
Abstract
Hepatocyte nuclear factor alpha (HNF1α), endoplasmic reticulum (ER) stress, and hepatocyte apoptosis contribute to severe acute exacerbation (SAE) of liver injury. Here, we explore HNF1α–ER stress-hepatocyte apoptosis interaction in liver injury. LO2, HepG2 and SK-Hep1 cells were treated with thapsigargin (TG) or tunicamycin (TM) to induce ER stress. Carbon tetrachloride (CCl4) was used to induce acute liver injury in mice. Low-dose lipopolysaccharide (LPS) exacerbated liver injury in CCl4-induced mice. Significant apoptosis, HNF1α upregulation, and nuclear factor kappa B (NF-κB) activation were observed in human-derived hepatocytes during ER stress. Knockdown of Rela, NF-κB p65, inhibited the HNF1α upregulation. Following CCl4 treatment ER stress, apoptosis, HNF1α expression and RelA phosphorylation were significantly increased in mice. HNF1α knockdown reduced activating transcription factor 4 (ATF4) expression, and aggravated ER stress as well as hepatocyte apoptosis in vivo and in vitro. The double fluorescent reporter gene assay confirmed that HNF1α regulated the transcription of ATF4 promoter. LPS aggravated CCl4-induced liver injury and reduced HNF1α, and ATF4 expression. Therefore, in combination, HNF1α and ER stress could be mutually regulated forming a feedback loop, which helps in protecting the injured liver by down-regulating hepatocyte apoptosis. Low-dose LPS aggravates hepatocyte apoptosis and promotes the SAE of liver injury by interfering with the feedback regulation of HNF1α and ER stress in acute liver injury.
Similar content being viewed by others


Introduction
Severe acute exacerbation (SAE) of liver diseases is an early stage of the acute or chronic form of liver failure that often results from cirrhosis following viral infection, excessive alcohol consumption, or exposure to toxins1, 2. Despite advances in treatment strategies, patients with liver failure have high morbidity and mortality rates due to late diagnosis, severe complications in the advanced disease stage, and the limited availability of donor organs3. Therefore, understanding the mechanism of SAE in liver injury and its early symptoms could result in a timely diagnosis that could improve the patient prognosis. The SAE of liver diseases is usually attributed to various risk factors that include underlying medical conditions, bacterial or fungal infection, and alcohol consumption4, 5. In addition, the aggravation of hepatocyte damage is correlated to the severity of liver injury6. Hepatocyte death controls the development and outcome of most liver diseases7. Therefore, improving hepatocyte resistance to apoptotic signals could be crucial for maintaining liver function and delaying the SAE of liver injury. Hepatocyte nuclear factor 1α (HNF1α) and endoplasmic reticulum (ER) stress contribute to a protective response in hepatocytes8, 9, which might influence the SAE of liver injury.
ER stress is induced by various injury factors, and it is correlated to liver disease pathology10. ER stress involves the unfolded protein response (UPR) that is triggered by the aggregation of unfolded or misfolded proteins in the ER lumen11. Similarly, ER stress promotes an inflammatory response, such as the activation of nuclear factor kappa B (NF-κB)12, 13. Activated transcription factor 6 (ATF6), inositol requiring enzyme 1 (IRE1), and protein kinase R-like kinase (PERK) are crucial for the regulation of UPR14. On ATF6 activation, X-box binding protein-1 (XBP1) is upregulated15. Subsequently, XBP1 mRNA is alternatively spliced by the active IRE1 that results in the translation of the spliced XBP1 form (XBPls), which then promotes ER stress-related gene expression16. PERK activation phosphorylates the eukaryotic translational initiation factor 2 alpha (eIF2α), which attenuates the overall protein translation and decreases the ER burden17. In addition, eIF2α selectively initiates the expression of activated transcription factor 4 (ATF4), which induces glucose-regulated protein 78 (GRP78) expression. Under physiological conditions, GRP78 inhibits the activation of IRE1, PERK, and ATF6 signaling pathways18, 19. Previously, it has been reported that ER stress increases multidrug-resistance protein 2 (MRP2) expression by activating NF-κB signaling20. The upregulation of MRP2 controls acute liver injury through a negative feedback mechanism that reduces ER stress. In addition, the inhibition of eIF2α dephosphorylation reduced hepatocyte apoptosis by alleviating ER stress in acute liver injury21. In combination, the regulation of ER stress could alleviate the pathological progression of liver injury. Thapsigargin (TG) and tunicamycin (TM) are known ER stress inducers that interrupt the intracellular calcium balance and inhibit protein glycosylation in the ER cavity, respectively22.
HNF1, which is a transcription factor enriched in the liver, is crucial for glucose metabolism, detoxification, and plasma protein synthesis23. Following acute inflammation, HNF1α participates in the regulation and repair of acute liver inflammation by promoting the expression of C-reactive protein24. To date, the impact of HNF1α on ER stress in liver injury remains unclear. Therefore, the impact of HNF1α on ER stress and apoptosis in human-derived hepatocytes and mice will be investigated.
Results
Significant apoptosis, HNF1α expression, and NF-κB activation in human hepatocytes during ER stress
In vitro, the addition of 1.0 μmol/L TG significantly decreased the viability of LO2 cells at 24 and 48 h (Fig. 1A, p < 0.05). In addition, it increased the expression of HNF1α, UPR signaling proteins (ATF4, GRP78, ATF6, and XBP1s), cleaved caspase-3, and RelA phosphorylation in LO2 cells at 12, 24, and 48 h (Fig. 1B, p < 0.05). Treatment of LO2 cells with different TG concentrations (0.5, 1.0, and 2.0 μmol/L) for 24 h revealed that the TG significantly reduced the viability of the LO2 cells in a dose-dependent manner (Fig. 1C). However, the expression of HNF1α, UPR signaling proteins (ATF4, GRP78, ATF6, and XBP1s), cleaved caspase-3, and RelA phosphorylation increased (Fig. 1D, p < 0.05). In addition, the treatment of LO2 cells with 1.0 μg/mL TM, another ER stress inducer, reduced LO2 viability (Fig. 1E, p < 0.05) and significantly upregulated HNF1α, ATF4, and cleaved caspase-3 expression and RelA phosphorylation (Fig. 1F, p < 0.05). Similarly, treatment of HepG2 cells with 1.0 μmol/L TG significantly decreased HepG2 viability (Fig. 1G, p < 0.05) and increased HNF1α, ATF4, and cleaved caspase-3 expression and RelA phosphorylation (Fig. 1H, p < 0.05). Similar results were observed in SK-Hep1 cells after treatment with 0.25 μmol/L TG (Fig. 1I and J, p < 0.05).

Significant apoptosis, HNF1α expression, and NF-κB activation in human-derived hepatocytes during ER stress. Human-derived hepatocytes were treated with TG in a time and dose-dependent manner and treated with dimethyl sulfoxide (DMSO) as a control: (A) LO2 cell viability as detected by MTS assay following treatment with TG (1.0 μmol/L) at different time points; (B) western blotting showing different protein expression and bar chart for relative protein expression following treatment with TG at different time points; (C) LO2 cell viability following ER stress induction by TG for 36 h in a dose-dependent manner; (D) western blotting showing the dose-dependent induced protein expression and bar chart for relative protein expression following TG treatment for 36 h; (E) cell viability in TM-induced LO2 cells; (F) western blotting and bar chart showing relative protein expression in TM-induced LO2 cells; (G) cell viability in TG-induced HepG2 cells; (H) western blotting and bar chart showing relative protein expression in TG-induced HepG2 cells; (I) cell viability in TG-induced SK-Hep1 cells; and (J) bar chart and western blotting showing relative protein expression in TG-induced SK-Hep1 cells. *p < 0.05, **p < 0.01 versus the control group. ††p < 0.01 versus the 0.5 μmoL/L TG group. ##p < 0.01 versus the 1.0 μmol/L TG group. μM: μmol/L.
Knockdown of HNF1A aggravates apoptosis and reduces ATF4 as well as GRP78 expression during ER stress in LO2 cells
To determine the role of HNF1α in hepatocyte apoptosis and ER stress in vitro, LO2 cells were transfected with HNF1A short hairpin RNA (shRNA) and related protein expression was analyzed 48 h later. Transfection of HNF1A shRNA significantly reduced the expression of HNF1α protein (Fig. 2A, p < 0.05). HNF1A shRNA reduced the viability of LO2 cells (Fig. 2B, p < 0.05), which was more obvious after 36-h TG treatment. In addition, the expression of HNF1α, ATF4, and GRP78 proteins decreased with or without TG (Fig. 2C, p < 0.05). However, it significantly increased the expression of ATF6, XBP1s, and cleaved caspase-3, and RelA phosphorylation.

Knockdown of HNF1A aggravates apoptosis and reduces ATF4 and GRP78 expression during ER stress in LO2 cells. LO2 cells were pretreated with control shRNA or HNF1A shRNA for 48 h, then incubated with DMSO or TG (1.0 μmol/L) for 36 h: (A) bar chart and representative western blotting showing the alteration of HNF1α expression after HNF1A shRNA transfection; (B) bar chart representing MTS cell viability among the different experimental groups in LO2 cells; and (C) bar chart representing the relative protein expression and representative Western blotting for the different experimental groups. *p < 0.05, **p < 0.01, versus the control group (control shRNA, or control shRNA + DMSO) or TG group (control shRNA + TG).
A dual-luciferase reporter gene was used to detect the transcriptional regulation activity of HNF1α on ATF4 promoter. Compared with the control group, HNF1α had an enhanced transcriptional regulation on either the wild-type ATF4 promoter or mutant ATF4 promoter (p < 0.01). Of interest, the transcriptional activity of HNF1α on wild-type ATF4 promoter was stronger than that on mutant ATF4 promoter (p < 0.01, Table 1).
Knockdown of ATF4 aggravates apoptosis and downregulates the expression of GRP78 in TG-treated LO2 cells
The downregulation of ATF4 protein was confirmed 48 h post-ATF4 shRNA transfection in LO2 cells (Fig. 3A, p < 0.01). The knockdown of ATF4 significantly reduced LO2 viability (Fig. 3B, p < 0.01), downregulated the expression of ATF4, GRP78 proteins, and increased the expression of cleaved caspase-3 in LO2 cells with or without TG (Fig. 3C, p < 0.01). However, knockdown of ATF4 did not alter HNF1α expression and RelA phosphorylation.

Knockdown of ATF4 aggravates apoptosis and downregulates the expression of GRP78 in TG-treated LO2 cells. LO2 cells were pretreated with control shRNA or ATF4 shRNA for 48 h and then incubated with DMSO or TG (1.0 μmol/L) for 36 h: (A) bar chart and representative Western blotting showing the alteration of ATF4 expression in different experimental; (B) bar chart representing LO2 cell viability between the different experimental groups; and (C) bar chart representing the relative protein expression and representative western blotting.**p < 0.01, versus the control (control shRNA, or control shRNA + DMSO) or TG groups (control shRNA + TG).
Knockdown of RELA inhibits HNF1α expression in TG-treated LO2 cells
Knockdown of ATF6 expression in LO2 cells by ATF6 shRNA did not alter the expression of HNF1α protein (Fig. 4A, p > 0.05). However, knockdown of RELA reduced the expression of HNF1α protein (Fig. 4B, p < 0.01). Of note, knockdown of RELA reduced the viability of LO2 cells with or without TG treatment (Fig. 4C, p < 0.01), and decreased TG-induced the expression of HNF1α, ATF4, and GRP78 proteins, but increased the expression of cleaved caspase-3 (Fig. 4D, p < 0.01). In addition, bioinformatic analysis using JASPAR (http://jaspar.genereg.net/) predicted the presence of fifteen binding sites in HNF1A promoter for human RELA, based on the relative profile score of ≥ 80% (Table 2).

Knockdown of RELA inhibits TG-induced HNF1α expression in LO2 cells. LO2 cells were pretreated with control shRNA, ATF6 shRNA, or RELA shRNA for 48 h, and then incubated with or without TG (1.0 μmol/L) for 36 h: (A) bar chart and representative western blotting showing the alteration in ATF6 and HNF1α expression after ATF6 shRNA transfection in LO2 cells; (B) bar chart and representative Western blotting showing the alteration in RelA and HNF1α expression after RELA shRNA transfection in LO2 cells; (C) bar chart representing the impact of RELA knockdown on LO2 cell viability; and (D) bar chart representing the relative protein expression and representative western blotting between the different experimental groups. **p < 0.01, versus the control (control shRNA, or control shRNA + DMSO) or TG group (control shRNA + TG).
HNF1α expression, ER stress, and apoptosis are associated with the severity of liver injury in CCl4-induced mice
Compared with the control group (olive oil), mice that were injected with 1.0 mL/kg carbon tetrachloride (CCl4) had significantly increased levels of serum alanine aminotransferase (ALT) and total bilirubin (TBil) at different time points (12, 24, and 48 h; Fig. 5A and B, p < 0.01). The area of necrotic tissue in the liver increased significantly at 12, 24, and 48 h (Fig. 5C, p < 0.01). Compared with the control group, HNF1α expression, protein levels in the UPR pathway (ATF4, GRP78, and caspase-12), cleaved caspase-3 expression, and the phosphorylation of RelA significantly increased at 12, 24, and 48 h after CCl4 injection (Fig. 5D, p < 0.01). The peak expression of HNF1α was observed at 48 h, and the peak expression of proteins involved in the ER stress pathway was observed 24 h after CCl4 injection. Similarly, the apoptotic index was significantly elevated at 12, 24, and 48 h after CCl4 injection, peaking at 24 h (Fig. 5E, p < 0.01).

Expression of HNF1α, ER stress, and apoptosis signaling in CCl4-induced mice: (A) enzymatic rate method was used to detect the time-dependent changes in serum ALT levels in a CCl4-induced liver injury mouse model: (B) diazonium method to detect the time-dependent changes in serum TBil levels in mice; (C) bar charts representing the proportion of necrotic liver tissue area and H&E staining representing the pathological changes in the liver; (D) Western blotting reflecting the time-dependent changes in protein expression in CCl4-induced liver injury mouse model; (E) hepatocyte apoptotic index as measured by TUNEL staining; (F) dose-dependent changes in serum ALT levels detected by the enzymatic rate method; (G) dose-dependent changes in serum TBil levels detected by the diazonium method; (H) bar charts representing the proportion of necrotic liver tissue area and H&E staining representing pathological changes in the liver; (I) bar chart representing the relative protein expression levels of each protein; (J) bar charts representing the proportion of hepatocyte apoptotic index and TUNEL staining representing apoptotic changes in liver tissue; (K) immunohistochemical staining representing HNF1α expression; and (L) GEP78 expression. *p < 0.05, **p < 0.01 versus the control group. ††p < 0.01 versus the 0.5 mL/kg CCl4 group. ##p < 0.01 versus the 1.0 mL/kg CCl4 group. IHC-P: immunohistochemistry-paraffin.
Then, the impact of different CCl4 doses (0.5, 1.0, and 2.0 mL/kg CCl4) was analyzed at 24 h post-CCl4 injection. Compared with the control group, serum ALT and TBil levels as well as the necrotic tissue area significantly increased in a dose-dependent manner (Fig. 5F–H, respectively; p < 0.05). The protein expression of HNF1α, UPR signaling (ATF4, GRP78, ATF6, XBP1s, and caspase-12), cleaved caspase-3, and RelA phosphorylation increased in a dose-dependent manner in liver tissue (Fig. 5I, p < 0.01). Similarly, the apoptotic index was significantly elevated after CCl4 injection (Fig. 5J, p < 0.05). The expression of HNF1α and GRP78 was significantly upregulated in the injured liver tissue (Fig. 5K and L).
Knockdown of Hnf1a aggravates CCl4-induced liver injury, hepatocyte apoptosis and ER stress in mice
Mice were transfected with Hnf1a shRNA and rAAV8 vectors. Western blotting demonstrated the downregulation of HNF1α protein in mice liver 6 weeks after the transfection time (Fig. 6A, p < 0.01). Compared with the CCl4 group (control shRNA + CCl4), Hnf1a downregulation resulted in a significant reduction in the serum ALT level (Fig. 6B, p < 0.01), increased the serum TBil level (Fig. 6C, p < 0.01) and the necrotic liver tissue area (Fig. 6D, p < 0.01) in Hnf1a shRNA + CCl4 group. In addition, Hnf1a knockdown before CCl4 injection decreased the expression of HNF1α, ATF4, and GRP78, but significantly increased the expression of caspase-12, cleaved caspase-3, and p-RelA (Fig. 6E, p < 0.01), as well as the elevated apoptotic index (Fig. 6F, p < 0.01).

Knockdown of Hnf1a aggravates CCl4-induced liver injury, hepatocyte apoptosis, and ER stress in mice: (A) bar chart and western blotting showing the alteration in HNF1α expression in the untreated, or treated with control shRNA, or Hnf1a shRNA groups; (B) impact of Hnf1a knockdown on serum ALT levels in mice model of liver injury induced by CCl4; (C) impact of Hnf1a knockdown on serum TBil levels; (D) bar chart showing the proportion of necrotic liver tissue area and histopathological changes in mouse liver in the different experimental groups; (E) bar chart representing the relative protein expression and representative western blotting for the different experimental groups; and (F) hepatocyte apoptotic index measured by TUNEL staining. **p < 0.01 versus control (control shRNA, or control shRNA + olive oil) or CCl4 group (control shRNA + CCl4).
LPS induces liver injury, hepatocyte apoptosis, ER stress and reduces HNF1α expression in mice
The dose-dependent impact of the intraperitoneal administration of lipopolysaccharide (LPS; 0.1, 0.5, 2.5, or 5.0 mg/kg for 24 h) on hepatic injury was examined. The low dosage of LPS (0.1 and 0.5 mg/kg) did not significantly alter the serum levels of ALT and TBil (Fig. 7A and B, p > 0.05), the necrotic liver tissue area (Fig. 7C, p > 0.05), the expression of ATF4, GRP78, caspase-12, cleaved caspase-3, and p-RelA (Fig. 7D, p > 0.05), and the apoptotic index (Fig. 7E, p > 0.05) in mice. However, 2.5 and 5.0 mg/kg LPS significantly increased the serum levels of ALT and TBil post-injection (Fig. 7A and B, p < 0.01). In addition, the necrotic liver tissue area significantly increased in the 2.5 or 5.0 mg/kg LPS (Fig. 7C, p < 0.01). Similarly, the same LPS doses increased the expression of ATF4, GRP78, caspase-12, cleaved caspase-3, and RelA phosphorylation (Fig. 7D, p < 0.01), and the apoptotic index (Fig. 7E, p < 0.01), but decreased the expression of HNF1α in the liver.

LPS induces liver injury, hepatocyte apoptosis and ER stress and reduces HNF1α expression in mice: (A) chart representing the impact of different LPS doses on the level of serum ALT in mice; (B) bar chart representing the impact of different LPS doses on the level of serum TBil in mice; (C) histopathological changes in mice liver in the different experimental groups; (D) bar chart and representative western blotting detecting the impact of different LPS doses on protein expression between the different experimental groups; and (E) hepatocyte apoptotic index measured by TUNEL staining. #p < 0.05 versus the control group. **p < 0.01 versus the 0.5 mg/kg group. ††p < 0.01 versus the 2.5 mg/kg LPS group.
Low dose LPS reduces CCl4-induced HNF1α expression and increases liver injury as well as hepatocyte apoptosis in mouse liver
Compared with the CCl4 group, the addition of LPS induced a slight increase in the serum ALT level (Fig. 8A, p < 0.05), a significant increase in the serum TBil level (Fig. 8B, p < 0.01), and the necrotic liver tissue area (Fig. 8C, p < 0.01) in the LPS + CCl4 group. This combination significantly downregulated the expression of HNF1α, ATF4, and GRP78 increased the expression of caspase-12 and cleaved caspase-3, the phosphorylation of RelA (Fig. 8D, p < 0.01), and the apoptotic index in model mice (Fig. 8E, p < 0.01).

Low-dose LPS reduces CCl4-induced HNF1α expression and aggravates hepatocyte apoptosis in mouse liver: (A) effect of 0.1 mg/kg LPS on the level of serum ALT in a mouse model of liver injury induced by 1.0 mL/kg CCl4; (B) effect of 0.1 mg/kg LPS on the level of serum TBil; (C) histopathological changes in mice livers in the different experimental groups; (D) bar chart and western blotting detecting the impact of 0.1 mg/kg LPS on the expression of relative proteins; and (E) hepatocyte apoptotic index measured by TUNEL staining. *p < 0.05, **p < 0.01 versus the CCl4 group.
Discussion
In this study, we examined the regulatory mechanism of HNF1α and ER stress and their impact on apoptosis in SAE of liver injury. Our results show that ER stress inducer TG or TM treatment induced apoptosis, and increased HNF1α expression in LO2, HepG2, and SK-Hep1 cells. Similarly, the upregulation of HNF1α expression, ER stress, and hepatocyte apoptosis were observed in the liver injury mouse model induced by CCl4. In addition, knockdown of RelA significantly inhibited the upregulation of HNF1α in vitro. Taken together, these results suggest that ER stress enhanced HNF1α expression in liver injury and might be involved in activating NF-κB signaling. Furthermore, the downregulation of HNF1α in vitro showed that apoptosis was aggravated. Of interest, the proteins involved in ER stress signaling were differentially expressed: the expression of ATF4 and GRP78 was significantly downregulated while ATF6 and XBP1 expression was upregulated. Similar results were observed in vivo, as well as enhanced ER stress-related apoptosis. In addition, the double fluorescent reporter gene assay confirmed that HNF1α regulated the transcription of ATF4 promoter; knockdown of ATF4 decreased GRP78 expression and aggravated apoptosis in TG-treated LO2 cells. This interesting result implied potential crosstalk between HNF1α and ER stress through a feedback loop to alleviate hepatocyte apoptosis in liver injury.
Spontaneous peritonitis aggravated liver injury by LPS was partially simulated. The results demonstrated that LPS induced liver injury, hepatocyte apoptosis, and ER stress, but inhibited the expression of HNF1α in a dose-dependent manner. Treatment with low dosage LPS (0.1 mg/kg) selectively decreased CCl4-induced HNF1α, ATF4, and GRP78 expression and further aggravated ER stress-related hepatocyte apoptosis and liver injury. The ability of low-dosage LPS in inducing apoptosis and hence promoting the SAE of liver injury could be possibly attributed to the interference with the feedback loop between HNF1α and ER stress.
Hepatocyte degeneration and necrosis are fundamental in the pathogenesis of various liver diseases. This can cause the leakage or release of ALT, and irregular bilirubin metabolism in hepatocytes25, 26. In this study, liver injury was induced by CCl4 or LPS. Liver injury was evaluated by the levels of serum ALT and TBil, and the necrotic area of the liver tissue. In the liver, CCl4 is converted into carbon trichloride that causes oxidative stress, inflammation, and ER stress27, 28. LPS causes liver injury by activating inflammatory pathways or directly damaging hepatocytes29. In this study, CCl4 and LPS increased serum ALT and TBil levels, as well as the area of liver tissue necrosis, in a dose-dependent manner. This suggests the successful establishment of different severity degrees of liver injury models.
Hepatocyte apoptosis is closely related to liver injury30. Caspase plays a crucial role in apoptosis signal transduction31. Caspase-12 is related to ER-stress-mediated apoptosis32. Degradation of DNA into fragments of about 180–200 bp is a prominent morphological change marking apoptosis. In this study, hepatocyte apoptosis was comprehensively evaluated by analyzing the protein expression of caspase-12 and cleaved caspase-3. The apoptosis index was measured by TUNEL staining. Our results demonstrated that hepatocyte apoptosis significantly increased in CCl4-induced liver injury. Aggravation of liver injury by LPS increased hepatocyte apoptosis. This agreed with previous reports, where hepatocyte apoptosis was associated with the severity of liver injury33. Therefore, enhancing the resistance of hepatocytes towards apoptosis might be a feasible strategy to prevent liver injury.
ER stress promotes intracellular homeostasis through UPR response, but excessive ER stress activates apoptosis signaling pathways34. Simultaneously, ER stress inhibits the overall protein synthesis through PERK/eIF2α signaling and upregulates the expression of molecular chaperones, such as GRP78 through ER stress-related transcription factors, such as ATF6, ATF4, and XBP121. In this study, the expression of ATF4, ATF6, XBP1s, GRP78, and caspase-12 were analyzed to monitor ER stress. Results demonstrated that ER stress was associated with hepatocyte apoptosis and liver injury that was induced by CCl4 or LPS. In addition, TG or TM induced ER stress and apoptosis in LO2, HepG2, and SK-Hep1 cells. Accumulating evidence suggests that targeted regulation of ER stress may change the progression of liver injury by altering hepatocyte apoptosis35.
HNF1α is a transcription regulator that is essential for normal liver function36. In rats, downregulating HNF1α promoted the development of liver fibrosis and the overexpression of HNF1α significantly reduced liver fibrosis in rats37. Following acute inflammation, HNF1α regulates the repair of acute liver inflammation by promoting C-reactive protein expression38, 39. In this study, it was demonstrated that HNF1α expression increased in TG-induced ER stress. Similarly, the upregulation of HNF1α expression in CCl4-induced liver injury in mice and HNF1α expression was positively correlated with the severity of liver injury. Immunohistochemistry indicated elevated HNF1α expression and ER stress. These results imply that ER stress can induce HNF1α expression, as well as hepatocyte apoptosis, in liver injury both in vivo and in vitro.
In ER stress, IRE1, PERK/eIF2α/ATF4, and ATF6 signaling are activated which promotes the downstream signaling cascades19. ATF4 plays an essential role in stress signaling, which includes ER stress, hypoxia, amino acid deletion, and oxidative stress40. In particular, ATF4 is involved in the transcriptional regulation of amino acid synthesis, protein folding and degradation, redox balance, autophagy, and apoptosis39, 41, 42. Mutations in ATF4 significantly altered glucose homeostasis and energy consumption43 .The activation of ATF6 upregulates the expression of ER stress-related proteins, which include XBP1 and GRP78, and enhances the ability of cells to eliminate misfolded proteins44, 45. NF-κB signaling refers to a family of nuclear transcription factors, which includes RelA (NF-κB p65), RelB, c-Rel, NF-κB1/p50, and NF-κB2/p529. In this study, ATF4, ATF6 and RelA were knocked down to analyze the impact of ER stress on HNF1α. The knockdown of RelA reduced the expression of HNF1α in TG-treated LO2 cells; however, ATF4 and ATF6 knockdown did not downregulate HNF1α expression. These results suggest that the activation of NF-κB might be one of the ways by which ER stress mediates the upregulation of HNF1α.
However, to the best of our knowledge, the regulatory role of HNF1α in ER stress and apoptosis in hepatocytes is unknown. In this study, the knockdown of HNF1α increased TG-induced apoptosis in vivo. On the other hand, HNF1α knockdown aggravated the CCl4-induced liver damage as indicated by increased TBil levels, apoptosis, and necrotic liver area. Of note, serum ALT levels did not increase, which might be attributed to the regulatory role of HNF1α on numerous liver-specific genes46. ALT is a metabolic enzyme; therefore, knocking down HNF1α might decrease liver ALT levels and decrease serum ALT These results suggest that HNF1α upregulation mitigates liver injury by reducing hepatocyte apoptosis. Further, knockdown of HNF1α differentially affects the expression of ER stress-related proteins. Specifically, it significantly downregulated the expression of ATF4 and GRP78, but increased ATF6, XBP1s, and caspase-12 expression in vivo and in vitro. In addition, this confirmed that HNF1α regulated the transcription of ATF4 promoter. These results suggest that HNF1α may reduce hepatocyte apoptosis through mitigating ER stress during acute phase liver injury.
The knockdown of ATF4 reduced GRP78 levels and increased apoptosis in TG-treated LO2 cells. This suggested that ATF4-mediated GRP78 expression in ER stress is beneficial to alleviate apoptosis, which agrees with previous research47,48,49,50. These results suggest that HNF1α may reduce ER stress-mediated hepatocyte apoptosis through upregulating the expression of ATF4 and GRP78.
Viral and bacterial infections are common risk factors that are associated with severe liver injury51, 52. Endotoxins, which are the main toxic effect component is LPS, are a cell wall component of Gram-negative bacteria. Under normal physiological conditions, a small amount of LPS produced by Gram-negative bacteria in the intestine can reach the liver via the portal circulation53. Most LPS is cleared by Kupffer cells without damaging hepatocytes. Compromised intestinal barrier function increases LPS leakage and this eventually leads to hepatocyte damage54. LPS causes liver inflammation, hepatocyte apoptosis, and aggravates liver cell injury55, 56. Blood endotoxin levels are positively correlated with the degree of hepatocyte apoptosis57, 58. In this study, the effect of spontaneous peritonitis aggravated liver injury by LPS injection was partially simulated. Our results demonstrated that LPS dose-dependently induced liver injury, hepatocyte apoptosis, and ER stress, but inhibited the expression of HNF1α during liver injury. In addition, low-dose LPS did not cause liver damage, and its preintervention could alleviate the liver injury59. However, low-dose LPS aggravates the liver injury, hepatocyte apoptosis and ER stress, inhibits the mutual regulation of HNF1α and ATF4, and reduces GRP78 expression in the CCl4-induced liver injury model. Therefore, LPS treatment can have different outcomes depending on the baseline status of the liver. Under physiological conditions, low-dose LPS is not enough to cause liver damage but it significantly increases the hepatocyte sensitivity towards apoptotic signaling mediated by ER stress after liver damage has occurred. Intestinal endotoxemia is common in severe liver injury60. However, the role of low-level LPS in promoting the SAE of the liver in future studies needs to be determined.
It has been demonstrated that HNF1α expression was not correlated with chronic liver failure61. The results of this study agree with previous research and support the existence of multiple regulatory modes of HNF1α in liver injury. The expression level of HNF1α might not correlate with the degree of injury, but the effective upregulation of HNF1α in liver injury could be beneficial in controlling liver injury.
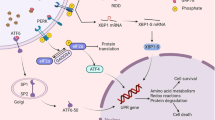
In conclusion, following liver injury, hepatocyte HNF1α and ER stress are mutually regulated to form a feedback loop, which can help reduce the severity of liver injury by regulating hepatocyte apoptosis. Whereas ER stress upregulates the expression of HNF1α by activating NF-κB signaling and the upregulated HNF1α reduces ER stress through selective regulation of ATF4 increasing GRP78 expression. In addition, high-dose LPS can directly cause liver injury, hepatocyte apoptosis as well as ER stress and inhibit the expression of HNF1α. Low-dose LPS alone cannot cause liver damage; however, it reduces the tolerance of hepatocytes to apoptosis in liver injury and leads to the deterioration of liver injury through interfering with the feedback regulation of HNF1α and ER stress (Fig. 9).

Schematic diagram representing the mechanism of suggested hepatocyte HNF1α–ER stress feedback loop on apoptosis in acute liver injury. Following liver injury, ER stress upregulates the expression of HNF1α by activating NF-κB signaling; the upregulated HNF1α reduces ER stress through positive feedback regulation with ATF4, which results in upregulation of GRP78 expression and reduction of hepatocyte apoptosis and liver injury. Low-dose LPS aggravates the CCl4 induced hepatocyte apoptosis and liver injury by inhibiting the mutual regulation of HNF1α and ER stress.
Methods
Cell culture and UPR induction
Human-derived hepatocytes LO2, HepG2, and SK-Hep1 cells were purchased from The Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China) and were maintained in RPMI-1640 that was complemented with 10% fetal bovine serum and 1% antibiotics. To induce UPR, LO2, HepG2, or SK-Hep1 cells were treated for 12, 24, and 48 h with either dimethyl sulfoxide (DMSO; TG solvent), phosphate buffer saline (PBS; TM solvent), TG (Sigma), or TM (Sigma). Alternatively, LO2 cells were treated with different concentrations of TG (0.5, 1.0, and 2.0 μmol/L) and samples were analyzed after 24 h. To analyze the impact of HNF1α downregulation in vitro, LO2 cells were transfected with the target shRNA or control shRNA using a plasmid vector according to the manufacturer protocols and protein expression was analyzed 48 h later (Table 3). Similarly, we analyzed the impact of ATF4, ATF6 and RelA downregulation using their respective target shRNA (Table 3). Then, cells were treated with TG (1.0 μmol/L) for 36 h to induce hepatic ER stress. Previous studies demonstrated that SK-Hep1 cells were more sensitive to ER stress inducers; therefore, an induction dose of 0.25 μmol/L TG was used62.
Cell viability assay
The percentage of living cells was assessed using an MTS assay, [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] assay (Promega, USA) according to the manufacturer protocol. Briefly, 100 μL of LO2, HepG2, or SK-Hep1 cells (104–105 cells/mL) were seeded in the inner wells of a 96 well plate. Each experimental condition was seeded in quadruplicate and wells around the edges were filled with sterile PBS to maintain humidity in the 96-well plate. The following day, cells underwent different interventions. At the end of the experiment, MTS assay solution was added to each well and incubated at 37 °C for 4 h. Finally, the colorimetric signal was observed at 490 nm on a microplate reader (Bio-Rad model 680; Bio-Rad, Hercules, CA, USA). The activity of the experimental control group was set to100%.
Animals and induction of liver injury
A total of 168 male BALB/c mice (25.0 ± 3.0 g) were obtained from the Animal Center of Zunyi Medical University (Guizhou, China). Mice were maintained under temperature and humidity-controlled conditions (18–22 °C and 50–60%, respectively) with food and water available ad libitum. Following acclimatization, mice were randomly assigned to different experimental groups (untreated, control, model group, and combination group; n = 12). The experimental protocol was reviewed and approved by the Animal Experimental Ethics Committee, Zunyi Medical University (ZMC-LS [2020] No. 2-321) according to the animal care and research guidelines63. This study is reported in accordance with ARRIVE guidelines.
Acute liver injury was induced by a single intraperitoneal injection of CCl4 dissolved in olive oil (CCl4: olive oil ratio = 1:4). Mice in the CCl4 model group received 1.0 mL/kg CCl4 injection for 12, 24, or 48 h whereas mice in the control group received the same volume of olive oil. Otherwise, mice in the CCl4 model group received either 0.5, 1.0, or 2.0 mL/kg CCl4 injections and the control group mice received the same dose of olive oil. Mice were not allowed to consume food or water 6 h before the injection. Alternatively, acute liver injury was induced by LPS; whereas mice in the model group were given an intraperitoneal injection of LPS (0.1, 0.5, 2.5, and 5.0 mg/kg) dissolved in PBS for 24 h. Mice in the solvent control group received the same volume of PBS and mice in the normal control group were untreated. Mice in the combination group (LPS and CCl4) received LPS (0.1 mg/kg) and CCl4 (1.0 mL/kg) for 24 h. Mice were not allowed to consume food or water 6 h before injection. The degree of liver injury was evaluated by detecting biochemical liver function indices, and histopathological changes in liver tissues. Further, liver injury was monitored at 12, 24, and 48 h. Mice were sacrificed by CO2 euthanasia. Mice were placed in a 4000-mL euthanasia box and the box chamber air was replaced with 100% CO2 at a rate of 30% of chamber volume/min64. Following the loss of consciousness, tissue and blood were harvested when blood circulation was maintained.
Knockdown of HNF1α in mice liver
Recombinant serotype 8 adeno-associated virus (rAAV8) that carried Hnf1a shRNA (target sequence: 5'-GCGATGAGCTGCCAACTAAGA-3'; Syngentech, Beijing, China; Table 4) was injected through the tail vein. Each mouse received an injection of 2 × 1010 viral genome copies that were dissolved in 0.1 mL PBS. Successful knockdown of Hnf1a was confirmed by Western blotting after 6 weeks of viral injection. Mice were randomly assigned to an untreated (normal control group; n = 12), control shRNA group (n = 12), and Hnf1a shRNA group (n = 12). Then, the role of HNF1α in liver injury was explored. Mice were randomly assigned to a control (control shRNA + olive oil; n = 12), Hnf1a shRNA (Hnf1a shRNA + olive oil; n = 12), CCl4 (control shRNA + CCl4; n = 12), Hnf1a shRNA + CCl4 group (n = 12). Mice in the control and Hnf1a shRNA groups received olive oil injections (same volume of CCl4 as previously); whereas mice in the control shRNA + CCl4 and Hnf1a shRNA + CCl4 groups received 1.0 mL/kg CCl4 injection for 36 h.
Western blotting
LO2, HepG2, SK-Hep1 cells, or liver tissues were homogenized in immunoprecipitation assay lysis buffer (R0010, Solarbio, Beijing, China). Liver lysates (40 µg) were separated on a 10% sodium dodecyl sulfate–polyacrylamide gel and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). Following blocking, membranes were probed with mouse monoclonal antibodies against ATF6 (sc-166659, 1:1000, Santa Cruz Biotechnology), eIF2α (sc-133132, 1:1000), GAPDH (glyceraldehyde-3-phosphate dehydrogenase; sc-365062, 1:1000), HNF1α (sc-393668, 1:1000), p-RelA (sc-166748, 1:1000), or RelA (sc-514451, 1:1000), rabbit monoclonal antibodies against ATF4 (11,815, 1:1000, Cell Signaling Technology), cleaved caspase-3 (9664, 1:1000, Cell Signaling Technology), GRP78 (ab108615, 1:10,000, abcam), XBP1s (40,435, 1:1000, Cell Signaling Technology), or p-eIF2α (3398, 1:1000, Cell Signaling Technology), or rabbit polyclonal antibody against caspase-12 (2202, 1:1000, Cell Signaling Technology), protein bands were detected with enhanced chemiluminescent and images were processed using Quantity One software (Bio-Rad, Hercules, CA, USA). Densitometric analysis was used to detect the level of each protein relative to the control.
Histochemical and immunohistochemical analysis
Following fixation, liver tissues were dehydrated, paraffin-embedded and sliced at 4 μm thickness. Tissue samples were stained with hematoxylin–eosin (H&E) according to standard protocols and scanned on a sliced Panoramic scanner (Pannoramic DESK/MIDI/250/1000, 3DHISTECH, Hungary) and CaseViewer2.2 software (3DHISTECH, Hungary).
Otherwise, sections were immunostained using monoclonal antibodies against HNF1α (ab272708, 1:200, abcam) and GRP78 (ab108615, 1: 200, abcam). Then, sections were visualized under a light microscope (OLYMPUS CX31). Each H&E section was independently scored by two experienced pathologists using the Histology Activity Index–Knodell score as detailed previously65.
TUNEL staining
Hepatocyte apoptosis was detected using a TUNEL kit (Roche, 11684817910). Deoxyribonucleotide terminal transferase (TdT) and deoxyribonucleotide derivative digoxigenin [(digoxigenin)-11-dUTP] were mixed at 2:28, added to the paraffin-embedded liver sections and incubated in a humidified chamber for 2 h according to standard protocols66. Positive staining was examined using a fluorescent microscope with 4′,6-diamidino-2-phenylindole (DAPI) counterstaining. The apoptotic index was calculated from six randomly selected fields according to the following formula: apoptotic index = number of positive cells/total number of cells × 100%.
Liver function indexes
Serum ALT and TBil levels were measured using the rate method and diazonium, respectively according to the standard protocols (Beckman Coulter auto-analyzer, AU5800, USA)19, 67.
Dual luciferase reporter assay
The search engine of the University of California, Santa Cruz Genomics Institute was used to find the promoter sequence of ATF4 (https://genome.ucsc.edu/). The binding site of HNF1α to the ATF4 promoter sequence was predicted through JASPAR (http://jaspar.genereg.net/). Then, the ATF4 promoter sequence was synthesized. Next and cloning it on the firefly luciferase gene, constructing as an ATF4-promoter-wt plasmid. In addition, the HNF1α binding site on the ATF4 promoter sequence was mutated to serve as the ATF4-promoter-mt plasmid. The nucleotide sequence that corresponded to HNF1A was cloned into the pcDNA3.1 vector. The pcDNA3.1 empty vector was used as a control. HEK 293FT cells (ATCC) were cultured and seeded into 24-well plates and grown for 10–24 h (80% confluence). Cells were cotransfected with the reporter gene plasmid, and the transcription factor expression plasmid, and the Renilla luciferase plasmid (an internal reference). Following cell lysis and protein extraction, luciferase activity was detected by firefly luciferase and Renilla luciferase detection reagents (Biyuntian Biotechnology, Shanghai, China) to determine the relative light unit (RLU). The RLU value (Fluc) obtained by the firefly luciferase assay was divided by the RLU (Rluc) value obtained by the Renilla luciferase assay as an internal reference. According to the obtained ratio, the activation degree of the target reporter gene between the different groups was compared68.
Statistical analysis
One sample Kolmogorov-Smirnow test was used to test whether the continuous variables satisfied a normal distribution. Quantitative data that satisfied a normal distribution were shown as mean ± standard deviation (\(\overline{\chi }\) ± SD). Differences between groups were evaluated using one-way ANOVA. If the difference was statistically significant, the Student–Newman–Keuls test was used for further comparison between the two groups. A p-value < 0.05 was statistically significant.
Ethics approval
This study was reviewed and approved by the Animal Experimental Ethics Committee, Zunyi Medical University (ZMC-LS [2020] No. 2-321) according to the animal care and research guidelines. All procedures were performed following the relevant guidelines and regulations.
Data availability
The datasets generated and analyzed during the current study are not publicly available due to none of the data types requiring uploading to a public repository but are available from the corresponding author on reasonable request.
References
Collaborators, G. B. D. C. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 5, 245–266 (2020).
Wong, G. L. et al. Increased liver stiffness measurement by transient elastography in severe acute exacerbation of chronic hepatitis B. J Gastroenterol Hepatol. 24, 1002–1007 (2009).
Samuel, D. & Coilly, A. Management of patients with liver diseases on the waiting list for transplantation: a major impact to the success of liver transplantation. BMC Med. 16, 113 (2018).
Fernandez, J. et al. Bacterial and fungal infections in acute-on-chronic liver failure: prevalence, characteristics and impact on prognosis. Gut 67, 1870–1880 (2018).
Brahmania, M. et al. Alcohol, tobacco and coffee consumption and liver disease severity among individuals with Chronic Hepatitis B infection in North America. Ann Hepatol. 19, 437–445 (2020).
Rutherford, A. & Chung, R. T. Acute liver failure: mechanisms of hepatocyte injury and regeneration. Semin Liver Dis. 28, 167–174 (2008).
Schwabe, R. F. & Luedde, T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol. 15, 738–752 (2018).
Maiers, J. L. & Malhi, H. Endoplasmic reticulum stress in metabolic liver diseases and hepatic fibrosis. Semin Liver Dis. 39, 235–248 (2019).
Tronche, F. & Yaniv, M. HNF1, a homeoprotein member of the hepatic transcription regulatory network. BioEssays 14, 579–587 (1992).
Lai, E., Teodoro, T. & Volchuk, A. Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology 22, 193–201 (2007).
Song, M. J. & Malhi, H. The unfolded protein response and hepatic lipid metabolism in non alcoholic fatty liver disease. Pharmacol Ther. 203, 107401 (2019).
Nakajima, S. & Kitamura, M. Bidirectional regulation of NF-κB by reactive oxygen species: a role of unfolded protein response. Free Radic Biol Med. 65, 162–174 (2013).
Kitamura, M. Control of NF-κB and inflammation by the unfolded protein response. Int Rev Immunol. 30, 4–15 (2011).
Johnston, B. P. & McCormick, C. Herpesviruses and the unfolded protein response. Viruses. 12, (2019).
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T. & Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 (2001).
Bae, D., Moore, K. A., Mella, J. M., Hayashi, S. Y. & Hollien, J. Degradation of Blos1 mRNA by IRE1 repositions lysosomes and protects cells from stress. J Cell Biol. 218, 1118–1127 (2019).
Proud, C. G. eIF2 and the control of cell physiology. Semin Cell Dev Biol. 16, 3–12 (2005).
Hughes, A., Oxford, A. E., Tawara, K., Jorcyk, C. L. & Oxford, J. T. Endoplasmic reticulum stress and unfolded protein response in cartilage pathophysiology; Contributing factors to apoptosis and osteoarthritis. Int J Mol Sci. 18, (2017).
Hetz, C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 13, 89–102 (2012).
Huang, W. G. et al. Endoplasmic reticulum stress increases multidrug-resistance protein 2 expression and mitigates acute liver injury. Curr Mol Med. 20, 548–557 (2020).
Tang, Y. J. et al. Inhibition of eIF2alpha dephosphorylation protects hepatocytes from apoptosis by alleviating ER stress in acute liver injury. Biomed Res Int. 2020, 2626090 (2020).
Abdullahi, A., Stanojcic, M., Parousis, A., Patsouris, D. & Jeschke, M. G. Modeling acute ER stress in vivo and in vitro. Shock 47, 506–513 (2017).
Pontoglio, M. Hepatocyte nuclear factor 1, a transcription factor at the crossroads of glucose homeostasis. J Am Soc Nephrol. 11(Suppl 16), S140-143 (2000).
Lau, H. H., Ng, N. H. J., Loo, L. S. W., Jasmen, J. B. & Teo, A. K. K. The molecular functions of hepatocyte nuclear factors - In and beyond the liver. J Hepatol. 68, 1033–1048 (2018).
Johnson, S. E. & Sherding, R. G. Diseases of the liver and biliary tract. Saunders Manual Small Anim Pract. 1, 747–809 (2006).
Liu, Z., Que, S., Xu, J. & Peng, T. Alanine aminotransferase-old biomarker and new concept: a review. Int J Med Sci. 11, 925–935 (2014).
Scholten, D., Trebicka, J., Liedtke, C. & Weiskirchen, R. The carbon tetrachloride model in mice. Lab Anim. 49, 4–11 (2015).
Shah, M. D., Gnanaraj, C., Khan, M. S. & Iqbal, M. Dillenia suffruticosa L. Impedes carbon tetrachloride-induced hepatic damage by modulating oxidative stress and inflammatory markers in rats. J Environ Pathol Toxicol Oncol. 34, 133–152 (2015).
Su, G. L. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am J Physiol Gastrointest Liver Physiol. 283, G256-265 (2002).
Canbay, A., Friedman, S. & Gores, G. J. Apoptosis: the nexus of liver injury and fibrosis. Hepatology 39, 273–278 (2004).
Szegezdi, E., Fitzgerald, U. & Samali, A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci. 1010, 186–194 (2003).
Riedl, S. J. & Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 5, 897–907 (2004).
Cao, L., Quan, X. B., Zeng, W. J., Yang, X. O. & Wang, M. J. Mechanism of hepatocyte apoptosis. J Cell Death. 9, 19–29 (2016).
Xie, R. J. et al. Calpain-2 activity promotes aberrant endoplasmic reticulum stress-related apoptosis in hepatocytes. World J Gastroenterol. 26, 1450–1462 (2020).
Olivares, S. & Henkel, A. S. Hepatic Xbp1 gene deletion promotes endoplasmic reticulum stress-induced liver injury and apoptosis. J Biol Chem. 290, 30142–30151 (2015).
Odom, D. T. et al. Control of pancreas and liver gene expression by HNF transcription factors. Science 303, 1378–1381 (2004).
Qian, H. et al. An HNF1alpha-regulated feedback circuit modulates hepatic fibrogenesis via the crosstalk between hepatocytes and hepatic stellate cells. Cell Res. 25, 930–945 (2015).
Sucajtys-Szulc, E. et al. Hepatocyte nuclear factors as possible C-reactive protein transcriptional inducer in the liver and white adipose tissue of rats with experimental chronic renal failure. Mol Cell Biochem. 446, 11–23 (2018).
Armendariz, A. D. & Krauss, R. M. Hepatic nuclear factor 1-alpha: inflammation, genetics, and atherosclerosis. Curr Opin Lipidol. 20, 106–111 (2009).
Ameri, K. & Harris, A. L. Activating transcription factor 4. Int J Biochem Cell Biol. 40, 14–21 (2008).
Zhao, X. et al. Reduction in activating transcription factor 4 promotes carbon tetrachloride and lipopolysaccharide/Dgalactosaminemediated liver injury in mice. Mol Med Rep. 18, 1718–1725 (2018).
Zhang, Z. et al. Activating transcription factor 4 increases chemotherapeutics resistance of human hepatocellular carcinoma. Cancer Biol Ther. 13, 435–442 (2012).
Hillary, R. F. & FitzGerald, U. A lifetime of stress: ATF6 in development and homeostasis. J Biomed Sci. 25, 48 (2018).
Shi, W. et al. Unravel the molecular mechanism of XBP1 in regulating the biology of cancer cells. J Cancer. 10, 2035–2046 (2019).
Oeckinghaus, A. & Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 1, a000034 (2009).
Tang, J. et al. CD147 induces UPR to inhibit apoptosis and chemosensitivity by increasing the transcription of Bip in hepatocellular carcinoma. Cell Death Differ. 19, 1779–1790 (2012).
Malhi, H. & Kaufman, R. J. Endoplasmic reticulum stress in liver disease. J Hepatol. 54, 795–809 (2011).
Kammoun, H. L. et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 119, 1201–1215 (2009).
Luo, S., Baumeister, P., Yang, S., Abcouwer, S. F. & Lee, A. S. Induction of Grp78/BiP by translational block: activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J Biol Chem. 278, 37375–37385 (2003).
Thalheimer, U., Triantos, C. K., Samonakis, D. N., Patch, D. & Burroughs, A. K. Infection, coagulation, and variceal bleeding in cirrhosis. Gut 54, 556–563 (2005).
Sun, J. et al. Gut-liver crosstalk in sepsis-induced liver injury. 24, 614 (2020).
Perez-Hernandez, E. G., Delgado-Coello, B., Luna-Reyes, I. & Mas-Oliva, J. New insights into lipopolysaccharide inactivation mechanisms in sepsis. Biomed Pharmacother. 141, 111890 (2021).
Guerville, M. & Boudry, G. Gastrointestinal and hepatic mechanisms limiting entry and dissemination of lipopolysaccharide into the systemic circulation. Am J Physiol Gastrointest Liver Physiol. 311, G1–G15 (2016).
Carpino, G. et al. Increased liver localization of lipopolysaccharides in human and experimental NAFLD. Hepatology 72, 470–485 (2020).
Nolan, J. P. & Leibowitz, A. I. Endotoxins in liver disease. Gastroenterology 75, 765–766 (1978).
Gandhi, C. R. Pro- and anti-fibrogenic functions of gram-negative bacterial lipopolysaccharide in the liver. Front Med (Lausanne). 7, 130 (2020).
Liu, D. et al. Nuclear import of proinflammatory transcription factors is required for massive liver apoptosis induced by bacterial lipopolysaccharide. J Biol Chem. 279, 48434–48442 (2004).
Kudo, H. et al. Lipopolysaccharide triggered TNF-alpha-induced hepatocyte apoptosis in a murine non-alcoholic steatohepatitis model. J Hepatol. 51, 168–175 (2009).
Kono, T. et al. Protective effect of pretreatment with low-dose lipopolysaccharide on D-galactosamine-induced acute liver failure. Int J Colorectal Dis. 17, 98–103 (2002).
Nolan, J. P. The role of intestinal endotoxin in liver injury: a long and evolving history. Hepatology 52, 1829–1835 (2010).
Guzman-Lepe, J. et al. Liver-enriched transcription factor expression relates to chronic hepatic failure in humans. Hepatol Commun. 2, 582–594 (2018).
Chen, H. et al. Alpha-fetoprotein/endoplasmic reticulum stress signaling mitigates injury in hepatoma cells. Neoplasma 68, 983–993 (2021).
Simmonds, R. C. Bioethics and animal use in programs of research, teaching, and testing. In: nd, Weichbrod RH, Thompson GAH, Norton JN, eds. Management of Animal Care and Use Programs in Research, Education, and Testing. Boca Raton (FL), 2018; 35–62.
Boivin, G. P., Hickman, D. L., Creamer-Hente, M. A., Pritchett-Corning, K. R. & Bratcher, N. A. Review of CO(2) as a Euthanasia agent for laboratory rats and mice. J Am Assoc Lab Anim Sci. 56, 491–499 (2017).
Knodell, R. G. et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology 1, 431–435 (1981).
Uddin, M. I. et al. In vivo imaging of retinal hypoxia in a model of oxygen-induced retinopathy. Sci Rep. 6, 31011 (2016).
Lippi, G. et al. Evaluation of the analytical performances of the novel Beckman Coulter AU5800. Clin Biochem. 45, 502–504 (2012).
Guo, B. et al. A ratiometric dual luciferase reporter for quantitative monitoring of pre-mRNA splicing efficiency in vivo. J Biol Chem. 297, 100933 (2021).
Funding
This study was supported by the National Natural Science Foundation of China (81560110), Tian Qing Liver Disease Research Foundation of the Chinese for Hepatitis Prevention and Control (TQGB20200001), and the Science and Technology Foundation of Guizhou Province (QKH·ZC [2019] 2803, QKH·PTRC [2017]5733-013, QKHJC-ZK[2022]YB642, and gzwjkj2020-1-041).
Author information
Authors and Affiliations
Contributions
Y.H.H., S.Y.L., and J.X.R. conceived and designed the study; S.Y.L., J.X.R., J.D., G.J.Z., X.L.J., J.C., and H.C. performed the experiments; Z.G.J. and D.L.X. provided intellectual input and supervised the study; Z.G.J., S.Y.L. and D.L.X. analyzed and interpreted the data; S.Y.L., J.X.R., and Y.H.H. wrote the manuscript, Y.H.H., Z.G.J. and D.L.X. revised the manuscript; Y.H.H. polished and finalized the manuscript. All authors made intellectual contributions to the study and approved the final version of this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, SY., Rao, JX., Deng, J. et al. Feedback loop between hepatocyte nuclear factor 1α and endoplasmic reticulum stress mitigates liver injury by downregulating hepatocyte apoptosis. Sci Rep 12, 11602 (2022). https://doi.org/10.1038/s41598-022-15846-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-15846-8
This article is cited by
-
Effects of toxicants on endoplasmic reticulum stress and hepatic cell fate determination
Toxicological Research (2023)
-
Endoplasmic reticulum stress-mediated cell death in liver injury
Cell Death & Disease (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
