
Abstract
Endoplasmic reticulum (ER) stress-associated cell death is prevalent in various liver diseases. However, the determinant mechanism how hepatocytes survive unresolved stress was still unclear. Interleukin-24 (IL-24) was previously found to promote ER stress-mediated cell death, and yet its expression and function in the liver remained elusive. Here we identified an antiapoptotic role of IL-24, which transiently accumulated within ER-stressed hepatocytes in a X-box binding protein 1 (XBP1)-dependent manner. Disruption of IL-24 increased cell death in the CCL4- or APAP-challenged mouse liver or Tm-treated hepatocytes. In contrast, pharmaceutical blockade of eukaryotic initiation factor 2α (eIF2α) or genetical ablation of C/EBP homologous protein (CHOP) restored hepatocyte function in the absence of IL-24. In a clinical setting, patients with acute liver failure manifested a profound decrease of hepatic IL-24 expression, which was associated with disease progression. In conclusion, intrinsic hepatocyte IL-24 maintains ER homeostasis by restricting the eIF2α-CHOP pathway-mediated stress signal, which might be exploited as a bio-index for prognosis or therapeutic intervention in patients with liver injury.
Similar content being viewed by others


Introduction
The liver, one of the most vital organs in metabolic homeostasis, has a unique potential to fully recover from acute liver injury. Despite recent studies elucidating various molecular pathways involved in liver damage1, a further understanding of the pivotal life-and-death decision mechanism is needed to improve current therapeutics. Endoplasmic reticulum (ER) content is rich in hepatocytes and participates in the processes of synthesizing, folding and trafficking of proteins2,3. Environmental stimuli or nutrient fluctuations disrupt the ER protein-folding procedure, referred to as ER stress4. With an accumulation of misfolded proteins in ER lumen, the unfolded protein response (UPR), a collection of intracellular signal pathways, is activated to increase protein-folding capacity and reduce global protein synthesis. Once the molecular adaption fails in resolving the protein-folding defect, hepatocytes enter persistent ER stress, which results in apoptosis5. ER stress-related apoptosis has been found in fatty liver disease, viral hepatitis, and alcohol or drug-induced liver injury3,6,7. The transcription factor C/EBP homologous protein (CHOP) mediates the most well-characterized proapoptotic pathway resulted from unresolved ER stress. CHOP induces the expression of proapoptotic BH3-only protein Bim, the cell surface death receptor TRAIL receptor 2, and inhibits Bcl2 transcription8,9,10,11. As previously reported, CHOP-deficient mice were protected from acetaminophen (APAP)-induced liver damage and conferred a survival advantage12.
Interleukin-24 (IL-24) was first identified as a negative regulator in human melanoma13,14. As an IL-10 superfamily member, IL-24 has been reported to exert a bystander anti-cancer function, but has no deleterious effect toward noncancerous cell13,15,16,17. Like other secretory proteins, IL-24 precursor, which is 206 amino acids in length, translocates to the ER lumen before it proceeds to the secretory pathway. Independently of its cognate receptors, adenovirus-mediated IL-24 overexpression in melanoma cells led to induction of apoptosis by interaction with glucose-regulated protein 78 (GRP78) and upregulation of GADD family genes, including CHOP18,19. Nonetheless, little is known about IL-24 expression and its correlation with ER stress in noncancerous cells. Interestingly, IL-24 production was elevated in diabetic pancreatic islets, where it induced beta cell ER stress and impaired glucose tolerance20. But it remains unclear whether IL-24 adapts ER homeostasis in epithelial cells.
Given the abundant IL-24 expression in the normal mouse or human liver detected in our preliminary experiments, the role of hepatocyte IL-24 in liver diseases has yet to be deciphered. To search for a possible link between IL-24 and ER stress within hepatocytes, we employed two mouse models characterizing IL-24 in the duration of acute liver injury. Unexpectedly, IL-24 deficiency did not alleviate liver damage but sensitized ER stressed hepatocytes to death. By monitoring tunicamycin (Tm)-stimulated mouse hepatocytes in vitro or manipulating the IL-24 level or UPR pathway in vivo, we further confirmed antiapoptotic function of intracellular IL-24. Indeed, we revealed that hepatocyte IL-24 governs the intrinsic adaption to ER stress by control of PERK-eIF2α-CHOP pathway. Collectively, these results highlight profound implications for understanding hepatocyte ER homeostasis and identify IL-24 as a critical anti-stress factor in the liver.
Results
Hepatocyte IL-24 transiently increases during ER stress-related acute liver injury
First, we detected the expression of IL-24 among different organs in normal wild type (WT) mice and found it was most highly expressed in the liver (Supplementary Fig. 1A). To explore whether liver IL-24 is linked to ER stress, we treated WT mice with a single dose of CCL4 (2 ml/kg) as reported previously19. Serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were markedly elevated and peaked at 48 h post treatment, then returned to baseline at 72 h time point (Supplementary Fig. 1B). In context, exposure to CCL4 unaffected the ER chaperon GRP78 but tremendously evoked CHOP expression in the liver (Fig. 1a, b). Noticeably, IL-24 mRNA level was transiently increased at 24 h, then decreased and reverted to normal level 72 h post CCL4 administration. A same trend was observed in IL-24 protein level (Fig. 1a). Regarding the potential inflammatory responses caused by CCL4, we also measured the serum IL-24 protein level. Nonetheless, CCL4-treated mice exhibited undetectable serum IL-24, which was comparable to that in none-treated WT or IL-24 KO mice (data not shown). Given the fact that hepatocytes take up the majority of hepatic cells, we asked whether the fluctuation of IL-24 expression was occurred in ER stressed hepatocytes. To answer this question, we investigated IL-24 expression in murine hepatocyte cell line AML12 in the presence of an ER stress inducer Tm. Consistent with the in vivo observation, a transient increase of IL-24 level was recapitulated in AML12 upon ER stress, as accompanied by accumulating CHOP expression (Fig. 1c, d). These results suggested a potential role of nonsecreted IL-24 and prompted us to understand how IL-24 was involved in hepatocyte ER stress.

a, b WT mice were intraperitoneally injected with a single dose of CCL4 (2 ml/kg). a GRP78, IL-24 and CHOP protein levels in the liver tissues were evaluated representatively by western blot at indicated time points. Results are normalized to β-actin. n = 3 independent experiments. b IL-24 and CHOP mRNA levels in the liver tissues were assessed by qRT-PCR at indicated time points. Results are normalized to β-actin. n = 3 biological replicates. AML12 cells were exposed to Tm (5 µg/ml) for the indicated time period. IL-24 and CHOP levels were evaluated by western blot (c) and qRT-PCR (d) at indicated time points. n = 2 independent experiments (c) or three biological replicates (d). e AML12 cells were transfected with indicated siRNAs or negative control (NC) 24 h prior to Tm treatment. IL-24 protein levels at indicated time points were assessed by western blot. n = 3 independent experiments. f Il24 promoter activity in AML12 cells expressing indicated siRNAs, as quantified using luciferase assay. Renilla luciferase activity was normalized to firefly activity and presented as relative luciferase activity. n = 3 biological replicates. g ChIP analysis of the Il24 promoter in Vector and sXBP1 overexpressing cells (Tm 0 h and 6 h) using flag antibodies. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. P values were determined by two-tailed t test.
The transcription factors activating transcription factor 4 (ATF4), ATF6, sliced X-box binding protein 1 (sXBP1) and CHOP regulate UPR-related gene expression5. Like CHOP, other three molecules were also upregulated in the CCL4-exposed mouse liver or ER stressed-AML12 cells, among which sXBP1 was the first to peak (Supplementary Fig. 1C, D). Intriguingly, the murine Il24 promoter harbors conserved binding motifs for ATF6/XBP1 and CHOP21,22(Supplementary Fig. 2A). To explore how IL-24 expression was affected in response to ER stress, we transfected AML12 cells with small interfering RNAs (siRNAs) targeting ATF4, ATF6, XBP1 and CHOP prior to Tm stimulation. IL-24 mRNA levels in these siRNA-expressing cells all decreased as compare to the negative control (NC) (Supplementary Fig. 2B), suggesting a regulatory relation between hepatocyte IL-24 and UPR pathways. Importantly, siXBP1 most significantly inhibited IL-24 mRNA level and blocked its upregulation in response to ER stress. Silencing of XBP1 (but not CHOP or ATF6) repressed IL-24 protein level as well as Il24 promoter activity in Tm-stimulated AML12 cells (Fig. 1e, f and Supplementary Fig. 2C). Besides, ChIP assays showed sXBP1 binding to Il24 promoter in AML12 cells at a basal level, which could be further enhanced by Tm treatment (Fig. 1g), Furthermore, we isolated primary hepatocytes from conditional XBP1 KO (Xbp1f/w;AlbCre) mice. XBP1 depletion unaffected cell viability under ER stress, but reduced IL-24 in both mRNA and protein levels (Supplementary Fig. 2D–F).
Hepatocyte IL-24 deficiency promotes ER stress-related liver injury
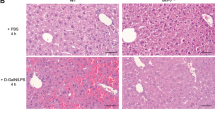
To further dissect the underlying impact of IL-24 fluctuation, IL-24 KO mice, in which 3306 bp of IL-24 allele was depleted, were subjected to CCL4-induced liver injury. In comparison to the WT mice, IL-24-null littermates were more susceptible to CCL4-induced liver injury, exhibiting a relatively higher ALT and AST level and a lower survival rate (Fig. 2a, b). The exacerbated liver damage in IL-24-null mice was visualized by hematoxylin and eosin (H&E). Meanwhile, a marked increase in the percentage of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive hepatocyte was observed in IL-24-null mice with respect to WT mice (Fig. 2c, d). Besides, proliferating cell nuclear antigen staining showed an increase of cell proliferation in IL-24 KO mice (Supplementary Fig. 3A), possibly due to compensatory liver regeneration. We then checked the inflammatory status and detected higher IL1A and IL6 and lower TNFA mRNA expression in the IL-24-deficient mouse liver (Supplementary Fig. 3B). Overdose of APAP, an analgesic and antipyretic drug, is the leading cause of drug-induced acute liver injury23. Accordingly, we subjected IL-24-null mice to oral administration of APAP, which was evident for inducing ER stress-related liver damage12. In context, worse liver function and survival rate and extensive hepatocyte death were manifested in IL-24-deficient group (Supplementary Fig. 3C–E). Given the possibility that the extracellular IL-24 might be implicated in liver injury, we treated WT mice with recombinant IL-24 (rIL-24) 1 h before administration with CCL4. Nonetheless, the levels of transaminases, percentages of hepatocyte death and expression of P-PERK and CHOP showed no statistical differences between vehicle and cytokine-treated mice (Supplementary Fig. 4A–C).

Sex- and age-matched WT and IL-24-null mice were intraperitoneally injected with vehicle or CCL4, randomly. a Mouse liver function was assessed by measuring serum ALT (left) and AST (right) levels. n = 5–8. b Mouse survival rate after CCL4-treatment was determined via Log-rank (Mantel–Cox) analysis. n = 11–13. Representatively H&E (c) and TUNEL (d) staining of the liver tissues from vehicle or CCL4-treated mice. n = 5–8 mice. Scale bar, 100 µm. e Hepatocyte apoptosis after CCL4-treatment was assessed by counting TUNEL positive cells. n = 5–8 mice. f AML12 cells were transfected with lentiviral vectors expressing IL-24-targeted sgRNA (left, referred to as IL-24 KO) or IL-24 cDNA (right, referred to as IL-24 OE). An empty vector was transfected as a negative control, respectively. Cell viability of indicated AML12 cells after Tm stimulation was assessed by CCK8 assay. n = 3 biological replicates. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. P values were determined by two-tailed t test.
To obtain a closer insight into the intrinsic IL-24 function, we genetically depleted IL-24 in AML12 cells by using clustered regularly interspersed short palindromic repeats (CRISPR)/CRISPR associated protein 9 (Cas9) strategy and treated with Tm to mimic the pathological process in vivo. Cell Counting Kit-8 (CCK8) assays indicated that loss of intrinsic IL-24 impaired cell viability. Reciprocally, overexpression of IL-24 in AML12 benefited cell survival upon ER stress (Fig. 2f). Furthermore, annexin-V and propidium iodide staining showed that IL-24 attenuated late phase of apoptosis (Supplementary Fig. 5A, B). In addition, we detected the intracellular reactive oxygen species (ROS) level. Despite ROS accumulation over time, no difference was observed between vector and IL-24 KO groups (Supplementary Fig. 8A, B). Together, these results suggested that hepatocyte IL-24 plays a fundamental role in protecting ER stress-mediated cell death.
Hepatocyte IL-24 deficiency activates PERK-eIF2α-CHOP pathway
To understand the mechanism behind hepatocyte stress, we assessed CHOP expression in both two acute liver injury models and Tm-exposed AML12 cells. Remarkably, loss of IL-24 in the mouse liver or hepatocytes unleashed CHOP expression, while introduction of IL-24 into AML12 efficiently diminished CHOP level (Fig. 3a, b and Supplementary Fig. 6A–C). In line with these findings, IL-24 depletion upregulated the expression of proapoptotic factors such as Bim and TRIB3, and yet downregulated antiapoptotic molecule Bcl2 (Supplementary Fig. 6D). To analyze how IL-24 was linked to ER stress, we further examined the UPR branches in the upstream of CHOP. Unexpectedly, either IRE1α phosphorylation or ATF6 expression appeared no difference between WT and IL-24 KO mice (Supplementary Fig. 6E). Noticeably, phosphorylation of PERK was selectively upregulated in the IL-24-deficient mouse liver or AML12 cells, and conversely downregulated upon IL-24 overexpression (Fig. 3a, b and Supplementary Fig. 6A). Accordingly, we evaluated the downstream molecules of PERK and found IL-24 deficiency robustly reinforced phosphorylation of eIF2α and expression of ATF4 and GADD34 in the stressed liver or AML12 cells, while overexpression of IL-24 in AML12 cells inhibited these molecules (Fig. 3a–e and Supplementary Fig. 6A–C). Immunohistochemical staining further confirmed higher levels of P-eIF2α and CHOP in IL-24-deficient liver (Fig. 3c). In aligned with these results, primary mouse hepatocytes isolated from IL-24-null mice manifested excessive activation of PERK-CHOP signal upon Tm-induced unresolved ER stress (Fig. 3f). Similarly, we also confirmed the activation of PERK-CHOP pathway in IL-24-deficient cells under in vitro CCL4 treatment (Supplementary Fig. 6F).

a, b P-PERK, P-eIF2α, CHOP and GRP78 protein levels in CCL4-treated WT and IL-24 KO mice, representatively (a). Tm-stimulated control and IL-24 KO AML12 cells (b), as evaluated by western blot at indicated time points. n = 3 independent experiments. c Representatively immunohistological staining of P-eIF2α and CHOP in the liver tissues from CCL4-treated WT and IL-24 KO mice. n = 5-8 mice. Scale bar, 100 µm. Results were represented in median integrated optical density (IOD) value. d GADD34, ATF4 and CHOP mRNA levels in the liver tissues from vehicle or CCL4-treated mice. n = 4-7 mice. e ATF4 and CHOP mRNA levels in indicated AML12 cells after Tm stimulation as assessed by qRT-PCR at indicated time points. f P-PERK and CHOP protein levels in Tm-stimulated primary hepatocytes from WT and IL-24 KO mouse representatively, as evaluated by western blot. n = 3 independent experiments. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. P values were determined by two-tailed t test.
Hepatocyte IL-24 selectively limits CHOP-mediated death signal
To explore whether CHOP is indispensable for IL-24 deficiency-related hepatocyte damage, we utilized siRNA targeting CHOP (siCHOP) to comprehend its executive role in ER-stressed hepatocytes. Administration of siCHOP to AML12 cells offset the marginal cell death caused by IL-24 depletion (Fig. 4a). In addition, we crossed IL-24-null mice with a CHOP-null strain to generate a double knockout (DKO) strain. In contrast to IL-24 KO counterparts, both CHOP KO and DKO mice rejected to CCL4-induced liver injury (Fig. 4b). As evident in histological staining, disruption of CHOP eliminated hepatocyte death in IL-24 KO mice (Fig. 4c). Therefore, these results suggest that IL-24 deficiency promotes hepatocyte death dependently on CHOP in the context of unresolved ER stress.

a AML12 cells were treated with siCHOP or negative control (NC). Cell viability of indicated AML12 cells after Tm stimulation was assessed by CCK8 assay. n = 3 biological replicates. b Serum ALT (upper) and AST (lower) levels in WT, IL-24 KO, CHOP KO and IL-24/CHOP double KO (DKO) mice treated with CCL4 for 24 h. n = 5–8 mice. c Representatively TUNEL staining (left) of the liver tissues from CCL4-treated mice and quantification of TUNEL positive cells (right). Scale bar, 100 µm. d IL-24 KO mice were intravenously injected with AAV particles expressing an empty vector or mouse IL-24 8 weeks prior to CCL4 administration. Liver injury was assessed by serum ALT and AST levels. n = 4 mice. e Representatively IL-24, P-PERK, P-eIF2α, CHOP and GRP78 protein levels in the liver tissues from CCL4-exposed IL-24 KO mice with or without IL-24 re-expression. n = 3 independent experiments. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. P values were determined by two-tailed t test.
To better understand the intrahepatic function of IL-24, we treated the IL-24 null mice with IL-24-expressing adeno-associated viral (AAV) particles 8 weeks prior to CCL4 administration. As shown in Fig. 4d, e, re-expression of IL-24 in the liver markedly reduced the levels of serum transferases and P-PERK, P-eIF2α and CHOP. In addition, we checked the liver inflammation and detected lower IL1A and IL6 and higher TNFA mRNA expression in the IL-24-re-expression group (Supplementary Fig. 4F).
Hepatocyte IL-24 attenuates liver damage by restricting the PERK-eIF2α branch
To ascertain the importance of PERK-eIF2α UPR branch, we built on an observation made with the inhibitor of the integrated stress response (ISRIB), which specifically blocks PERK-eIF2α signaling but unaffects ATF6 or inositol-requiring enzyme 1 (IRE1α) pathway24. Strikingly, pretreating AML12 with ISRIB compensated the viability loss for the lack of IL-24 but did not change the viability of WT cells (Fig. 5a). Next, we treated IL-24-null mice with vehicle or ISRIB 1 h prior to CCL4 administration. As indicated by serum aminotransferases, ISRIB reversed the deterioration of liver damage in IL-24-null mice but showed no profound impact on WT mice (Fig. 5b). H&E and TUNEL staining visualized an amelioration in hepatocyte death intensified by IL-24 depletion (Fig. 5c), which could be explained by the reduction of CHOP expression after ISRIB treatment (Fig. 5d).

a Indicated AML12 cells were pre-treated with 200 nM ISRIB or DMSO (Vehicle) 24 h prior to Tm exposure. Cell viability of indicated AML12 cells after Tm stimulation was assessed by CCK8 assay. n = 3 biological replicates. b, c WT and IL-24 KO mice were intraperitoneally injected with vehicle or ISRIB (0.25 mg/kg) 60 min prior to CCL4 administration. Liver injury was assessed by serum ALT and AST levels (b) and representatively TUNEL positive cell ratios (c). n = 5 mice. Scale bar, 100 µm. d Representatively immunoblotting of CHOP in the liver tissues from CCL4-exposed mice with or without ISRIB treatment. n = 3 independent experiments. e Immunoblotting of PERK in the precipitates obtained by immunoprecipitation of endogenous GRP78 in indicated AML12 cells. n = 3 independent experiments. f IL-24 KO mice were intravenously injected with AAV particles expressing an empty vector or mouse GRP78 8 weeks prior to CCL4 administration. Liver injury was assessed by serum ALT and AST levels. n = 5 mice. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. P values were determined by two-tailed t test.
It is known that the ER protein chaperon GRP78 binds to the cytoplasmic and ER luminal domains of PERK to prevent its activation25. Accordingly, we asked whether hepatocyte IL-24 affected the interaction between GRP78 and PERK. By performing immunoprecipitation, we pulled down GRP78 in AML12 cells and detected a significant binding of PERK after 6 h of Tm stimulation. In concert with the UPR degrees, its association with PERK was significantly enhanced in IL-24-deficient cells and was weakened in IL-24-overexpressed cells (Fig. 5e and Supplementary Fig. 7A). To obtain a functional relevance in vivo, we replenished chaperon expression in the mouse liver by intravenous injection of GRP78-expressing AAV. As shown in Fig. 5f and Supplementary Fig. 7B, C, overexpression of GRP78 strongly mitigated liver damage and PERK-CHOP activation in the CCL4-treated IL-24-null mice.
Hepatocyte IL-24 predicts prognosis for patients with acute liver injury
To examine the biological significance of IL-24 in clinical situations, we collected liver tissue and serum samples from 9 healthy donors, 9 patients with liver cirrhosis and 22 patients with acute liver failure (ALF). Serum IL-24 levels in both two groups of liver injury patients were as low as that in heathy donors (data not shown), which was aligned with what we found in CCL4-induced liver injury mouse models. Nonetheless, immunohistochemistry, immunoblots and qRT-PCR indicated that IL-24 expression was reduced in patients with liver cirrhosis and even lower in those with ALF (Figs. 6a–c, f and 7), whereas CHOP expression was concomitantly escalated in cirrhosis and ALF patients (Fig. 6a, f), suggestive of a strong correlation between IL-24 expression and hepatocyte stress. In a further analysis of ALF patients, we obtained the individual liver function test before liver transplant and found IL-24 expression in hepatocytes was negatively related to serum ALT level as well as liver CHOP expression (Fig. 6d, e). Taken together, hepatocyte IL-24 may function as a prognosis predictor for patients with acute liver injury.

a Representatively immunohistological staining of IL-24 and CHOP in the liver tissues from healthy donors (n = 9), cirrhosis (n = 9) and acute ALF (n = 22) patients. Scale bar, 100 µm. b Quantification of IL-24 protein levels in a, as represented in median integrated optical density (IOD) value. c IL-24 mRNA levels in the human liver tissues indicated in a, as assessed by qRT-PCR. Results are normalized to β-actin. n = 9–22 patients. Pearson correlation analysis between liver IL-24 protein level and serum ALT level (d) or liver CHOP protein level (e). n = 22. f Representatively immunoblotting of IL-24, P-eIF2α and CHOP in the liver tissues from healthy donors and acute ALF patients. n = 9 patients. Data are presented as means ± SEM. *P < 0.05, **P < 0.01. P values were determined by two-tailed t test.

Hepatocyte ER stress engages sXBP1 for upregulating IL-24 transcription, which in turn improves ER homeostasis and represses CHOP-mediated cell death by harnessing PERK-eIF2α branch reaction.
Discussion
UPR is executed through three ER transmembrane stress sensors: IRE1α, PERK and activating transcription factor 6 (ATF6)5. Activated IRE1α splices XBP1 mRNA, which encodes transcription factor to increase the protein-folding capacity and degrade misfolded proteins. While IRE1α engages STAT3 pathway to promote liver regeneration upon liver injury26, XBP1 switches pro-survival to proapoptotic signal cascades through multiple gene regulation27,28. It has been reported that ATF6 exerts a pro-inflammatory effect on ischemia-reperfusion liver injury29. PERK antagonizes UPR by reducing the flux of protein translocating and phosphorylating eIF2α, a pervasive translation initiation factor, which inhibits ribosome assembly and translation. However, eIF2α selectively upregulates the transcription factor ATF4 and its downstream target CHOP. Sustained ER stress engages CHOP to enhance UPR and inflammation signaling and lead to apoptosis22,30 (Fig. 7). The mouse models in our study manifested activation of three UPR sensors and CHOP. The latter was also found to be upregulated in human cirrhosis and ALF patients. Given the recovery of liver function in most cases, we hypothesized an unknown machinery restoring hepatocyte ER homeostasis.
A recent review has summarized the functions of IL-20 family cytokines in liver diseases31. IL-24 was suggested to function as a protective cytokine during liver inflammation through binding to its corresponding receptors. In the present study, however, administration of recombinant IL-24 did not ameliorate CCL4-induced liver damage. Noticeably, we detected abundant IL-24 expression in the mouse or human liver under normal condition. Since exogenous IL-24 in melanoma cells cause ER stress through a secretion-independent manner19, we asked whether the endogenous IL-24 in hepatocytes interacts with ER components under normal or ER-stressed conditions. In the CCL4 model, hepatocyte IL-24 increases instantaneously and then returns to baseline as the liver function recovers. Nonetheless, serum IL-24 was undetectable in CCL4-treated mice and human cirrhosis and ALF patients, excluding its engagement as a “hepatokine” in UPR condition. Importantly, targeting ER stress-related transcription factors (ATF4, ATF6, XBP1 and CHOP) significantly reduced the mRNA level of hepatocyte IL-24. However, only silencing of XBP1 deprived IL-24 expression under either normal or deleterious circumstance. As anticipated, sXBP1 directly binds to Il24 promoter and reinforces its transcription. This reflects a physiological function of hepatic XBP1 and also raises a possibility that intrinsic IL-24 may regulate stress signals.
Since adenovirus-mediated overexpression causes protein synthesis overload and induces potential UPR, we employed gene knockout mice to improve our understanding of cytosolic IL-24 in ER stress and liver damage. Remarkably, we found IL-24-null mice were more sensitive to CCL4-induced liver injury than WT counterparts. While either ATF6 expression or IRE1α phosphorylation was unaffected, P-PERK, P-eIF2α, CHOP and GADD34 exhibited excessive expression in the IL-24-deficient mouse liver. In addition, our results showed elevated levels of Bim and TRIB3 and a reduced level of Bcl2 in IL-24-null mice, which might be a consequence of CHOP activation3,11. Effects of hepatocyte IL-24 on PERK-eIF2α-CHOP branch and cell death were corroborated by depleting or introducing IL-24 in AML12 cells. Furthermore, knocking out CHOP in IL-24-null mice or knocking down CHOP in IL-24-null AML12 protected ER stress-associated hepatocyte damage. It has been reported that eIF2α phosphorylation acts as a central event sensitizing stressed cells to death12,27,29. Accordingly, we found that the extensive liver damage in IL-24-null mice could be alleviated by administration of ISRIB, which selectively reverses the effects of eIF2α phosphorylation24. Clinically, we observed a concomitant downregulation of IL-24 and upregulation of CHOP in the cirrhosis and ALF tissues as compared with the healthy liver. Taken together, these findings demonstrated the protective role of IL-24 in resolving hepatocyte ER stress is implemented by perturbation of PERK-eIF2α-CHOP pathway. However, other downstream targets of IL-24 remain to be investigated.
As one of the most important ER chaperons, GRP78 is essential in conjunction with misfolded proteins and is important for maintaining ER homeostasis8,32. Given the reinforced ER stress in IL-24-null hepatocytes, GRP78 conserved its affinity to combine and sequester overreacted PERK. Despite this, redundant PERK phosphorylated itself and triggered the downstream signaling. Previous study indicated that in vivo overexpression of GRP78 using an adenovirus vector could attenuate ER stress-associated liver steatosis33. In this study, introduction of GRP78 in the IL-24-deficient mouse liver by AAV infection attenuated PERK-facilitated hepatocyte stress. This may provide a potential therapeutic opportunity for UPR-related human liver diseases, especially those with low hepatocyte IL-24 expression.
To our knowledge, a variety of cytokines, including those expressed or secreted by hepatocytes, evoke inflammatory responses and promote cell death in liver diseases. In a diet-induced steatohepatitis mouse model, hepatocyte IL-1α was found to be upregulated in response to ER stress, which in turn enhanced CHOP expression; IL-1α released from necrotic hepatocytes accelerates steatohepatitis via induction of inflammatory cytokines34,35. In lipopolysaccharide (LPS)-induced liver injury, hepatocyte-derived IL-7 augmented CD8+T cell cytotoxic activity and promoted the development of autoimmune diseases36. In the present study, we showed that intracellular IL-24 uniquely benefited hepatocyte ER homeostasis, exerting an anti-inflammatory effect. However, it has not been defined whether hepatocyte can secrete IL-24 under pathological conditions and how it affects hepatocytes or immune cell populations in an autocrine or paracrine fashion.
Conclusively, we uncovered that cytosolic IL-24 is critical for protecting ER stressed hepatocytes from death, which may be a good diagnostic and therapeutic target for clinical liver diseases.
Materials and methods
Patients
Cirrhosis and liver failure tissues were collected from liver transplant recipients treated in Department of Liver Surgery, Renji Hospital, School of Medicine, Shanghai Jiaotong University. Normal liver tissues were collected from the healthy transplant donors through liver biopsy. Eligibility Criteria: All patients were aged ≥20 years. Liver cirrhosis criteria include: based on the patients’ laboratory tests and the findings from imaging studies, (a) ultrasonography, computed tomography and pathological evidence that revealed liver cirrhosis, splenomegaly, esophageal varices, and/or ascites; and (b) no hepatic encephalopathy, hepatocellular carcinoma or low estimated glomerular filtration rate (eGFR; <15 mL/min/1.73 m2), or acute renal failure37. ALF criteria include: (a) hepatic encephalopathy of any degree; (b) evidence of moderately severe coagulopathy (international normalized ratio (INR) greater than or equal to 1.5); (c) presumed acute illness within 26 weeks of onset of symptoms; and (d) no pathologically cirrhosis38. All samples were collected randomly with informed consent, and the experiments were approved by the ethical review committee of the World Health Organization Collaborating Center for Research in Human Production (authorized by the Shanghai Municipal Government).
Mice
All mice used in this study were in C57BL/6J background, collected randomly and no blinding was done. IL-24 knockout (KO) mice were generated in ShanghaiTech University. The targeted Embryonic stem (ES) cells were ordered from The Knockout Mouse Project (KOMP) Repository, in which the insertion of Velocigene cassette ZEN-Ub1 created a deletion of size 3306 bp between positions 132779010-132782315 of Chromosome 1 (Genome Build37). These ES cells were injected into albino C57BL/6J blastocysts and the following inbred strain was generated by backcrossing breeding. The following PCR primers were used to identify WT (571 bp) and KO (338 bp) alleles: 5′- GTACCCACTCCAATGCATACATT -3′, 5′- GCTCATCCAGGATGAAGCTACAC -3′, and 5′-GAAACCAGGCAAATCTCCACTCC -3′. The CHOP KO mice and AlbCre transgenic mice were purchased from Jackson Laboratories. We crossed the CHOP KO and IL-24 KO strains to produce the double knockout (DKO) strain. The Xbp1F/F strain, a gift from Dana Farber Cancer Institute, USA, was previously described39. Mice devoid of XBP1 selectively in hepatocytes were generated by breeding the Xbp1F/F mice with AlbCre strain. For acute liver injury model, CCL4 dissolved in olive oil was injected intraperitoneally into 8-week-old female mice at a dose of 2 ml/kg26. Besides, mice were fasted overnight and administered 500 mg/kg APAP (Sigma) by oral gavage12. In some settings, mice were intraperitoneally injected with 0.25 mg/kg ISRIB (Selleck) or recombinant IL-24 (R&D) at a dose of 5 μg per mouse 1 h prior to CCL4 administration. All mice were maintained under specific pathogen-free (SPF) conditions, on a 12 h light-dark cycle. All mouse experiments were approved by the Shanghai Administrative Committee for Laboratory Animals.
Statistical analysis
At least three biological replicates were used in each experiment unless otherwise stated. Data were analyzed with GraphPad Prism 7 and were presented as the mean ± standard error of the mean (SEM). Two-tailed Student’s t tests were performed to assess the statistical significance of differences between groups. Pearson correlation coefficients (r) were calculated to assess correlation and statistical significance was assessed by a two-tailed t test of r = 0.
Supplementary methods
More detailed methods including Isolation of primary mouse hepatocytes and cell cultures, siRNA, sgRNA and gene transfection, Recombinant AAV construction and In vivo transduction, Immunohistochemistry, Immunofluorescence, Immunoprecipitation, Western blot, Quantitative PCR, dual luciferase assay, chromatin immunoprecipitation, apoptosis and ROS analysis are provided in supplementary information.
References
Schwabe, R. F. & Luedde, T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat. Rev. Gastroenterol. Hepatol. 15, 738–752 (2018).
Dara, L., Ji, C. & Kaplowitz, N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 53, 1752–1763 (2011).
Malhi, H. & Kaufman, R. J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 54, 795–809 (2011).
Hiramatsu, N., Chiang, W.-C., Kurt, T. D., Sigurdson, C. J. & Lin, J. H. Multiple mechanisms of unfolded protein response-induced cell death. Am. J. Pathol. 185, 1800–1808 (2015).
Walter, P. & Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 (2011).
He, C. et al. ER stress regulating protein phosphatase 2A-B56gamma, targeted by hepatitis B virus X protein, induces cell cycle arrest and apoptosis of hepatocytes. Cell Death Dis. 9, 762 (2018).
Lebeaupin, C. et al. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 69, 927–947 (2018).
Mccullough, K. D., Martindale, Jl, klotz, L.-o, aw, T.-y & Holbrook, N. J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell Biol. 21, 1249–1259 (2001).
Chen, B. P. C., Wolfgang, C. D. & Hai, T. Analysis of ATF3, a transcription factor induced by physiological stresses and modulated by gadd153. Mol. Cell Biol. 16, 1157–1168 (1996).
Sylvester, S. L., Rhys, C. M. Ja, Luethy-Martindale, J. D. & Holbrook, N. J. Induction of GADD153, a CCAAT/enhancer-binding protein (C/EBP)-related gene, during the acute phase response in rats. J. Biol. Chem. 269, 20119–20125 (1994).
Puthalakath, H. et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129, 1337–1349 (2007).
Uzi, D. et al. CHOP is a critical regulator of acetaminophen-induced hepatotoxicity. J. Hepatol. 59, 495–503 (2013).
Jiang, H., Lin, J., Su, Z.-Z., Goldstein, N. I. & Fisher, P. B. Subtraction hybridization identifies a novel melanoma differentiation associated gene,mda7,modulated during human melanoma differentiation,growth and progression. Oncogene 11, 2477–2486 (1995).
Jiang, H. et al. The melanoma differentiation-associated gene mda-7 suppresses cancer cell growth. Proc. Natl. Acad. Sci. USA 93, 9160–9165 (1996).
Saeki, T. et al. Inhibition of human lung cancer growth following adenovirus-mediated mda-7 gene expression in vivo. Oncogene 21, 4558–4566 (2002).
Su, Z.-Z. et al. The cancer growth suppressor gene mda-7 selectively induces apoptosis in human breast cancer cells and inhibits tumor growth in nude mice. Proc. Natl. Acad. Sci. USA 95, 14400–14405 (1998).
Chada, S. et al. MDA-7/IL-24 is a unique cytokine-tumor suppressor in the IL-10 family. Int. Immunopharmacol. 4, 649–667 (2004).
Sarkar, D. et al. MDA-7/(IL-24) Mediates selective apoptosis in human melanoma cells by inducing the coordinated overexpression of the GADD family of genes by means of p38 MAPK. Proc. Natl. Acad. Sci. USA 99, 10054–10059 (2002).
Gupta, P. et al. BiP/GRP78 is an intracellular target for MDA-7/IL-24 induction of cancer-specific apoptosis. Cancer Res. 66, 8182–8191 (2006).
Hasnain, S. Z. et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat. Med. 20, 1417–1426 (2014).
Yamamoto, K., Yoshida, H., Kokame, K., Kaufman, R. J. & Mori, K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 136, 343–350 (2004).
Hong, F. et al. CNPY2 is a key initiator of the PERK-CHOP pathway of the unfolded protein response. Nat. Struct. Mol. Biol. 24, 834–839 (2017).
Larson, A. M. et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 42, 1364–1372 (2005).
Sidrauski, C. et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife 2, e00498 (2013).
Wang, M. & Kaufman, R. J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 14, 581–597 (2014).
Liu, Y. et al. Role for the endoplasmic reticulum stress sensor IRE1alpha in liver regenerative responses. J. Hepatol. 62, 590–598 (2015).
Han, C. Y., Lim, S. W., Koo, J. H., Won Kim & Kim, S. G. PHLDA3 overexpression in hepatocytes by endoplasmic reticulum stress via IRE1-Xbp1s pathway expedites liver injury. Gut 65, 1377–1388 (2016).
Fink, E. E. et al. XBP1-KLF9 axis acts as a molecular rheostat to control the transition from adaptive to cytotoxic unfolded protein response. Cell Rep. 25, 212–223 e214 (2018).
Rao, J. et al. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am. J. Transpl. 14, 1552–1561 (2014).
Hotamisligil, Gk. S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917 (2010).
Caparrós, E. & Francés, R. The interleukin-20 cytokine family in liver disease. Front. Immunol. 9, 1155 (2018).
Dent, P. et al. The development of MDA-7/IL-24 as a cancer therapeutic. Pharm. Ther. 128, 375–384 (2010).
Nakagawa, H. et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343 (2014).
Werman, A. et al. The precursor form of IL-1α is an intracrine proinflammatory activator of transcription. Proc. Natl. Acad. Sci. USA 101, 2434–2439 (2004).
Kandel-Kfir, M. et al. Interleukin-1alpha deficiency attenuates endoplasmic reticulum stress-induced liver damage and CHOP expression in mice. J. Hepatol. 63, 926–933 (2015).
Sawa, Y. et al. Hepatic interleukin-7 expression regulates T cell responses. Immunity 30, 447–457 (2009).
Uojima, H. et al. Furosemide dose changes associated with furosemide/tolvaptan combination therapy in patients with cirrhosis. Dig. Dis. 19, 1–8 (2019).
Speiser, J. L. et al. Predicting daily outcomes in acetaminophen-induced acute liver failure patients with machine learning techniques. Comput. Methods Prog. Biomed. 175, 111–120 (2019).
Song, M. et al. IRE1alpha-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 562, 423–428 (2018).
Acknowledgements
We thank all patients and donors for participating in this study. We thank Ms Jiaxin Li (Renji Hospital, Shanghai, China) for helping with clinical sample collection, Dr Indrajit Das (QIMR Berghofer Medical Institute, QLD, Australia) for the advice on ER stress antagonism, and Dr Laurie H. Glimcher (Dana-Farber Cancer Institute, MA, USA) for providing Xbp1F/F mice as a kind gift. This study was supported by National Natural Science Foundation of China (81672801 to J.H., 81670598 and 81972205 to Q.X. and 81700498 to Haiyan Z.), Chen Guang Project in Shanghai Municipal Education Commission and Shanghai Education Development Foundation (15CG13 to J.H. and 17CG20 to Haiyan Z.) and National Key Research and Development Program of China (2016YFC0905901 to X.H.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. M. Fimia
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Hu, B., Zhao, Z. et al. Intracellular XBP1-IL-24 axis dismantles cytotoxic unfolded protein response in the liver. Cell Death Dis 11, 17 (2020). https://doi.org/10.1038/s41419-019-2209-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-019-2209-6
This article is cited by
-
The immunological mechanisms and therapeutic potential in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity
Cell & Bioscience (2022)
-
IL-24 deficiency protects mice against bleomycin-induced pulmonary fibrosis by repressing IL-4-induced M2 program in macrophages
Cell Death & Differentiation (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}