
Abstract
Despite increased early diagnosis and improved treatment in breast cancer (BRCA) patients, prognosis prediction is still a challenging task due to the disease heterogeneity. This study was to identify a novel gene signature that can accurately evaluate BRCA patient survival. The gene expression and clinical data of BRCA patients were collected from The Cancer Genome Atlas (TCGA) and the Molecular Taxonomy of BRCA International Consortium (METABRIC) databases. Genes associated with prognosis were determined by Kaplan–Meier survival analysis and multivariate Cox regression analysis. A prognostic 16-gene score was established with linear combination of 16 genes. The prognostic value of the signature was validated in the METABRIC and GSE202203 datasets. Gene expression analysis was performed to investigate the diagnostic values of 16 genes. The 16-gene score was associated with shortened overall survival in BRCA patients independently of clinicopathological characteristics. The signalling pathways of cell cycle, oocyte meiosis, RNA degradation, progesterone mediated oocyte maturation and DNA replication were the top five most enriched pathways in the high 16-gene score group. The 16-gene nomogram incorporating the survival‐related clinical factors showed improved prediction accuracies for 1-year, 3-year and 5‐year survival (area under curve [AUC] = 0.91, 0.79 and 0.77 respectively). MORN3, IGJ, DERL1 exhibited high accuracy in differentiating BRCA tissues from normal breast tissues (AUC > 0.80 for all cases). The 16-gene profile provides novel insights into the identification of BRCA with a high risk of death, which eventually guides treatment decision making.
Similar content being viewed by others

Introduction
BRCA (BRCA) is the most prevalent female malignancy in US and China. An estimated 284,200 cases will be diagnosed and 44,130 patients will die of the disease in 2021, accounting for more than 15% of newly diagnosed cancer cases and 7.3% of cancer-related mortalities1,2. According to the molecular classifications, BRCA can be mainly divided into five subtypes: luminal A, luminal B/human epidermal growth factor receptor 2[Her2] negative, triple positive (ER+, Progesterone receptor [PR]+, Her2+), Her2-enriched, and triple negative (ER−, PR−, Her2−)3. With the significant progresses of medical technology, the prognosis of BRCA has been remarkably ameliorated. However, the prognosis is still not optimistic for BRCA patients diagnosed at late stages.
The development of methods for risk stratification in BRCA has been a hotspot of research. Several studies demonstrate that multigene signatures might be more accurate for risk stratification than the traditional approaches in BRCA4,5. The MammaPrint, a 70-gene signature, is a prognostic model to stratify node-negative BRCA patients with different survival probabilities4. Oncotype DX is a 21-gene signature that provides information of the likelihood of recurrence and weighs the potential benefits of chemotherapy in the node-negative, estrogen receptor positive BRCA5. These multigene assays show potential clinical utility, but still need to be validated in large, randomized trials6. Moreover, the established methods are applicable to only limited disease subtypes, there is still lack of an effective prognostic model that could be used for almost all BRCA subtypes.
In the current study, we aimed to develop a novel gene profile to accurately estimate disease prognosis. We first examined all genes for their association with overall survival (OS) using the gene expression and clinical data of The Cancer Genome Atlas (TCGA) dataset7 and validated the results in the Molecular Taxonomy of BRCA International Consortium (METABRIC)8 and GSE2022039 datasets. We next established a 16-gene score based on a linear combination of 16 gene expression levels and 16-gene nomogram to precisely predict the overall survival (OS) of BRCA patients. Lastly, we performed expression analysis of 16 genes and demonstrated their diagnostic values in BRCA.
Methods and materials
Data acquisition and processing
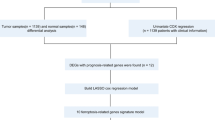
We obtained RNA-seq expression data and clinical data of BRCA patients from the two different sources, the first of which was the TCGA database (n = 1080 patients), the second source was the METABRIC study which was used to validate the associations between gene expression and OS (n = 1904 patients). Clinical features of BRCA patients are summarized and presented in Table1 respectively. As the gene expression unit of the TCGA dataset differs from that of the METABRIC cohort, normalization of gene expression was performed using the formula z = (x-\(\overline{x }\))/s. where x, \(\overline{x }\) and s are the gene expression value, mean and standard deviation of gene expression values. This study was in compliance with strict confidentiality guidelines and regulations regarding personal data protection, all methods were carried out in accordance with relevant guidelines and regulations. We also obtained 2913 breast cancer patients from the GEO database (GSE202203) to validate the association between gene expression, risk score and overall survival9.
Identification of survival-related clinical features and genes
We aimed to identify survival-related clinical features using different statistical methods. For quantitative variables, we utilized student t test to characterize their associations with OS. With respects to qualitative variables, we implemented fisher exact test to investigate their associations with OS. We followed Sha et al.’s methods10 to identify and classify survival-related genes. In brief, we firstly split BRCA samples into two subgroups, the low-expression and high-expression groups, based on the median expression value. We performed Kaplan–Meier survival analysis to evaluate the statistical significance of the differences in OS with the survival package11,12 and conducted multivariate Cox regression model to further validate the survival analysis. Survival-related genes with odd ratio [OR] > 1were considered as risk genes, while genes with 0 < OR < = 1 were defined as protective genes. To further evaluate the prognostic importance of the 16-gene score, we drew receiver operating characteristic (ROC) curves and computed the area under curve (AUC) values using the R package pROC13. To investigate the potential biological function of prognosis-related genes, we analyzed the enrichment of Gene Ontology (GO) term and Kyoto Encyclopedia of Genes and Genomes (KEGG) signalling pathway using the online tool g:profiler14.
Establishment and validation of the 16-gene score
We followed Lai et al.’s methods15 to choose the set of genes which performed best in prognosis prediction and develop the 16-gene risk score. In brief, the least absolute shrinkage and selection operator (LASSO) models comprising different number of genes were evaluated for prediction accuracies of OS using glmnet in the TCGA dataset16. The 16-gene score was created using the following formula: 16-gene score = − 1.91 + expression of gene 1 × β1 + expression of gene 2 × β2 + ⋯ + expression of gene n × βn. β values represented the coefficients generated from the optimal LASSO model. We then implemented Kaplan–Meier survival analysis, multivariate Cox regression analysis and stratification analysis to further investigate the association between the 16-gene score and OS in BRCA. We also analyzed the prediction capability of the 16-gene score for progression-free survival (PFS) and disease-free survival (DFS) in the TCGA cohort using Kaplan–Meier survival analysis. Lastly, we utilized linear regression model to investigate the correlations between clinical characteristics and the 16-gene score in the TCGA and METABRIC cohorts. P < 0.05 was considered statistically significant.
Gene set enrichment analysis
On the basis of the median 16-gene score, the BRCA patients were split into two subgroups: the high and low 16-gene score groups. Gene set enrichment analysis (GSEA)17 was implemented to determine the dysregulated signalling pathways related to the 16-gene score using the default parameters. Q value < 0.25 was considered statistically significant.
Construction and validation of the 16-gene nomogram
Nomogram was constructed using the rms package in R, and included patient’s age, tumor stage, menopause status, number of positive lymph nodes and 16-gene signature as they are significantly correlated with OS of BRCA. The performance of the nomogram developed was evaluated in the TCGA cohort and validated in the METABRIC cohort using the R package pROC. AUC values were computed accordingly for the nomogram in the prediction of 1-year, 3-year and 5-year survival.
Expression analysis of prognosis-related genes
The online server cbioportal18 was utilized to analyze the mutational profiles of the 16 genes in the TCGA cohort. Furthermore, the expression data of 779 BRCA tissues and 100 paired non-cancerous tissues were downloaded from the TCGA database. Differentially expressed genes were determined between BRCA tissues and paired normal tissues using student t test. To investigate the diagnostic values of the 16 genes, the pROC package was used to determine whether the gene expression could effectively distinguish cancer tissues from paired normal ones. P value was adjusted using false discovery rate. Adjusted P < 0.05 indicated statistical significance.
Ethical statement
As all the data were obtained from public databases, the study didn’t need to be approved by the Ethics committee of Ningbo Women and Children’s Hospital.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent to publish
The authors affirm that human research participants provided informed consent for publication of this study.
Results
Identification of survival-related clinical features in BRCA
We initially performed survival analysis between clinical features and OS and revealed higher patient’s age, more positive lymph nodes, higher cancer stage, clinical T stage, clinical N stage, clinical M stage, post-menopause were high risk prognosticators for OS in the TCGA cohort (P < 0.05 for all cases, Table1). Furthermore, the inverse correlations between overall survival and patient’s age, more positive lymph nodes, higher cancer stage, post-menopause, tumor size, radiotherapy were independently validated in the METABRIC cohort (P < 0.05 for all cases, Table1).
Identification and validation of survival-related genes in BRCA
We first examined the relation between gene expression and OS in the TCGA data set. The results showed that high expression levels of 1374 genes were related to significantly prolonged OS. While, high expression levels of 678 genes were related to significantly reduced survival in the TCGA cohort (P < 0.05 for all cases, log rank test, Fig. 1). Multivariate Cox regression analysis confirmed 432 protective prognostic genes and 219 risk prognostic genes following the adjustment of clinical characteristics. Furthermore, the association between 651 gene expression and OS was analyzed in the METABRIC dataset (n = 1904). The results validated 80 protective genes and 34 risk genes in the METABRIC cohort respectively (P < 0.05 for all cases, log rank test, Fig. 1). Then, we analyzed the functional involvement of the protective and risk genes with g.profiler and uncovered the 80 protective genes were significantly enriched in the KEGG pathway of focal adhesion. While, the risk genes were significantly over-represented in GO terms, such as nuclear division, organelle fission, DNA metabolic process and nucleic acid metabolic process (adjusted P value < 0.05 for all cases, supplementary Fig. 1).

The prognosis-related genes common to the TCGA and METABRIC datasets. (A) The protective prognostic genes common to the TCGA and METABRIC datasets. (B) The risk prognostic genes common to the TCGA and METABRIC datasets. KM_TCGA and multivariate_TCGA represent prognosis-related genes determined by the Kaplan–Meier survival analysis and multivariate Cox regression analysis respectively in the TCGA cohort. Similarly, KM_METABRIC and multivariate_METABRIC denote survival-related genes in the METABRIC cohort.
Construction of a 16-gene signature and its prognostic value in BRCA
We followed Lai et al.’s methods15 to choose the set of genes which performed best in prognosis prediction and develop the 16-gene risk score from 114 selected genes. The LASSO model comprising 16 genes showed the highest AUC value and was deemed the best model for survival prediction (Fig. 2A). Then we established the 16-gene score formula and computed the risk score for each BRCA patient, the coefficients of the 16 genes were presented in Fig. 2B. The Kaplan–Meier survival analysis and multivariate Cox regression analysis indicated that the high 16-gene score was indicative of worse OS in BRCA (P < 0.05 for all cases, OR: 3.47, 95% confidence interval: 2.08–5.78, Fig. 2C and supplementary Fig. 2A). We also analyzed the association between the 16-gene score and DFS and PFS in the TCGA cohort. Similarly, we demonstrated that the high 16-gene score was significantly associated with shorter DFS and PFS (P value < 0.05 for all cases, supplementary Fig. 3). For further verification, the 16-gene score was calculated in the METABRIC dataset. The results also confirmed the negative correlation between the 16-gene score and patient's OS (Fig. 2D, supplementary Fig. 2B). Furthermore, the 16-gene score (AUC = 0.72, 0.71, 0.73, respectively) outperformed cancer stage (AUC = 0.71, 0.69, 0.66, respectively, supplementary Fig. 4) in predicting 1-year survival, 3-year survival and 5-year survival in the TCGA cohort. The results were also validated in the METABRIC cohort (supplementary Fig. 4) and suggested the 16-gene score is superior to cancer stage in the prediction of prognosis of BRCA patients. We also obtained 2913 breast cancer patients from the GSE202203 dataset to validate the association between gene expression, risk score and overall survival. As expected, 12 of the ten survival-related genes showed positive correlation with prolonged survival and functioned as protective genes, PXDNL and DERL1 were shown as risk gene (supplementary Table 1). Furthermore, we recalculated risk score using the validation dataset and confirmed the risk score is a negative factor for overall survival in breast cancer (supplementary Fig. 5).

The 16-gene score is an indicator of infavorable survival in BRCA. (A) The relationship among AUC, log scaled lambda values and number of genes with non-zero coefficients in the LASSO model. The x axis represents the log value of the independent variable λ, whilst the y axis represents the partial likelihood deviance of the log value of each independent variable λ. (B) The coefficients of 16 genes in the LASSO model. (C) Kaplan–Meier survival curve of patients’ OS for BRCA patients with different 16-gene scores in the TCGA cohort, (D) Kaplan–Meier survival curve of patients’ OS for BRCA patients with different 16-gene scores in the METABRIC dataset.
Correlations between the 16-gene score and clinical factors in BRCA
The linear regression model analysis showed the 16-gene score was significantly positively associated with age, HER2 status, menopause status, clinical stage, clinical T stage, clinical M stage and negatively correlated with PR status, ER status, hormone therapy and radiotherapy in the TCGA cohort (p < 0.05 for all cases, Fig. 3A). Moreover, the 16-gene score also exhibited positive correlation with age, HER2 status, menopause status, clinical stage and negative correlation with PR status, ER status, hormone therapy and radiotherapy in the METABRIC cohort (p < 0.05 for all cases, Fig. 3B). Next, we split BRCA patients into subgroups according to the clinical characteristics and conducted the Kaplan–Meier survival analysis to assess the prognostic value of the 16-gene score in clinical factor-specific subgroups. Overall, the results demonstrated that the high-risk was significantly correlated with worse OS in the same clinical subgroup of the TCGA cohort (P < 0.05 for all cases, log rank test, supplementary Table 2 and supplementary Fig. 6). Similar findings were also observed in the METABRIC cohort (supplementary Table 3 and supplementary Fig. 7), suggesting that the implication of 16-gene score with OS is independent of clinicopathological characteristics.

The correlations between the clinical characteristics and the 16-gene score. (A) The associations between clinical characteristics and the 16-gene score in the TCGA cohort. (B) The associations between clinical characteristics and the 16-gene score in the METABRIC cohort.
Identification of signalling pathways associated with the 16-gene score
We performed the GSEA analysis to understand the biological functions related to the 16-gene score. The results exhibited thirteen signalling pathways were significantly over-represented in the high 16-gene score group of the TCGA cohort. Cell cycle, RNA degradation, oocyte meiosis, progesterone mediated oocyte maturation and DNA replication were the top five most enriched pathways (Fig. 4, q value < 0.25 for all cases, supplementary Table 4). While, up-regulation of arachidonic acid metabolism pathway genes were significantly associated with the low 16-gene score in the TCGA cohort (Fig. 4, q value < 0.25, supplementary Table 5). These results suggest that the aforementioned pathways probably are implicated in the association between 16-gene score and OS in BRCA.

GSEA analysis revealed three significantly enriched pathways related to the high 16-gene score, including oocyte meiosis (A), cell cycle (B), DNA replication (C), and the significantly up-regulated arachidonic acid metabolism (D) associated with the low 16-gene score. For each gene set, vertical bars along the x-axis represent where the genes locate within the ranked list. Negative enrichment score indicates down-regulation, while, positive value denotes up-regulation of the gene set.
Nomogram combined 16-gene signature and clinical‐related variables predicts patients’ OS
In the TCGA and METABRIC cohorts, patient’s age, tumor stage, menopause status, number of positive lymph nodes and 16-gene signature were significantly associated with OS. Then based on the above analysis results, we established a 16-gene nomogram that incorporated the survival‐related clinical factors and 16-gene signature (Fig. 5A). The nomogram predicted well the 1-year, 3-year and 5‐year survival for BRCA patients in the TCGA cohort, ROC plot revealed the 16-gene nomogram showed improved prediction accuracies for 1-year, 3-year and 5‐year survival as compared to the 16-gene score alone (AUC: 0.91, 0.79 and 0.77 respectively, Fig. 5B). The improved prognosis prediction was also validated in the METABRIC cohort (AUC: 0.83, 0.77 and 0.76 respectively, Fig. 5C), demonstrating the clinical value and validity of the 16-gene nomogram for OS evaluation of BRCA patients.

The 16-gene nomogram to predict the risk of disease in patients with BRCA. (A) mRNA nomogram to predict disease‐free survival. 1, 2, 3 for the menopausal status denote pre-menopause, peri-menopause and post-menopause respectively. 0 and 1 for the 16-gene score represent high and low 16-gene scores respectively which were divided by the median 16-gene score. (B) The ROC plot for the nomogram in predicting of 1-year, 3-year and 5-year survival in the TCGA cohort. (C) The ROC plot for the nomogram in predicting of 1-year, 3-year and 5-year survival in the METABRIC cohort.
Assessment of diagnostic value
We utilized the online server cBioPortal to investigate the genomics variants of 16 genes from the TCGA datasets. The results showed that DERL1, TNN, PXDNL, PCSK6 and KLRB1were the top five most frequently mutated genes, with mutation frequencies of 19%,10%, 9% 4%, 3% respectively in BRCA (supplementary Fig. 8). Similar mutation distribution was observed in the METABRIC cohort (supplementary Fig. 9). By comparing expression levels of 16 genes between 779 BRCA samples and 100 paired normal breast tissues, 7 genes expression, such as C7orf63, C9orf103, IGJ, ZNF385B and TNN, was significantly lower in tumor tissues as compared with those in normal tissues. In contrast, 9 genes, such as PXDNL, PCSK6, MORN3 and DERL1, were significantly higher expressed in BRCA tissues (adjusted P < 0.05 for all cases, student t test, Fig. 6A). ROC curves analysis further showed MORN3, IGJ, DERL1 particularly were able to differentiate BRCA tissues from normal breast tissues with high accuracy (Fig. 6B, adjusted P values < 0.05, AUC > 0.80 for all cases).

Gene expression analysis of prognosis-related genes. (A) The heatmap shows the expression differences of 16 prognosis-related genes between BRCA tissues and paired normal breast tissues. (B) The ROC curves for the top eight prognosis-related genes showing the highest diagnostic capability.
Discussion
BRCA is a heterogeneous disease with several molecular subtypes, each of which has its distinct biological and clinical characteristics19. The identification of reliable prognostic biomarkers would enable to prioritize patients at high risk for death and relapse and guide treatment. The traditional methods for the risk stratification include tumor size, tumor stage, lymph node metastasis and molecular subtype, which could be applicable to certain subgroup of BRCA, however, there is still lack of a prognostic model that could be applicable to almost all BRCA subtypes. Recent studies have shown gene expression profiles could serve as prognostic biomarkers in BRCA20,21. However, the accuracies of the previous gene profiles are still relatively low. In the current study, we have successfully established the 16-gene score which is correlated with poor OS, DFS and PRS in BRCA. We also demonstrated that the prognostic value of the 16-gene score was independent of clinical factors and applied to all subtypes of BRCA patients, which is advantageous to the MammaPrint model and Oncotype DX that show applicability to limited disease subtypes. Furthermore, we established a 16-gene nomogram that incorporated the survival‐related clinical factors and 16-gene signature. As compared to established gene profiles, the 16-gene nomogram (AUC = 0.91, 0.79 and 0.77, respectively) performed better than the Teschendorff's22 (AUC = 0.44, 0.47, 0.50, respectively) and Bianchini's23 immune-related gene signatures (AUC = 0.53, 0.56, 0.51, respectively)20 and cancer stage (AUC = 0.71, 0.69, 0.66, respectively) in predicting the 1-year, 3-year and 5-year survival of BRCA patients. Therefore, the 16-gene nomogram might be a reliable and useful prognostic tool for OS evaluation and will promote tailored therapy for all subtypes of BRCA patients.
The mechanisms by which the higher 16-gene score is associated with poor prognostic implication remain to be poorly understood. The GESA analysis uncovered cell cycle, RNA degradation, oocyte meiosis, progesterone mediated oocyte maturation and DNA replication were significantly over-represented in the high 16-gene score group. Cell cycle checkpoints are critical for ordered cell cycle progression, which ensures genomic stability and inhibits the process of carcinogenesis24. The deregulation of the cyclin-dependent kinase inhibitors p21 and p27, cyclins D1 and E frequently exerts negative impacts on BRCA outcome and response to therapy25. We believe the dysregulation of cell cycle signalling pathway largely contribute to the prognostic value of 16-gene score in BRCA.
Of the 16 prognosis-related genes, many have been shown to play key roles in the development and progression of various cancers. High expression of SERPINA1 gene encoding acute phase protein, alpha1-antitrypsin (AAT), is associated with various tumors. Experiments in vitro revealed that external AAT and/or overexpressed SERPINA1 gene significantly enhanced cancer cell migration, colony formation and resistance to apoptosis26. The SERPINA1 gene functions as key prognostic gene for patients with colon cancer27 and non-small-cell lung cancer patients26. SERPINA1 has also a significant predictive value for the OS of ER+/HER2+ patients. ER is constitutively activated, leading to an E2-independent ER binding to the SERPINA1 gene and upregulation of SERPINA1 expression in breast cancer28. KLRB1 is encoded by killer cell lectin-like receptor B1 gene and a new candidate inhibitor of tumour-infiltrating T cells. KLRB1 was differentially expressed and associated with better overall survival in a variety of tumour types. In addition, KLRB1 expression was significantly associated with immunoregulatory interactions between lymphoid and non-lymphoid cells, T cell infiltration, immune checkpoints, immune activating genes, immunosuppressive genes, chemokines, and chemokine receptors29. KLRB1 (coding for CD161) gene expression shows a positive association with favorable outcome in non-small-cell lung cancer, independently of the size of T and B cell infiltrates, making CD161-expressing CD4+ T cells ideal candidates for anti-tumor recall responses30. Elongation of very-long chain fatty acid protein 2 (ELOVL2) was hypermethylated and downregulated in tamoxifen-resistant breast cancer patients as compared with the tamoxifen-sensitive patients. ELOVL2 was shown to increase tamoxifen sensitivity up to 70% in the MCF-7/tamoxifen-resistant cells and in a xenograft mouse model31. Of note, elevated ELOVL2 expression levels were observed in renal cell carcinoma and significantly associated with a poor prognosis of patients with renal cell carcinoma. Furthermore, ELOVL2 promotes cancer progression by repressing cell apoptosis in renal cell carcinoma32.
Though we have established a risk score that is predictive of overall survival of BRCA patients independently of clinical characteristics, this study shows a number of limitations. First of all, the prediction accuracy of the nomogram incorporating clinical factors and risk scores is relatively low for 3-year and 5‐year survival, therefore, future studies may need to add more useful features to increase the performance of our model. Secondly, the functions of the identified prognosis-associated genes have not been fully illustrated, more experimental investigation should be performed to further characterize their biological functions in breast cancer.
Conclusion
Taken together, this study identified a novel 16 gene signature that could serve as an independent factor for predicting BRCA prognosis independently of clinical characteristics. The gene set related to the high-risk group participated in the cell cycle signal pathway.
Data availability
The datasets generated and/or analysed during the current study are available in the figshare repository (figshare ID: 15048003, https://figshare.com/s/df0ee21997f1aa0da4bd).
Code availability
The codes in the current study are available upon reasonable request.
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2019. CA. Cancer J. Clin. 69, 7–34 (2019).
Zheng, R. S. et al. Report of cancer epidemiology in China, 2015. Zhonghua Zhong Liu Za Zhi 41, 19–28 (2019).
Kast, K. et al. Impact of breast cancer subtypes and patterns of metastasis on outcome. Breast Cancer Res. Treat. 150, 621–629 (2015).
Buyse, M. et al. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J. Natl. Cancer Inst. 98, 1183–1192 (2006).
Toole, M. J., Kidwell, K. M. & Van Poznak, C. Oncotype Dx results in multiple primary breast cancers. Breast Cancer Basic Clin. Res. 8, 1–6 (2014).
Kwa, M., Makris, A. & Esteva, F. J. Clinical utility of gene-expression signatures in early stage breast cancer. Nat. Rev. Clin. Oncol. 14, 595–610 (2017).
Hoadley, K. A. et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291-304.e6 (2018).
Pereira, B. et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 7, 1–16 (2016).
Dalal, H. et al. Clinical associations of ESR2 (estrogen receptor beta) expression across thousands of primary breast tumors. Sci. Rep. 12, 4696 (2022).
Lai, Y., Sheng, L., Wang, J., Zhou, M. & OuYang, G. A novel 85-gene expression signature predicts unfavorable prognosis in acute myeloid leukemia. Technol. Cancer Res. Treat. 20, 15330338211004932 (2021).
Therneau, T. Survival analysis. Cran https://doi.org/10.1007/978-1-4419-6646-9 (2016).
Fox, J. Cox Proportional-hazards regression for survival data the Cox proportional-hazards model. Most 2008, 1–18 (2002).
Robin, X. et al. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 12, 77 (2011).
Reimand, J., Kull, M., Peterson, H., Hansen, J. & Vilo, J. G:Profiler-a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 35, 193–200 (2007).
Sha, K. et al. Identifying a novel 5-gene signature predicting clinical outcomes in acute myeloid leukemia. Clin. Transl. Oncol. https://doi.org/10.1007/s12094-020-02460-1 (2020).
Simon, N., Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for Cox’s proportional hazards model via coordinate descent. J. Stat. Softw. 39, 1–13 (2011).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 102, 15545–15550 (2005).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
Blows, F. M. et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: A collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 7, e1000279 (2010).
Xie, P. et al. Development of an immune-related prognostic signature in breast cancer. Front. Genet. 10, 1–14 (2020).
Shimizu, H. & Nakayama, K. I. A 23 gene-based molecular prognostic score precisely predicts overall survival of breast cancer patients. EBioMedicine 46, 150–159 (2019).
Teschendorff, A. E. et al. Improved prognostic classification of breast cancer defined by antagonistic activation patterns of immune response pathway modules. BMC Cancer 10, 604 (2010).
Bianchini, G. et al. Prognostic and therapeutic implications of distinct kinase expression patterns in different subtypes of breast cancer. Cancer Res. 70, 8852–8862 (2010).
Malumbres, M. & Carnero, A. Cell cycle deregulation: A common motif in cancer. Prog. Cell Cycle Res. 5, 5–18 (2003).
Caldon, C. E., Daly, R. J., Sutherland, R. L. & Musgrove, E. A. Cell cycle control in breast cancer cells. J. Cell. Biochem. 97, 261–274 (2006).
Ercetin, E. et al. Clinical significance of SERPINA1 gene and its encoded alpha1-antitrypsin protein in NSCLC. Cancers 11, 1306 (2019).
Xu, J., Dai, S., Yuan, Y., Xiao, Q. & Ding, K. A prognostic model for colon cancer patients based on eight signature autophagy genes. Front. Cell Dev. Biol. 8, 602174 (2020).
Chan, H. J. et al. SERPINA1 is a direct estrogen receptor target gene and a predictor of survival in breast cancer patients. Oncotarget 6, 25815–25827 (2015).
Zhou, X. et al. A pan-cancer analysis of CD161, a potential new immune checkpoint. Front. Immunol. 12, 688215 (2021).
Braud, V. M. et al. Expression of LLT1 and its receptor CD161 in lung cancer is associated with better clinical outcome. Oncoimmunology 7, e1423184 (2018).
Jeong, D. et al. ELOVL2: A novel tumor suppressor attenuating tamoxifen resistance in breast cancer. Am. J. Cancer Res. 11, 2568–2589 (2021).
Tanaka, K. et al. ELOVL2 promotes cancer progression by inhibiting cell apoptosis in renal cell carcinoma. Oncol. Rep. 47, 1–11 (2022).
Funding
This study was funded by Ningbo Natural Science Foundation (Project ID: 2018A610324) and Ningbo key medical disciplines (Project ID:2022-F27).
Author information
Authors and Affiliations
Contributions
F.F. conceived the study. Z.Z. and W.J. performed the survival analyses. Z.Z., W.J., J.Z. developed the 16-gene score and nomogram and implemented the validation analysis. Z.L., F.F. performed the GSEA analysis. Z.Z. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhong, Z., Jiang, W., Zhang, J. et al. Identification and validation of a novel 16-gene prognostic signature for patients with breast cancer. Sci Rep 12, 12349 (2022). https://doi.org/10.1038/s41598-022-16575-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-16575-8
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
