
Abstract
Inborn errors of immunity (IEI) are a heterogeneous group of monogenic disorders that include primary immunodeficiency’s and other disorders affecting different aspects of the immune system. Next-Generation Sequencing (NGS) is an essential tool to diagnose IEI. We report our 3-year experience in setting up facilities for NGS for diagnosis of IEI in Chandigarh, North India. We used a targeted, customized gene panel of 44 genes known to result in IEI. Variant analysis was done using Ion Reporter software. The in-house NGS has enabled us to offer genetic diagnoses to patients with IEI at minimal costs. Of 121 patients who were included pathogenic variants were identified in 77 patients. These included patients with Chronic Granulomatous Disease, Severe Combined Immune Deficiency, leukocyte adhesion defect, X-linked agammaglobulinemia, Ataxia Telangiectasia, Hyper-IgE syndrome, Wiskott Aldrich syndrome, Mendelian susceptibility to mycobacterial diseases, Hyper-IgM syndrome, autoimmune lymphoproliferative syndrome, and GATA-2 deficiency. This manuscript discusses the challenges encountered while setting up and running targeted NGS for IEI in our unit. Genetic diagnosis has helped our patients with IEI in genetic counselling, prenatal diagnosis, and accessing appropriate therapeutic options.
Similar content being viewed by others
Introduction
Inborn errors of immunity (IEI) are a group of phenotypically and genetically diverse disorders characterized by monogenic defects affecting human immunity1. Patients with IEI have an increased susceptibility to infections, autoimmunity, autoinflammation, allergy, and the development of malignancies2,3. Accurate diagnosis of these conditions is essential for tailoring management protocols. Population prevalence of IEI ranges from 1:1000 to 1:10,000. With the recent discovery of several novel genetic defects, the prevalence of IEI is now believed to be much higher4,5.
Wider usage of next-generation sequencing (NGS) platforms has resulted in increased recognition of monogenic forms of IEI in recent years. According to the 2019 International Union of Immunological Societies (IUIS) classification, 424 IEI have a genetic basis6.
Diagnosing IEI in the developing world is challenging due to lack of awareness, delays in clinical presentation, and limitations in the availability of necessary diagnostic techniques7. Variations in genotype and phenotype of IEI in different regions of the world make the diagnosis even more complex and challenging. While several monogenic defects have similar clinical phenotypes, monogenic defects in the same gene can result in varied clinical phenotypes depending on the type of the variant and its functional consequences. Molecular testing, however, is an indispensable tool for diagnosing IEI with atypical presentations8.
NGS has contributed significantly in terms of defining novel genes in patients where previous approaches have been unrewarding. NGS for diagnostic purposes needs high-quality sequencing data, clinically appropriate turn-around time, and affordable cost9.
Although NGS is routinely used in immunology laboratories the world over, commissioning, installation, and effectively running such a facility for patient care in the context of a developing country was very challenging. However, we were able to convince the hospital administration at our Institute of the urgent need to establish such a facility at the Advanced Pediatrics Centre. This required several rounds of deliberations. We herein report our preliminary experience of initiating a sequencing facility to diagnose IEI using targeted NGS.
Materials and methods
Participants
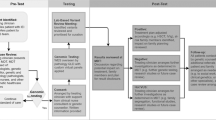
Patients were enrolled in Primary Immunodeficiency Clinic, Advanced Pediatrics Centre, Postgraduate Institute of Medical Education and Research (PGIMER), Chandigarh, India after obtaining informed assent from the children and informed consent from the parents or legal guardians. All experiments were performed in accordance with guidelines and regulations outlined by the departmental review board of Advanced Pediatrics Centre, PGIMER, Chandigarh (Vide DRB-104-21). Written Informed consent was obtained from all participants and considered mandatory for participation in the study. More than 700 patients have been diagnosed to have IEI at our centre. Patients fulfilling European Society of Immunodeficiency (ESID) diagnostic criteria for various primary immunodeficiency diseases and referred or being followed up at our centre were included in the study. Patients with both possible and probable diagnosis were included cases. Molecular diagnosis was made at our centre initially with the help of collaboration with international laboratories or by sending samples to commercial laboratories. We started performing Sanger sequencing for a few common genes for IEI in 2016. We then initiated an in-house targeted NGS facility at our centre from August 2018. We have since completed targeted NGS for 121 patients with different IEI (Fig. 1).

Evolution of molecular diagnosis of patients with IEIs at Allergy Immunology Laboratory, Advanced Pediatric Centre, Chandigarh, India.
We have included the patients in which the preliminary diagnosis of IEI was made based on clinical presentation and basic immunological investigations such as complete blood counts (CBCs), nephelometric assessment for immunoglobulins, antibody response to vaccine antigens (diphtheria and tetanus for protein antigens and pneumococcal polysaccharide for polysaccharide antigens) by enzyme-linked immunosorbent assays and lymphocyte subset analysis by flow cytometry.
Flow-cytometry evaluation
Flow-cytometry helps in the confirmation and categorization of IEIs. Multiparametric flow cytometry helps rapidly delineate and confirm many forms of IEIs in concert with genetic diagnoses. Flow cytometry for lymphocyte subsets in patients with suspected severe combined immunodeficiency (SCID) was carried out using markers—CD45, CD3, CD19, and CD56. Btk protein expression on monocytes labelled with CD14 was assayed in patients with XLA. Diagnosis of CGD was based on the dihydrorhodamine (DHR) testing using phorbol 12-myristate 13-acetate (PMA) as a stimulant. b558 (gp91phox/p22phox), p47phox and p67phox staining was subsequently carried out to sub-categorize the type of CGD. Estimation of naïve T cells (CD45RA + CD45RO–) and memory T cells (CD45RO + CD45RA−) in CD4 + and CD8 + T lymphocyte populations was done for patients with SCID. Intracellular staining of DOCK8 in lymphocytes was used to recognize patients with DOCK8 defect. The IFN-γR1 and IL12Rβ1 assays were carried out by surface staining using anti-human CD119 and CD212 (upon stimulation with a mitogen for 72 h) respectively in patients with suspected Mendelian susceptibility to mycobacterial disease (MSMD). CD18 was estimated on neutrophils in patients with leukocyte adhesion deficiency (LAD)10,11,12.
Targeted sequencing
Of the three major NGS strategies- (i.e. Whole Genome sequencing, Whole Exome sequencing and targeted panel gene sequencing), we chose the last option as it can be ubiquitously applied in clinical settings. Targeted NGS has the following advantages- (i) It provides disease-specific data with fewer variants of uncertain significance; hence, it simplifies bioinformatic analysis (ii) It ensures coverage and read depth for target genes of interest (iii) It obviates the need for expensive laboratory equipment and data storage facility13. We used the Ion Torrent S5 system from ThermoFisher Scientific for targeted NGS (details of genes in our targeted panel are listed in Table S1) in this study14,15.
We have recently paired the Ion S5 instrument with the Ion-Chef library preparation and chip-loading device in our setting. This has made sample processing less laborious, rapid and more accurate16.
Panel design
We selected genes in our panel based on available literature then, genetic databases for IEIs and gene defects most common in our cohort. We used Ion AmpliSeq Designer (ThermoFisher Scientific, USA) to design a 44 gene PID panel covering genes most commonly implicated in inborn errors of immunity. This custom panel comprised two pools of 672 amplicons (coverage summary of each gene in Table S1).
Targeted NGS library preparation and sequencing
Genomic DNA was quantified using a Qubit dsDNA HS Assay kit on QubitTM Fluorometer (ThermoFisher Scientific, USA). Five nanograms of gDNA were used for library preparation. Each sample was amplified using a custom Ion Ampliseq panel (PID 2X 2- primer pool) and HiFi mix (Thermo Fisher). PCR pools were combined for each sample and subjected to primer digestion with FuPa reagent (Thermo Fisher). The libraries were indexed; amplicons were ligated to adapters and barcodes using Ion Xpress Barcode Adapter Kit. Barcoded libraries were purified using Agencourt AMPure XP reagent (Beckman Coulter, CA) and quantified with QubitTM Fluorometer (ThermoFisher Scientific, USA). Samples were diluted and pooled for emulsion PCR.
Further, after combining the library, template preparation by emulsion PCR was done. The DNA fragment was immobilized on an Ion sphere particle (ISP) and clonally amplified. This was an automatic process performed on Ion One TouchTM 2 Instrument. This emulsion PCR results in beads with clonally amplified DNA fragments. Enrichment was done to eliminate empty beads using a robotic enrichment system (Ion One TouchTM ES). Finally, the beads containing clonal populations of DNA were obtained. Sequencing primer and polymerase were added to template positive ISPs and loaded onto Ion 530 Chip. Sequencing was done using Ion S5TM Instrument and simultaneously processed on the Ion torrent server for assembly and further analysis. The instrument was set for post-run clean-up after every run. The variant calling and analysis of results was made using Ion reporter software (ThermoFisher Scientific, USA)17,18. Large deletions/duplications were screened by Integrative Genomics Viewer (IGV) software using BAM files and confirmed by multiplex-ligation dependent probe amplification in selected cases.
Sanger sequencing
Sanger validation of the identified variants in NGS was done in 20 patients. PCR products from genomic DNA were sequenced on an automated fluorescence-based sequencer (ABI 3500, Applied BiosystemsTM; Thermo Fisher Scientific, USA) using BigDyeTM Terminator (V3.1 Applied Biosystems™). Sequencing primers were the same as those used for amplification PCR. Upon sequencing, the results were obtained in .abi format and were analyzed using Codon-code aligner for DNA sequence assembly (4.2.5/2013). The patient sequence was compared with the reference human sequence obtained from the Ensemble database (https://asia.ensembl.org/index.html)19. Variants were classified using multiple tools including Human Gene mutation Database, ClinVar, dbSNP, and VarSome20. While filtering variants, all the synonymous, intronic, common variants with 1% or higher population frequency were initially excluded. Rare variants were then individually evaluated. Multiplex ligation dependent probe amplification (MLPA) was also performed to validate the large deletion/duplication as described previously21,22. All the variants were categorized into the following five groups- Pathogenic, Likely pathogenic, Benign, Likely benign and variant of uncertain significance (VUS)23. All the variants classified as benign or pathogenic in databases were considered benign or pathogenic, respectively.
Ethics approval
The manuscript was approved by the departmental review board of the Advanced pediatrics Center, PGIMER, Chandigarh, India (Ref no. DRB-104–21).
Consent to participate
Written informed consent was obtained from participants in the manuscript, wherever required. In case of minors the consent was obtained from the legally authorised representative.
Consent for publication
Due consent taken for publication of clinical photographs and other clinical images. In case of minors the consent was obtained from the legally authorised representative.
Results
Identification and evaluation of potential variants- One hundred twenty-one (121) patients (91 males, 30 female) were analysed using targeted NGS. Representative Sanger validation data have been provided in Supplementary Fig. S1-2. We could identify pathogenic variants in 63.6% (n = 77/121) patients. Pathogenic variants identified include CYBB (n = 13), NCF2 (n = 7), CYBA (n = 1), BTK (n = 8), RAG1 (n = 3), RAG2 (n = 1), ADA (n = 3), JAK3 (n = 2), IL2RG (n = 4), ITGB2 (n = 11), ATM (n = 9), WAS (n = 2), DOCK8 (n = 1), STAT3 (n = 3), IFNGR1 (n = 2), IL12RB1 (n = 1), CD40LG (n = 2), CD40 (n = 1), GATA2 (n = 1), FAS (n = 2). Eight patients had biallelic variants in different PID genes (five ATM, one NCF2, RAG1 and JAK3 gene).
Of the thirty-three patients with suspected CGD, deleterious variants could be identified in 21 (63.6%). All had reduced or absent NADPH oxidase activity assessed by Nitroblue tetrazolium test or Dihydrorhodamine test. CYBB variants were present in 13, and NCF2 in 724.
We have analyzed 20 patients with suspected SCID. Lymphocyte subset was the first line of assessment for SCID patients. It delineated the immunological phenotype (T-B-NK+, T-B-NK-, T-B+ NK+) in these infants. Nearly 80% of infants died before genetic diagnosis. However, genetic counselling was done, and a prenatal diagnosis was offered for subsequent pregnancies. Pathogenic variants were detected in 13 patients- Four in IL2RG, three each in RAG1 and ADA, two in JAK3, and one in RAG2. This was because our panel had only seven common SCID genes. DCLER1C gene was intentionally not included in the panel as most of the patients with Artemis defect have large deletions (involving exons 1, 2 and 3 of the DCLER1C gene) likely to be missed on NGS25.
Nine patients with suspected XLA were analyzed. NGS revealed variants in 8 patients; no variant could be detected in one patient. We identified four missense, one nonsense, one frameshift, one large deletion (See Supplementary Fig. S3) and a splice-site mutation in BTK in XLA patients26.
Twelve patients with LAD were analyzed. All had been diagnosed based on clinical presentation (omphalitis, skin and soft tissue infections, delayed umbilical cord detachment, otitis media, sepsis, skin ulcer) and CD18 expression on peripheral blood leukocytes by flow cytometry. Eleven had pathogenic variants in the ITGB2 gene; 1 had no variant27. Nine patients with ataxia-telangiectasia were analyzed—all had defects in the ATM gene and presented with neurological defects and telangiectasia.
Five patients with WAS were analyzed—two had a defect in WAS gene (one stop-loss and another stop-gain variant, respectively); three had no variants. Twelve patients with suspicion of Hyper-IgE syndrome were examined for molecular defects—4 were found to have pathogenic variants (1 in DOCK8, 3 in STAT3); 7 had no variants. Laboratory investigations for patients with DOCK8 deficiency revealed eosinophilia and increased serum levels of IgE. Immunological features included low T and B cell numbers and decreased levels of serum IgM. pSTAT3 protein expression and Th17 cells were reduced in patients with STAT3 gene defects.
Nine patients with MSMD were analyzed—2 had variants in IFNGR1; 1 had IL12RB1 defect; 6 had no variants in any of the genes in our targeted panel. Four patients suspected to have Hyper-IgM were analyzed- 2 had CD40L; 1 had CD40 defect; 1 had no variant in any gene.
Four patients with ALPS were screened—2 had a germline FAS gene variant; 1 had a somatic variant in the FAS gene that was missed on initial analysis. The latter was detected when reanalyzed with a somatic pipeline. No variant was noted for the other two patients. A patient with Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) had an AIRE gene defect; this variant was not picked up by Ion reporter since there were no reads from the defined amplicon. Variant details of patients with various IEIs and corresponding flow-cytometry results have been provided in Table 1.
Discussion
Advances in genetic technology have rapidly changed healthcare delivery in low- and middle-income countries. NGS utilization has decreased the time to diagnosis, increased the diagnostic rate, and provided valuable insight into the genotype–phenotype correlation of IEI in a timely and cost-effective way28,29. IEI is not uncommon in India; however, their diagnosis is either missed or delayed due to a lack of awareness and a paucity of diagnostic facilities. There is an urgent need to increase testing capacity for early recognition, diagnosis, and management of IEI in our country30,31,32.
We have been diagnosing patients with IEI at our centre for the past 25 years. However, services for molecular diagnosis for IEI both in government and commercial sectors have not been available in India until 2016. For molecular diagnosis of IEI, we established academic collaboration with Service Hématologie, Immunologie et de Cytogénétique, Hôpital de Bicêtre, Le Kremlin Bicêtre, at France in the year 2007. Later, we established collaboration with institutes at Japan (National Defense Medical College, Saitama) and Hong Kong (Department of Paediatric and Adolescent Medicine, University of Hong Kong) in the years 2008 and 2010, respectively. This has facilitated molecular diagnosis for many of our patients with IEI. Our centre was designated as Centre for Advanced Research in diagnosis and treatment for primary immunodeficiency diseases by the Indian Council of Medical Research, Government of India, in 2015. Until 2016, tests available for diagnosis of IEI at our centre include immunoglobulin estimation, NBT, and flow cytometry for several surface and intracellular proteins10. With the increase in patients diagnosed with IEI in the last few years, we felt the need to establish molecular analysis at our centre4. We initiated Sanger sequencing for BTK, CYBB, and WAS genes in our centre in 2016 (Fig. 1).
Commercial laboratories in India came up with facilities (targeted exome) for molecular diagnosis of IEI in 2016. Costs incurred for sequencing in commercial laboratories were exorbitant (USD 400–500) in 2016 that later reduced in the subsequent years (USD 200 currently). The introduction of targeted NGS for IEI in 2018 at our centre has enabled us to offer this diagnostic modality to many of our patients who could not afford the costs of commercial testing. We have also been able to diagnose more IEIs each year and at a much faster pace than in previous years. The cost of targeted genetic sequencing at our setup is USD 83 per sample. This is much less than the costs incurred at commercial laboratories in India33. In addition, infants less than one year are covered under the JSSK (Janani Sishu Suraksha Karyakram) scheme of the Government of India. They are entitled to avail of NGS free of cost. Our Institute also provides free diagnostic services to patients from low-income groups who cannot afford the NGS charges, and charges are minimal for those who can afford this facility.
We have worked upon and improvised the standard protocol of NGS to suit our setup. We made some ingenious modifications to the recommended protocol to reduce the cost per sample and accommodate more patient samples in each run. Towards this end, we have successfully used half the recommended volume of reagents (however, concentration remained the same) at each successive step by starting with an initial DNA volume of 2.5µL instead of 5µL. So, a larger number of patient samples could be accommodated in each run. We have effectively run 42 patient samples with a 24-reaction reagent kit for 24 samples.
NGS sample preparation is a tedious and labour-intensive process requiring focus and concentration at each successive step34,35. After chip-loading and sequencing, we did not get results for two runs. On both these occasions, instead of repeating from the start, we started after the library quantification step as we were sure about the quality of the library preparation. So, restarting with the template preparation step instead of beginning from the start in the case of a failed run could be a helpful strategy if we are sure about the quality of library preparation.
We describe preliminary results of targeted NGS in 121 patients with different forms of IEIs diagnosed and managed at our centre. Our variant pick-up rate of 63.6% is much higher than previous studies- 25% by Yska et al. in 2019 and 29% by Vorsteveld et al. in 202128,36. The pick-up rate of variants in other studies were 16%7 (Gallo et al., Italy, 2016), 14% (Kojima et al., Japan, 2016)37, 2.1% (Sun et al., China in a cohort of infants)38, 28.6% (Cifaldi et al., Italy, 2020)18 and 42.4% (Arunachalam et al., India, 2020)33.
There are several reasons for a higher diagnostic yield in our study. Careful patient selection with a high pre-test probability based on clinical manifestations and preliminary immunological investigations was done. Patients with a high likelihood of having a pathogenic variant in one of 44 genes included in the gene panel are sorted out in consultation with clinicians trained in immunology and have broad experience in caring and managing patients with IEI. Currently more than 400 genes are implicated in various IEI. However, we selected 44 genes based on the most common diseases we encounter at our centre and also since we aimed to provide genetic diagnosis to maximum number of patients at an affordable cost. A large panel although more desirable would be costlier to design and in addition fewer samples would be accommodated in each run. Samples of patients who are very likely to have genetic variants in the genes included in the panel were included based on clinical history and initial immunological investigations. Patients with IEI not clearly delineated upon initial immunological investigations are referred for a clinical exome or whole-exome analysis. This analysis is outsourced to commercial laboratories providing these services at an affordable cost.
NGS has facilitated the early diagnosis of patients with IEI in situations where flow cytometry was either not conclusive or did not match the clinical presentation. For instance, patient 56 was clinically suspected of having an autosomal recessive hyper-IgM was found to have biallelic variants in the ATM gene. Hence, relying solely on typical manifestations of the IEI may not be ideal, and a rapid genetic diagnosis is indispensable39.
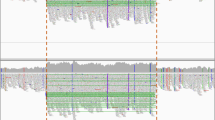
There have also been instances when the initial analysis on the Ion Reporter did not reveal a pathogenic variant. In patient 8 with clinically suspected XLA, no pathogenic variant was detected at initial analysis. There was a strong clinical suspicion of XLA in this case; we manually visualized the data on Integrative Genomics Viewer (IGV). We found a large deletion of exon-10, 11 and 12 in the BTK gene (Fig. 2)40. Similarly, in another patient with suspected CGD (Pt.27), a large deletion was found in the CYBA gene, which was missed by the ion reporter software but was detected on manual reanalysis and visualization on the IGV. Patient 42 had an indel in IL2RG gene. In patient 42, analysis by the Ion reporter software revealed two IL2RG variants in close proximity, which was confusing. However, upon visualization of the BAM file on IGV, we realized that it was an indel (insertion of 3 nucleotides and deletion of 8 nucleotides) which was misinterpreted as two variants by the ion Reporter software.

Large deletion of Exon- 10 to 12 in BTK gene on Integrative Genome Viewer.
Hence, manual data visualization on IGV and manual analysis of annotated vcf files instead of relying on variants detected by initial analysis by software is crucial. We have been able to detect these variants in these cases using this strategy.
Detection of genetic variants in genes with known pseudogene is another problem that we encountered in our patient cohort. We faced this difficulty in patients with autosomal recessive CGD due to NCF1 gene defect. The targeted NGS panel systematically missed the most common pathogenic variant in NCF1, i.e., deletion of two nucleotides at the start of Exon-2. NCF1 gene has two flanking pseudogenes (ΨNCF1)41. We assume that the amplicon designed for exon-2 of the NCF1 gene was unable to bind to its target, and thus, there was no amplification of this region, resulting in no reads for exon-2 in these patients. We performed a gene scan in 3 patients who had no reads in Exon-2 of the NCF1 gene to check for this variant and confirmed NCF1 GT deletion in all 3 of these patients (Fig. 3A,B).

(A) IGV snapshot showing no reads from Exon-2 of NCF1 gene in 2 patients with AR-CGD (B) Gene Scan for Exon 2 NCF1 gene from control and an AR-CGD patient with no reads from exon 2 of NCF1 gene.
We have also been able to offer prenatal services to many patients. Patient 40 was clinically suspected of having SCID but had expired before a genetic defect could be established. His mother was pregnant at this time, and the period of gestation was 13 weeks. We were able to identify a splice-site variant in the IL2RG gene in this family with X-linked SCID, and the mother was offered prenatal diagnosis by chorionic villous sampling. Molecular confirmation of diagnosis helped the family to get timely antenatal testing and appropriate genetic counselling. For some patients, especially SCID, rapid diagnosis through targeted NGS has saved lives, or genetic counselling has prevented an affected child in the subsequent pregnancy.
Pt 76 was the mother of a deceased child suspected to have X-linked Hyper-IgM, but a genetic diagnosis could not be established during the child’s life. Targeted NGS revealed a synonymous variant in exon 1 of the CD40LG gene proximal to donor splice-site. In-silico prediction for this variant was found to be ‘damaging’ by Mutation Taster2. Synonymous variants involving canonical splice-sites can also be pathogenic and should not be filtered out.
Genetic findings were beneficial in providing genetic counselling to affected families, carrier screening, and prenatal diagnosis. Moreover, genetic information is required for devising appropriate transplantation related strategies. Genetic findings were also crucial in deciding the treatment modalities in a few cases. Cases harbouring defects leading to antibody deficiencies were placed on regular replacement intravenous immunoglobulin therapy.
Limitations
Some apparent limitations are intrinsic to these types of studies. The list of genes involved in the pathogenesis of immune-related diseases is continuously increasing at an exponential rate, so some of the recently discovered genes (e.g., RIPK1, ICOSLG, and CYBC1) were not included in our NGS panel. Copy number variations (CNVs), as NGS adaptation to CNV testing requires additional bioinformatics and analytical efforts. It is pertinent to mention that CNVs seem to be uncommon for PID patients. However, CNV changes were very well described for IL7R and DOCK8 genes42,43.
We have also missed few variants with low coverage or absence of reads in that particular amplicon. Semiconductor-based sequencing are also fraught with inaccuracies in sequencing genomic regions with homopolymer repeats of the same nucleotide. This stems from an erroneous measurement of the magnitude of the voltage pulse in stretches of homopolymer repeats in the genome44.
Heterozygous exonic deletions could not be detected reliably using an amplicon sequencing approach. Large deletions are also not detected by NGS and may be missed unless BAM files are visually inspected on Integrative Genomics Viewer.
Another limitation of the present study is that not all genetic variants detected by NGS were validated by Sanger sequencing. While analyzing the data, we have to be cautious as no reads in an exon can be confused with deletions. In some patients, we were not able to detect any pathogenic variants. This may be due to the presence of defects in genes that are not included in our panel.
Conclusion
The attainment of NGS use would require an amalgamation of knowledge based on clinical, immunological and molecular data and association among diverse experts in these fields. A clear description of clinical phenotype and immunological test results for NGS-based diagnostics is essential for several disease-specific features. The possibility of performing pedigree analysis and immunological follow-up is an important step relevant to understanding a given patient’s disease manifestations45.
A better clinical, immunological and genetic description of new IEI will meaningfully contribute to identifying diagnostic and prognostic markers and early individual therapeutic strategies with significant benefits for patients. In summary, this study describes our nascent experience in using NGS as a tool for the genetic diagnosis of IEI and discusses the expected and unexpected findings obtained. The cases described illustrate the heterogeneity and complexity encountered by professionals involved in the clinical management and genetic diagnosis of these disorders. We have also highlighted the difficulties encountered in setting up and running this facility in the context of a developing country.
References
Bucciol, G. & Meyts, I. Recent advances in primary immunodeficiency: From molecular diagnosis to treatment. F1000Res 9, 1 (2020).
Barreto, I., Barreto, B. A. P., Cavalcante, E. & Condino Neto, A. Immunological deficiencies: More frequent than they seem to be. J. Pediatr. (Rio J.) 97(1), S49–S58. https://doi.org/10.1016/j.jped.2020.10.009 (2021).
Notarangelo, L. D., Bacchetta, R., Casanova, J. L. & Su, H. C. Human inborn errors of immunity: An expanding universe. Sci. Immunol. 5, 1. https://doi.org/10.1126/sciimmunol.abb1662 (2020).
Jindal, A. K., Pilania, R. K., Rawat, A. & Singh, S. Primary immunodeficiency disorders in India-A situational review. Front. Immunol. 8, 714. https://doi.org/10.3389/fimmu.2017.00714 (2017).
Kobrynski, L., Powell, R. W. & Bowen, S. Prevalence and morbidity of primary immunodeficiency diseases, United States 2001–2007. J. Clin. Immunol. 34, 954–961. https://doi.org/10.1007/s10875-014-0102-8 (2014).
Bousfiha, A. et al. Human inborn errors of immunity: 2019 Update of the IUIS phenotypical classification. J. Clin. Immunol. 40, 66–81. https://doi.org/10.1007/s10875-020-00758-x (2020).
Gallo, V. et al. Diagnostics of primary immunodeficiencies through next-generation sequencing. Front. Immunol. 7, 466. https://doi.org/10.3389/fimmu.2016.00466 (2016).
Nijman, I. J. et al. Targeted next-generation sequencing: a novel diagnostic tool for primary immunodeficiencies. J. Allergy Clin. Immunol. 133, 529–534. https://doi.org/10.1016/j.jaci.2013.08.032 (2014).
Rudilla, F. et al. Expanding the clinical and genetic spectra of primary immunodeficiency-related disorders with clinical exome sequencing: Expected and unexpected findings. Front. Immunol. 10, 2325. https://doi.org/10.3389/fimmu.2019.02325 (2019).
Rawat, A. et al. Flow cytometry for diagnosis of primary immune deficiencies—A tertiary center experience from North India. Front. Immunol. 10, 2111. https://doi.org/10.3389/fimmu.2019.02111 (2019).
Rawat, A. et al. Infection profile in chronic granulomatous disease: A 23-year experience from a Tertiary Care Center in North India. J. Clin. Immunol. 37, 319–328. https://doi.org/10.1007/s10875-017-0382-x (2017).
Singh, S. et al. X-linked agammaglobulinemia: Twenty years of single-center experience from North West India. Ann. Allergy Asthma Immunol. 117, 405–411. https://doi.org/10.1016/j.anai.2016.07.044 (2016).
Henson, J., Tischler, G. & Ning, Z. Next-generation sequencing and large genome assemblies. Pharmacogenomics 13, 901–915. https://doi.org/10.2217/pgs.12.72 (2012).
Bewicke-Copley, F., Arjun Kumar, E., Palladino, G., Korfi, K. & Wang, J. Applications and analysis of targeted genomic sequencing in cancer studies. Comput. Struct. Biotechnol. J. 17, 1348–1359. https://doi.org/10.1016/j.csbj.2019.10.004 (2019).
Qin, D. Next-generation sequencing and its clinical application. Cancer Biol. Med. 16, 4–10. https://doi.org/10.20892/j.issn.2095-3941.2018.0055 (2019).
Mehrotra, M. et al. Versatile ion S5XL sequencer for targeted next generation sequencing of solid tumors in a clinical laboratory. PLoS ONE 12, e0181968. https://doi.org/10.1371/journal.pone.0181968 (2017).
Bukowska-Olech, E. et al. Adapting SureSelect enrichment protocol to the Ion Torrent S5 platform in molecular diagnostics of craniosynostosis. Sci. Rep. 10, 4159. https://doi.org/10.1038/s41598-020-61048-5 (2020).
Cifaldi, C. et al. Targeted NGS Platforms for Genetic Screening and Gene Discovery in Primary Immunodeficiencies. Front. Immunol. 10, 316. https://doi.org/10.3389/fimmu.2019.00316 (2019).
Heiner, C. R., Hunkapiller, K. L., Chen, S. M., Glass, J. I. & Chen, E. Y. Sequencing multimegabase-template DNA with BigDye terminator chemistry. Genome Res. 8, 557–561. https://doi.org/10.1101/gr.8.5.557 (1998).
Kopanos, C. et al. VarSome: the human genomic variant search engine. Bioinformatics 35, 1978–1980. https://doi.org/10.1093/bioinformatics/bty897 (2019).
Tyagi, R., Aggarwal, P., Mohanty, M., Dutt, V. & Anand, A. Computational cognitive modeling and validation of Dp140 induced alteration of working memory in Duchenne Muscular Dystrophy. Sci. Rep. 10, 11989. https://doi.org/10.1038/s41598-020-68381-9 (2020).
Tyagi, R. et al. Repurposing pathogenic variants of DMD gene and its isoforms for DMD exon skipping intervention. Curr. Genom. 20, 519–530. https://doi.org/10.2174/1389202920666191107142754 (2019).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Rawat, A. et al. Clinical, immunological, and molecular profile of chronic granulomatous disease: A multi-centric study of 236 patients from India. Front. Immunol. 12, 625320. https://doi.org/10.3389/fimmu.2021.625320 (2021).
Vignesh, P. et al. Clinical, immunological, and molecular features of severe combined immune deficiency: A multi-institutional experience from India. Front. Immunol. 11, 619146. https://doi.org/10.3389/fimmu.2020.619146 (2020).
Rawat, A. et al. Clinical and genetic profile of X-linked Agammaglobulinemia: A multicenter experience from India. Front. Immunol. 11, 612323. https://doi.org/10.3389/fimmu.2020.612323 (2020).
Kambli, P. M. et al. Clinical and genetic spectrum of a large cohort of patients with leukocyte adhesion deficiency type 1 and 3: A multicentric study from India. Front. Immunol. 11, 612703. https://doi.org/10.3389/fimmu.2020.612703 (2020).
Yska, H. A. F. et al. Diagnostic yield of next generation sequencing in genetically undiagnosed patients with primary immunodeficiencies: A systematic review. J. Clin. Immunol. 39, 577–591. https://doi.org/10.1007/s10875-019-00656-x (2019).
Horton, R. H. & Lucassen, A. M. Recent developments in genetic/genomic medicine. Clin. Sci. (Lond.) 133, 697–708. https://doi.org/10.1042/CS20180436 (2019).
Qureshi, S. et al. The spectrum of primary immunodeficiencies at a tertiary care hospital in Pakistan. World Allergy Organ J. 13, 100133. https://doi.org/10.1016/j.waojou.2020.100133 (2020).
Chinn, I. K. et al. Diagnostic interpretation of genetic studies in patients with primary immunodeficiency diseases: A working group report of the Primary Immunodeficiency Diseases Committee of the American Academy of Allergy, Asthma & Immunology. J. Allergy Clin. Immunol. 145, 46–69. https://doi.org/10.1016/j.jaci.2019.09.009 (2020).
McCusker, C., Upton, J. & Warrington, R. Primary immunodeficiency. Allergy Asthma Clin. Immunol. 14, 61. https://doi.org/10.1186/s13223-018-0290-5 (2018).
Arunachalam, A. K. et al. Primary immunodeficiencies in India: Molecular diagnosis and the role of next-generation sequencing. J. Clin. Immunol. 41, 393–413. https://doi.org/10.1007/s10875-020-00923-2 (2021).
Hess, J. F. et al. Library preparation for next generation sequencing: A review of automation strategies. Biotechnol. Adv. 41, 107537. https://doi.org/10.1016/j.biotechadv.2020.107537 (2020).
Rothberg, J. M. et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 475, 348–352. https://doi.org/10.1038/nature10242 (2011).
Vorsteveld, E. E., Hoischen, A. & van der Made, C. I. Next-generation sequencing in the field of primary immunodeficiencies: Current yield, challenges, and future perspectives. Clin. Rev. Allergy Immunol. 61, 212–225. https://doi.org/10.1007/s12016-021-08838-5 (2021).
Kojima, D. et al. Application of extensively targeted next-generation sequencing for the diagnosis of primary immunodeficiencies. J. Allergy Clin. Immunol. 138, 303–305. https://doi.org/10.1016/j.jaci.2016.01.012 (2016).
Sun, J. et al. Screening for primary immunodeficiency diseases by next-generation sequencing in early life. Clin. Transl. Immunol. 9, e1138. https://doi.org/10.1002/cti2.1138 (2020).
Raje, N. et al. Utility of next generation sequencing in clinical primary immunodeficiencies. Curr. Allergy Asthma Rep. 14, 468. https://doi.org/10.1007/s11882-014-0468-y (2014).
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinf. 14, 178–192. https://doi.org/10.1093/bib/bbs017 (2013).
Dekker, J., de Boer, M. & Roos, D. Gene-scan method for the recognition of carriers and patients with p47(phox)-deficient autosomal recessive chronic granulomatous disease. Exp. Hematol. 29, 1319–1325. https://doi.org/10.1016/s0301-472x(01)00731-7 (2001).
Rae, W. et al. Clinical efficacy of a next-generation sequencing gene panel for primary immunodeficiency diagnostics. Clin. Genet. 93, 647–655. https://doi.org/10.1111/cge.13163 (2018).
Suspitsin, E. N. et al. Next generation sequencing analysis of consecutive Russian patients with clinical suspicion of inborn errors of immunity. Clin. Genet. 98, 231–239. https://doi.org/10.1111/cge.13789 (2020).
Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351. https://doi.org/10.1038/nrg.2016.49 (2016).
Elsink, K. et al. National external quality assessment for next-generation sequencing-based diagnostics of primary immunodeficiencies. Eur. J. Hum. Genet. 29, 20–28. https://doi.org/10.1038/s41431-020-0702-0 (2021).
Acknowledgements
We would acknowledge all our patients and their family members for participating in the present study. We thank our present and pass-out DM residents, Research scholars, PhDs and technical staff for extending their cooperation and support in sample selection. We extend our gratitude to Dr Osamu Ohara (Kazusa DNA Research Institute, Chiba, Japan), Dr Yu Zhang (NIH, USA) and Dr Sudhir Gupta (University of California at Irvine, CA, USA) for motivating and helping us starting this type of study at our centre. We are thankful to Dr Andrew Oler, NIAID, NIH for his support in this work. The authors gratefully acknowledge the support provided by the Indian Council of Medical Research for fellowship grant [3/1/3/JRF-2016/LS/HRD-56(10589)] and Department of Health Research, Government of India.
Funding
The study was supported by the Department of Health Research, Government of India (GIA/48/2014-DHR).
Author information
Authors and Affiliations
Contributions
M.S.: Performed experiments, analysed data and prepared initial draft of manuscript. V.P., D.S., A.J.: patient management, clinical evaluation and enrolment of patients. J.D., V.J., R.T., J.S., G.K.: Performed experiment and prepared the manuscript. Y.L.L., K.I., S.N., M.L.: scientific discussion, data interpretation and revised the manuscript. A.R., S.S.: Initiated, designed, critical revision and intellectual inputs in the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rawat, A., Sharma, M., Vignesh, P. et al. Utility of targeted next generation sequencing for inborn errors of immunity at a tertiary care centre in North India. Sci Rep 12, 10416 (2022). https://doi.org/10.1038/s41598-022-14522-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-14522-1
This article is cited by
-
Clinical and Immunological Features, Genetic Variants, and Outcomes of Patients with CD40 Deficiency
Journal of Clinical Immunology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
