the bald notothen, Trematomus borchgrevinki. G3 Genes Genom. Genet. 12, 25 (2022).
Sarker, B., Khare, N., Devignes, M. D. & Aridhi, S. Improving automatic GO annotation with semantic similarity. BMC Bioinform. 23, 14 (2022).
Törönen, P. & Holm, L. PANNZER—a practical tool for protein function prediction. Protein Sci. 31, 118–128 (2022).
Tang, X. F. et al. Genomic insight into the scale specialization of the biological control agent Novius pumilus (Weise, 1892). BMC Genom. 23, 25 (2022).
Asalone, K. C. et al. Regional sequence expansion or collapse in heterozygous genome assemblies. PLoS Comput. Biol. 16, 25 (2020).
Wang, Y. et al. Biological characteristics and pathogenicity of Acanthamoeba. Front. Microbiol. 14, 25 (2023).
Siqueira-Neto, J. L. et al. Cysteine proteases in protozoan parasites. PLoS Negl. Trop. Dis. 12, 25 (2018).
Madero-Ayala, P. A., Mares-Alejandre, R. E. & Ramos-Ibarra, M. A. In silico structural analysis of serine carboxypeptidase Nf314, a potential drug target in Naegleria fowleri infections. Int. J. Mol. Sci. 23, 25 (2022).
Huang, J. M. et al. Pathogenic Acanthamoeba castellanii secretes the extracellular aminopeptidase m20/m25/m40 family protein to target cells for phagocytosis by disruption. Molecules 22, 25 (2017).
Lotonin, K., Bondarenko, N., Nassonova, E., Rayko, M. & Smirnov, A. Balamuthia spinosa n. sp. (Amoebozoa, Discosea) from the brackish-water sediments of Nivå Bay (Baltic Sea, The Sound)—a novel potential vector of Legionella pneumophila in the environment. Parasitol. Res. 121, 713–724 (2022).
Akbar, N. et al. Azole and 5-nitroimidazole based nanoformulations are potential antiamoebic drug candidates against brain-eating amoebae. J. Appl. Microbiol. 134, 25 (2023).
Thomas, C. & Gwenin, C. D. The role of nitroreductases in resistance to nitroimidazoles. Biology 10, 25 (2021).
Shing, B., Balen, M., McKerrow, J. H. & Debnath, A. Acanthamoeba keratitis: An update on amebicidal and cysticidal drug screening methodologies and potential treatment with azole drugs. Expert Rev. Anti Infect. Ther. 19, 1427–1441 (2021).
Lamsisi, M. & Ennaji, M. M. Involvement and roles of long noncoding rnas in the molecular mechanisms of emerging and reemerging viral infections. In Emerging and Reemerging Viral Pathogens 71–92 (Elsevier, 2019).
Sun, L. et al. Whole transcriptome analysis of HCT-8 cells infected by Cryptosporidium parvum. Parasit. Vectors 15, 25 (2022).
Acknowledgements
We are grateful to the CIIDIR-IPN Sinaloa Unit for support with the ooream cluster to perform bioinformatics analysis. The authors thank the Consejo Nacional de Humanidades, Ciencia y Tecnología (CONAHCYT) postgraduate scholarship program, the Cátedras-CONAHCYT program, and the Instituto Tecnológico de Sonora.
Funding
This article was funded by Consejo Nacional de Humanidades, Ciencia y Tecnología (Grant no. 840834), Program for the Promotion and Support of Research Projects (Grant no. PROFAPI_2023_118).
Author information
Authors and Affiliations
Contributions
Design of the work, L.Z.R.A.; writing—original draft preparation, A.O.R.; review and editing, L.Z.R.A., F.L.V., J.R.G.G., A.C.M., L.F.L.V., L.F.L.A.B.; interpretation of data, A.O.R., A.C.M., L.F.L.A.B.; project administration, L.Z.R.A., F.L.V.; funding acquisition, L.Z.R.A. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Otero-Ruiz, A., Rodriguez-Anaya, L.Z., Lares-Villa, F. et al. Functional annotation and comparative genomics analysis of Balamuthia mandrillaris reveals potential virulence-related genes. Sci Rep 13, 14318 (2023). https://doi.org/10.1038/s41598-023-41657-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-41657-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
