
Abstract
Impacts of climate changes are particularly severe in polar regions where warmer temperatures and reductions in sea-ice covers threaten the ecological integrity of marine coastal ecosystems. Because of their wide distribution and their ecological importance, mussels are currently used as sentinel organisms in monitoring programs of coastal ecosystems around the world. In the present study, we exploited the concept of liquid biopsy combined to a logistically friendly sampling method to study the hemolymphatic bacterial microbiome in two mussel species (Aulacomya atra and Mytilus platensis) in Kerguelen Islands, a remote Subantarctic volcanic archipelago. We found that the circulating microbiome signatures of both species differ significantly even though their share the same mussel beds. We also found that the microbiome differs significantly between sampling sites, often correlating with the particularity of the ecosystem. Predictive models also revealed that both species have distinct functional microbiota, and that the circulating microbiome of Aulacomya atra was more sensitive to changes induced by acute thermal stress when compared to Mytilus platensis. Taken together, our study suggests that defining circulating microbiome is a useful tool to assess the health status of marine ecosystems and to better understand the interactions between the sentinel species and their habitat.
Similar content being viewed by others
Introduction
Climate changes are known to alter biodiversity at a global scale1,2. This is particularly true for Subantarctic regions, which have undergone significant changes in recent years due to global warming3,4,5. Sea surface temperatures in the Western Antarctic Peninsula have increased by ~ 1 °C since the industrial revolution and is expected to accelerate with the constant augmentation of global CO2 concentrations, as suggested by predictive models6,7. The consequences of the global warming include alterations of the interactions between hosts and their associated microbiomes (i.e., assemblages of microorganisms)8,9,10. Such dysbiosis could not only affect marine ecosystems per se, but also favors the emergence and proliferation of opportunistic pathogens11,12. Emerging infectious disease outbreaks in marine ecosystems would not only impact directly on the host populations but can also lead to mass mortalities of keystone species9,13. This is particularly true for sedentary and filtrating marine organisms such as mussels and oysters that are more prone to frequent, episodic mortality events during heat waves in summer14,15,16. Moreover, bivalves, which play a central role in an ecosystem integrity, are the most widely used sentinel species to monitor coastal marine environments as they are sedentary and have high filtering capacities leading to accumulation of pathogens and pollutants17,18. Their high filtering activity links them to their surrounding environment. Many marine mussel-based biomarkers have thus been developed over the years for monitoring environmental quality and for assessing environmental variations, particularly in remote areas such as polar regions17,19,20.
The development of ethical (non-lethal) and logistically friendly sampling methods combined with the concept of liquid biopsy offers, however, a new window of opportunity to develop highly sensitive and predictable biomarkers21. Liquid biopsy, combined with high throughput DNA sequencing of circulating plasmatic DNA fragments, is commonly used in the biomedical field for decision-making in patients with cancer22. More recently, it has also been shown to be a useful mean to measure the presence of DNA fragments of non-self origin even in absence of overt diseases23. This has led to a novel concept, the “circulating microbiome DNA” (cmDNA)24. In contrast to tissue-specific microbiome analysis, characterization of the cmDNA allows to obtain a signature that reflects bacterial populations derived from classical niches (such as the gut, skin, oral cavity, etc.). This is also true in sentinel species, such as bivalves, which hemolymphatic cmDNA contains a rich and diverse microbiome and can be used as biomarkers to reflect host fitness and health status25,26,27,28,29. In fact, characterization of microbiomes in general has become an important tool to measure the impact of environmental and anthropogenic stressors in health and resilience of marine coastal ecosystems29. For example, in the case of M. galloprovincialis, in vivo exposure to polystyrene nanoplastics significantly affects different hemolymph immune parameters and a shift the ciruclating microbiome27.
The coastal marine ecosystems of the Subantarctic Islands of Kerguelen (49°26′S, 69°50′E) are inhabited by two mussel species: the Subantarctic population of blue mussels, Mytilus platensis (M. platensis), and the ribbed mussel Aulacomya atra (A. atra). The isolated archipelago of Kerguelen is part of an oceanic submerged plateau that was built 35 million years ago following continuous volcanic activity. This archipelago is home to a maritime nature reserve classified as a UNESCO World Heritage Site since 2019. It is also a central hub for research on marine biodiversity in the Southern Ocean30. Recent observations indicate that Kerguelen Plateau is also characterized by the submarine gas emission that may represents active volcanic activity associated with cold seeps and hydrothermal vents31. This makes Kerguelen a unique site to measure the impact of climate changes on marine coastal habitats. In the present work, we report our findings of the first study of the hemolymphatic circulating microbiome collected in the two mussel species that inhabit mixed mussel beds located at different sites of the Kerguelen Islands.
Results
Species-dependent variations in bacterial communities
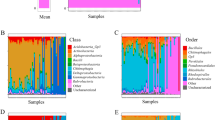
A total of 150 samples of M. platensis (all from the intertidal zone) and A. atra (intertidal and subtidal) were analyzed. For each sample, the cmDNA was profiled by sequencing the 16S rRNA gene amplicons of the V3–V4 hypervariable region. A total of 6,489,570 paired-end sequences passed quality filtering (43,264 ± 27,652 per sample). Amplicon sequence variants (ASVs) were generated from 21,389 high-quality reads. Overall, the relative abundance of bacterial phyla in both mussel species were dominated by Proteobacteria (56.0% and 41.6% for A. atra and M. platensis respectively), Bacteroidetes (14.8% and 14.2% respectively), and Parcubacteria (7.2% and 8.3% respectively), which accounted for more than 60% of all reads (Fig. 1). This composition is consistent with a recent metagenomic study showing that bacteria enriched in seawater in polar regions were mostly Proteobacteria, Actinobacteria, Bacteroidetes and Parcubacteria32. Multivariate analysis (PERMANOVA) on a weighted UniFrac showed, however, that the global composition of the microbiome was significantly different between A. atra and M. platensis (weighted UniFrac PERMANOVA, F(1, 154) = 5.02, p < 0.001). A case in point at the phylum level is the presence of Acidobacteria (4%), Chloroflexi (4%), Spirochaetes (2%), Tenericutes (3%), and Verrucomicrobia (3%) in M. platensis but not in A. atra. In contrast, Fusobacteria (4%) were more abundant in A. atra in the intertidal zone. This phylum is commonly found in marine sediment environments but has been reported to constitute the major phylum in oil-contaminated anaerobic niche of seawater following the deep-water horizon oil spill and other marine reservoirs33,34. Interestingly, the highest abundance of Fusobacteria was detected at Port-aux-Français, the capital settlement of Kerguelen Islands and the only site with active port activity (Supplementary Figure S1). Furthermore, no significant differences in the composition of the microbiome were found in A. atra between subtidal and intertidal zones (weighted UniFrac PERMANOVA, F(1, 88) = 1.49, p = 0.103)).

Phylum-level analysis of hemolymphatic bacterial DNA. (A) M. platensis and (B) A. atra collected in intertidal zones. (C) of A. atra collected in subtidal zones. Phylum with a relative abundance of ≤ 1.5% are represented as "Other".
Site-dependent beta diversity analysis
We next examined spatial variations in the cmDNA profiles of both mussel species. For this, we collected samples from 16 sites, including 5 mixed mussel beds (Port-aux-Français, Port-Couvreux, Île Haute, Île Haute-Baie des Bergers, and Anse-aux-Écueils) and 5 subtidal sites where samples of A. atra were collected (Fig. 2). To compare β-diversity between sampling sites, principal coordinate analysis (PCoA) based on the weighted UniFrac distance was carried out for both species (Fig. 3). Multivariate analysis revealed a significant separation of samples according to sampling sites. An ADONIS on weighted UniFrac analysis showed a significant difference according to sampling sites (p < 0.001) for both species. For M. platensis, distances to group centroids were significantly different (Permutest, F(10, 55) = 4.83, p = 0.002) in samples collected at Anse de Saint-Malo and Anse Sablonneuse (Fig. 3A). In the case of A. atra, the betadisper analysis highlights four distinct microbial sites where the community structure was significantly higher (Permutest, F(13, 75) = 3.32, p = 0.002); Port-Couvreux (intertidal), Îlot Channer (intertidal), Anse-aux-Écueils (intertidal) and Anse-du-Halage (subtidal) showed a strong separation with other sites.

Map of the French Subantarctic Kerguelen Islands showing the sampling sites. The map was generated with the ggmap package in R programming language (R version 4.0.3, R Core Team88).

Principal Coordinates Analysis (PCoA) of bacterial DNA bacterial communities at different sites. Weighted UniFrac-based of in samples collected in M. platensis (A), A. atra in intertidal (B) and subtidal (C) zones. Centroids for each site are illustrated by larger circles. Black ellipses represent 90% confidence interval.
Site-dependent alpha diversity analysis
We next compared the alpha diversity for each site using three diversity indices including richness, Shannon index, and Pielou’s evenness. Globally, we found no significant differences in the cmDNA for both mussel species collected during two successive campaigns at Kerguelen (2017 and 2018) (Supplementary Figure S2). A comparative analysis of the alpha diversity according to sites revealed, however, significant differences between sites for both species (Fig. 4). In the case of M. platensis, the richness estimator showed a significant variation (χ2 = 18.977, p = 0.0401, df = 10). The lowest richness value was observed at Anse Sablonneuse (65.7 ± 9.9), a mussel bed occurring in mid to lower shore sand. In the case of A. atra, richness estimator (χ2 = 38.779, p < 0.001), Shannon index (χ2 = 37.996, p < 0.001) and Pielou’s evenness (χ2 = 36.647, p < 0.001) varied greatly between sites for both intertidal and subtidal zones. Lowest alpha diversity indices in A. atra sites were observed at Port-Couvreux (intertidal) and Île Longue (subtidal).

Alpha diversity analysis. Box plots of alpha diversity of (A) M. platensis collected in intertidal zone and A. atra in intertidal (B) and subtidal (C) zones. Species evenness observed richness and Shannon diversity indexes were calculated for each site. Significant p values (p < 0.05) were obtained using permutation test as a non-parametric test following by a post-hoc pairwise comparisons.
Site-dependent variations at the genus level
Differences between sites for both species were further documented at the genus level, focusing first on the top 30 genus, which represent 32% of all genera. Among the notable findings, we found strong abundance of Pseudoalteromonas and Psychromonas in M. platensis collected at the Anse Sablonneuse (Fig. 5). The cmDNA of M. platensis, but not A. atra, collected in the intertidal zone of the Îlot Channer, an islet formed of basaltic rocks surrounded by a belt of large brown algae (Durvillea and Macrocystis), was dominated by Aquabacterium, a common bacteria found within kelps35. In the case of A. atra, we found a relative abundance of Sphingomonas at several sites, including Port-Couvreux, Ile Longue, Île du Suhm and Portes-Noires which accounted for 31.5–57.7% of total reads (Supplementary Table S1). This genus dominance could be attributed to the significant occurrence of kelp (Macrocystis pyrifera) at these sampling sites. This is in agreement with Florez et al.36 who reported that epiphytic bacterial communities in macroalgae are associated with Sphingomonadales/Sphingomonas are known to degrade alginate as carbon source37,38. It was also worth noting that A. atra collected at the intertidal zone of the fjord of Portes Noires showed quite a unique abundance of Acidovorax, of which several species are phytopathogenic, and Parvularcula, compared to any other sites or to M. platensis. Interestingly, abundance of Vibrio was higher in A. atra in both intertidal and subtidal mussels of Port-aux-Français.

Heatmaps showing relative abundance (%) of the top 30 bacterial genera of hemolymphatic microbiota between in both mussel species. Red colors indicate higher abundance and blue colors indicate lower abundances.
The differences between sites were further studied at the genus level by focusing this time on the phyla that differed between both species. The most significant difference between M. platensis and A. atra was observed at Port-Couvreux (Supplementary Figure S3). We also found that bacterial DNA from the genus Kistimonas, a member of the Hahellacae family of Proteobacteria, was not detected in A. atra at any site but was present in M. platensis at Port-Couvreux, Île Haute, Île Haute-Baie du Berger, and Anse-aux-Écueils. Bacterial DNA from the genus Sulforimonas was also commonly found in M. platensis at several sites but rarely in A. atra. Overall, these results showed that both mussel species have distinct circulating microbiome profiles that vary according to the geographic locations of the mussel beds. Given the fact that season changes may play a major role in shaping bacterial community structure in bivalves, further investigations are needed to compare seasonal variability in the microbiome between both mussel species39,40.
Environmental microbiota can also be influenced by anthropogenic sources. This could explain why the relative abundance of bacteria of the Vibrio genus were more abundant at Port-aux-Français (in both intertidal and subtidal zones) in A. atra than at other sampling sites (Supplementary Table S1). LEfSe analysis indeed revealed that Vibrio was the top genus-level biomarkers that distinguished Port-aux-Français from all other sites (Supplementary Table S2). This finding is consistent with a previous study showing that anthropogenic impacts are higher at Port-aux-Français when compared to other sites41. In Mediterranean Sea, the proliferation of opportunistic pathogens affiliated to Vibrio spp. is suspected to be a major factor in disease outbreaks in bivalve species42,43. Moreover, Vibrio spp. encompasses a variety of pathogens whose presence within bivalve tissues and interactions with their immune system are well documented44. Taken together, our results indicate that hemolymph cmDNA profiles reflect the local environment and the impacts of anthropogenic stressors.
Functional analysis of the cmDNA
Predictive functional analysis from 16 s rRNA profiling is becoming a common tool to link the abundance of specific taxa with metabolic profiles45,46,47. To further explore the distinctive traits that distinguish the circulating microbiome of mussels at different sites, we studied its functional content predicted from the KEGG database using Piphillin tool. Our findings confirmed that the bacterial pathways varied significantly between A. atra and M. platensis and between sites (Fig. 6). A case in point is the abundance of the pathways involved in xenobiotics degradation, carbohydrate metabolism and amino acid metabolism in the hemolymph of A. atra at Port-Couvreux.

Predictive functional analysis. Heatmaps based on the main KEGG pathways predicted at the different sampling sites. Relative abundance (natural logarithm base) of metabolic pathways based on 16S rRNA data is shown for both mussel species. Enrichments correspond to the positive values (red color) while depletions correspond to negative values (blue).
Effect of thermal stress on cmDNA
Environmental conditions, particularly temperature, are known to affect the hemolymphatic microbiome in many marine invertebrates26,48,49. In fact, alteration of microbiota by thermal stress can induce mortality in some organisms50. To determine whether the circulating microbiome of both species was modulated equally to thermal stress, both mussel species was subjected to an acute thermal stress under laboratory conditions. Our results showed that Pseudoalteromonas spp., a bacterial genus found in the bacterioplankton in Antarctica51, was the most abundant genus obtained in both mussel specimens and its abundance increased significantly (p < 0.001) in both mussels exposed at a higher temperature (30 °C) (Fig. 7A, Supplementary Table S3). A significant increase in Cobetia, a facultative psychrotrophic bacteria that can grow at temperature ranging from 0 to 40 °C52, was also seen in A. atra but not in M. platensis. A Venn diagram showed that the hemolymphatic cmDNA in A. atra harbored 3–4 times more genera that that of M. platensis, suggesting a higher diversity (Fig. 7B). The higher diversity found in A. atra was confirmed upon analysis of richness and Shannon indices (Fig. 7C). Taken together, these results suggest that both mussel species react differently to thermal stress, consistent with results reported in an experimental model of acute thermal stress21.

Changes in bacterial cmDNA profiles in mussels following an acute thermal stress. (A) Heatmap outlining the presence and the relative abundance (%) of the top 30 bacterial genera of at different temperatures. The data were generated from 8 345 ASVs. Red colors indicate higher abundance and blue colors indicate lower abundances. Stars indicate taxa that were significantly different from four groups, as determined by LDA Effect Size analysis. (B) A Venn diagram showing the number of unique and shared bacterial genera in both species at 8 °C and 30 °C. (C) Alpha diversity of A. atra and M. platensis hemolymphatic cmDNA at 8 °C and 30 °C. Species evenness, observed richness and Shannon diversity indexes were calculated for each group. Note: *0.01 < p ≤ 0.05; **0.001 < p ≤ 0.01; ***p ≤ 0.001.
Discussion
In the present work, using hemolymphatic liquid biopsies, we compared the cmDNA of two mussel species that cohabit the coastal ecosystems of Kerguelen. More specifically, we found that: 1) The cmDNA bacterial profiles of both species differ significantly; 2) differences were site-dependent; 3) both species had distinct functional microbiota, and 4) the circulating bacterial microbiome of A. atra was more sensitive to changes induced by an experimental model of thermal stress. Overall, this study shows that both mussel species of Kerguelen Islands have distinctive bacterial cmDNA signatures that can be used for long term monitoring of the impact of climate changes on marine coastal ecosystems. We also show that defining the bacterial cmDNA signature in sentinel species is compatible with a minimally invasive, ethical and logistically friendly sampling method adapted for research in remote regions such as Kerguelen Islands.
Our study has revealed that hemolymphatic bacterial DNA in mussels are species-specific, even though both mussel species co-habit intimately in mussel beds of Kerguelen. These results suggest the existence of a selective bacterial retention between the two mussel species. Similar conclusions have been reported recently by Weingarten et al.53 who studied the gut microbiota of four different freshwater Unionidae mussels. Several non-exclusive hypotheses can be formulated to explain these findings. First, this could be related to intrinsic characteristics of filtration between species54. For instance, M. platensis is characterized by longer labial palps allowing to capture and reject finer material in comparison with other bivalves and have higher filtration rates than A. atra55,56. It has also been suggested that the circulating microbiome is not just dependent of particles ingestion but may be impacted by the interactions between environmental microbiota and host gills for selectively retaining certain taxa. Indeed, it has been demonstrated that ctenidia (i.e., paired gills) are responsible for particle sorting such as plankton, organic detritus and bacteria. Some studies have shown that picoplankton (0.2–2 µm) represents a significant contribution to the mussel diet and their ingestion was found to be cleared at higher rates than bacteria (~ 0.6 µm in size) by M. platensis57,58,59. This suggests that factors other than size affect capture of particles by ctenidia of bivalves, such as availability of the seston, might come into play. Marine aggregates (1–500 µm) may also contribute to enhance the uptake of picoplankton and bacteria60,61. Particle selection and ingestion in filter-feeding bivalves may thus depend on differential morphology of the ctenidia54,62. Particle discrimination can also be influenced by interactions between chemical constituents of the cohesive mucous of pallial organs and surface carbohydrates of captured particles59,63. Differences between microbiota in bivalves can also be modulated by the mucosal immunity64,65,66. Bivalve pallial mucus contains agglutinins that can interact with several bacterial species. It may also depends to immune humoral factors, such as antimicrobial peptides, that play a central role in defense, most notably in the extrapallial compartment where enriched nutrients such as proteins and polysaccharides that promote microbial proliferation67. Another immune mechanism that may explain the differential microbiome signature is the selective killing of bacteria bacteriophages65. For instance, two phages isolated in Pacific oyster larvae provide a non–host-derived immunity against pathogenic V. coralliilyticus68. Bacteriophage adherence to bivalve mucosal surfaces is central in this symbiotic relationship. Clearly, further comparative studies between M. platensis and A. atra are required to address this question.
Our data highlights that cmDNA bacterial profiles are site-specific and may provide critical information on the surrounding marine ecosystems. A good example is the presence sulfur-oxidizing bacteria (SOB) (e.g., Sulfitobacter spp. and Sulfurimonas spp.), which represent a significant part in hemolymph microbiota in both mussel species collected at different sites (Supplementary Table S1). SOB represent a large part of microbial communities in marine surface sediments where they play a key role in element cycling69,70. Coastal ecosystems around the Kerguelen islands are characterized by high wind speeds and strong tidal currents causing sediment dispersion along the coast71,72,73. In addition to symbionts, the region around the Kerguelen Plateau is characterized by the presence of cold seeps and hydrothermal vents emitting chemical-rich fluids31. It is likely that symbiotic SOB are present in the gills of the two mussels and contribute to provide organic carbon as energy source to the host by consuming sulfide from the Subantarctic vents. Another example of how the circulating microbiome can provide clues on local environmental conditions is the detection of DNA derived from thermophilic bacteria, such as Geobacillus spp. and Thermomonas spp., which were found in A. atra collected at Îlot-des-Trois-Bergers and Prince-de-Monaco (Supplementary Table S1). Both sites are located near geothermally heated habitats74. High abundance of kelps could also modulate the circulating microbiome signature. The coastlines of Kerguelen Islands are occupied by highly productive giant kelp beds forming undersea forests in hard bottom. Giving the fact that numerous strains of the genus Sphingomonas can decompose algal polysaccharides and be a dominant bacterium from a marine oligotrophic environment38,75, it is therefore not surprising to find a great abundance of this genus at sampling sites characterized by kelp forests. Taken together, these results illustrate the potential of defining the circulating microbiome signature in mussels for long-term monitoring of environmental changes occurring in coastal marine ecosystems.
Intertidal mussels are exposed to sea surface and air temperature variations reflecting the solar exposition, the wind speed, the sea level pressure, humidity index and the dual tides in Kerguelen76,77,78. In 2018, according to the PROTEKER’s registries from their temperature loggers at Îlot Channer site at a depth of 5 m, the temperature varied from 2.7 to 9.5 °C, where A. atra is found79. The situation is of course quite different in the intertidal zones where temperature changes is subjected to high variations within a single day and between tides. Considering the growth rate of some bacteria, it is logical to expect a microbiome shift after a thermal stress as we did observe in our experimental model of thermal stress. Such effect of thermal stress on the cmDNA has been documented in the past for bivalves26,40,48. However, this effect was more pronounced in A. atra when compared to M. platensis, as shown by the significant increase of the bacteria of the Cobetia genus in A. atra. These bacteria can grow at temperature up to 40 °C52. In addition, it is also important to pinpoint that the microbiome signature in mussels will also be affected by seasonal variations.
Overall, our data with the circulating microbiome suggests that A. atra is more sensitive to thermal stress when compared to M. platensis. These results are consistent with previous studies comparing the hemolymphatic component of both species in response to thermal stress80. Whether such this plays a role in its progressive disappearance for the profit of Mytilus spp. in many marine ecosystems is unclear now81. Clearly, monitoring of mixed beds may represent a unique opportunity to study the effect of climate change on marine coastal ecosystems in the Southern hemisphere, a particularly sensitive region to climate change.
Materials and methods
Sample collection
A. atra and M. platensis specimens (55–70 mm length) were collected at 16 sites of the French sub-Antarctic Kerguelen Islands during the 2017 and 2018 Summer campaigns (Fig. 2). Liquid biopsy samples were collected from samples collected in both intertidal or subtidal (~ 5 m depth) zones and immediately processed (within an hour) on site as previously described21. Briefly, intravalvular liquid was removed with the tip of a knife and hemolymph withdrawn from the adductor muscle using a syringe fitted with a 21-gauge needle. Samples were immediately transferred into 1.5 mL sterile Eppendorf tube and centrifuged on site for 3 min at 3000×g at ambient temperature using a portable, battery-operated TOMY Multi Spin centrifuge (TOMY, Japan). Samples were pooled to eliminate individual differences and to minimize small-scale variability25,82. After centrifugation, cell pellets were dispersed gently and a 50 µL aliquot was applied on individual discs of Whatman 903™ FTA® cards (Sigma-Aldrich, Oakville, ON, Canada). After a 30 min drying period at ambient temperature, individual cards were kept in zip-sealed sampling plastic bags containing one small desiccant. Ethanol was used to disinfect handling equipment, and gloves were always worn during the procedure to prevent contamination from human hands.
Thermal stress experiments
Adult specimens of M. platensis and A. atra were collected on the intertidal rocky shores of Port-aux-Français (49°21′4.682″ S, 70°13′22.496″ E) at Kerguelen Islands in November and December 2018. Mussels were transported to laboratory within the hour and immediately transferred in a temperature-controlled (8 °C) aerated aquarium containing filtered recirculating seawater maintained on a 12 h:12 h light/dark cycle for at least 24 h. Mussels were placed in a 30 °C seawater recipient for 90 min. This acute stress model is commonly used in other studies to investigate the effect of temperature stress between mussels congeners80,83,84,85. Controls included mussels incubated for the same period of time at 8 °C. After 90 min, hemolymph samples were immediately processed as described above and 70 µL of hemolymph was spotted on FTA cards kept in sample bags with a desiccant.
Preprocessing and sequencing
Individual discs were cut from the FTA cards using a sterile 5.0 mm single round hole punch and total DNA isolated using the QIAamp DNA Investigator Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Quantification of DNA was carried out in duplicate using Quant-iT™ PicoGreen® dsDNA detection kit (Molecular Probes, Eugene OR). Amplification of the 16S ribosomal RNA (rRNA) gene amplifications and 16S gene amplicon sequencing for all DNA samples were performed at Centre d'expertise et de services Génome Québec (Montréal, QC, Canada) using the universal primers 341F (5’-CCTACGGGNGGCWGCAG-3’) and 805R (5’-GACTACHVGGGTATCTAATCC-3’)86. Sequence libraries were prepared by Génome Québec with TruSeq® DNA Library Prep Kit (Illumina, San Diego, CA, USA) and quantified using KAPA Library Quantification Kit for Illumina platforms (Kapa Biosystems). Paired-end sequences were generated on a MiSeq platform PE300 (Illumina Corporation, San Diego, CA, USA) with the MiSeq Reagent Kit v3 600 cycles (Illumina, San Diego, CA, USA).
Bioinformatic analysis
Illumina sequence data (FASTQ files) were received as output from Génome Québec. For each primer pair dataset, reads from the raw sequencing data were trimmed using Cutadapt (version 2.8) tool implemented in Unix Shell (version 4.4.19; Ubuntu version 18.04). For data pre-processing, the DADA2 pipeline (version 1.16.0; Callahan et al.87) was used to generate 16S rRNA (V3-V4) amplicon sequence variants (ASVs). Briefly, forward and reverse reads were trimmed, filtered and truncated based on their quality scores. The error model was calculated for forward and reverse reads. After denoising and merging, chimeric sequences (bimeras) were removed from the datasets by following the consensus method as implemented in DADA2. All subsequent analyses were performed within the R environment (R version 4.0.3, R Core Team88). The final table obtained consisted of a tabulation of number of occurrences of non-chimeric ASVs in each sample. Taxonomy assignments of representative ASVs were performed using the naïve Bayesian classifier method with the latest RDP 16 database. Sequences (4.8% of reads) attributed to Archaea or unclassified at phylum level were removed from the dataset. Raw datasets analyzed during the current study are publicly available on NCBI Sequence Read Archive (PRJNA773369).
Statistical analysis
Bacterial community analysis were performed in the R environment using the phyloseq, microbiomeSeq, microbiomeMarker and vegan packages89,90,91,92. Alpha diversity indices, including the Shannon index, Pielou's evenness and richness estimator, were calculated using Wilcoxon/Kruskal–Wallis test between groups. Beta diversity between sample groups was determined based on the UniFrac distance and visualized by principal coordinates analysis (PCoA). Heatmaps were performed based on the relative abundance and constructed with the 30 most abundant genera. Bacterial biota composition differences among sites were studied using multivariate analysis of variance with permutation (PERMANOVA) with 9999 permutations. Permutation multivariate analysis of dispersion (PERMDISP) was also conducted with the function betadisper and permutest in order to test for homogeneity of multivariate dispersions (i.e., deviations from centroids) among sampling sites. The linear discriminant analysis (LDA) identifies the effect size (LEfSe) with which these taxa differentiate the samples. The threshold on the logarithmic score of LDA analysis was set to 2.0. Comparative metagenomic functional composition was predicted from the latest Kyoto Encyclopedia of Genes and Genomes (KEGG) database93 using a recently developed online tool Piphillin (http://secondgenome.com/Piphillin)45,94. The differential abundance analysis of gene abundance data was completed with the online tool MicrobiomeAnalyst95.
Abbreviations
- A. atra :
-
Aulacomya atra
- M. platensis :
-
Mytilus platensis
- cmDNA:
-
Circulating microbiome DNA, biodiversity
- PCoA:
-
Principal Coordinate Analysis
- ASVs:
-
Amplicon sequence variants
- PERMANOVA:
-
Multivariate analysis of variance with permutation
- LEfSe:
-
Linear discriminant analysis effect size
- SOB:
-
Sulphur-oxidizing bacteria
References
Brondizio, E. S., Settele, J., Díaz, S. & Ngo, H. T. (eds.) Global Assessment Report on Biodiversity and Ecosystem Services of the Intergovernmental Science–Policy Platform on Biodiversity and Ecosystem Services (IPBES Secretariat, 2019).
Weiskopf, S. R. et al. Climate change effects on biodiversity, ecosystems, ecosystem services, and natural resource management in the United States. Sci. Total Environ. 733, 137782. https://doi.org/10.1016/j.scitotenv.2020.137782 (2020).
Turner, J. & Marshall, G. J. Climate Change in the Polar Regions (Cambridge University Press, 2011).
Meredith, M. et al. Polar Regions. Chapter 3, IPCC Special Report on the Ocean and Cryosphere in a Changing Climate. https://www.ipcc.ch/srocc/chapter/chapter-3-2/ (2019).
Rignot, E. et al. Four decades of Antarctic Ice Sheet mass balance from 1979–2017. Proc. Natl. Acad. Sci. USA 116, 1095–1103. https://doi.org/10.1073/pnas.1812883116 (2019).
Siegert, M. et al. The Antarctic Peninsula under a 1.5°C global warming scenario. Front. Environ. Sci. 7, 102. https://doi.org/10.3389/fenvs.2019.00102 (2019).
Iz, H. B. Is the global sea surface temperature rise accelerating?. Geod. Geodyn. 9, 432–438. https://doi.org/10.1016/j.geog.2018.04.002 (2018).
Qiu, Z. et al. Future climate change is predicted to affect the microbiome and condition of habitat-forming kelp. Proc. R. Soc. B. 286, 20181887. https://doi.org/10.1098/rspb.2018.1887 (2019).
Burge, C. A., Kim, C. J., Lyles, J. M. & Harvell, C. D. Special issue Oceans and Humans Health: The ecology of marine opportunists. Microb. Ecol. 65, 869–879. https://doi.org/10.1007/s00248-013-0190-7 (2013).
Cavicchioli, R. et al. Scientists’ warning to humanity: Microorganisms and climate change. Nat. Rev. Microbiol. 17, 569–586. https://doi.org/10.1038/s41579-019-0222-5 (2019).
Harvell, C. D. et al. Emerging marine diseases–climate links and anthropogenic factors. Science 285, 1505–1510. https://doi.org/10.1126/science.285.5433.1505 (1999).
Egan, S. & Gardiner, M. Microbial dysbiosis: Rethinking disease in marine ecosystems. Front. Microbiol. 7, 991. https://doi.org/10.3389/fmicb.2016.00991 (2016).
Wilkins, L. G. E. et al. Host-associated microbiomes drive structure and function of marine ecosystems. PLoS Biol. 17, e3000533. https://doi.org/10.1371/journal.pbio.3000533 (2019).
Seuront, L., Nicastro, K. R., Zardi, G. I. & Goberville, E. Decreased thermal tolerance under recurrent heat stress conditions explains summer mass mortality of the blue mussel Mytilus edulis. Sci. Rep. 9, 17498. https://doi.org/10.1038/s41598-019-53580-w (2019).
Tsuchiya, M. Mass mortality in a population of the mussel Mytilus edulis L. caused by high temperature on rocky shores. J. Exp. Mar. Biol. Ecol. 66, 101–111. https://doi.org/10.1016/0022-0981(83)90032-1 (1983).
Malham, S. K. et al. Summer mortality of the Pacific oyster, Crassostrea gigas, in the Irish Sea: The influence of temperature and nutrients on health and survival. Aquaculture 287, 128–138. https://doi.org/10.1016/j.aquaculture.2008.10.006 (2009).
Beyer, J. et al. Blue mussels (Mytilus edulis spp.) as sentinel organisms in coastal pollution monitoring: A review. Mar. Environ. Res. 130, 338–365. https://doi.org/10.1016/j.marenvres.2017.07.024 (2017).
Ladeiro, M. P. et al. Mussel as a tool to define continental watershed quality. In Organismal and Molecular Malacology (ed Ray, S.), IntechOpen. https://doi.org/10.5772/67995 (2017).
Bonacci, S. et al. Esterase activities in the bivalve mollusc Adamussium colbecki as a biomarker for pollution monitoring in the Antarctic marine environment. Mar. Pollut. Bull. 49, 445–455. https://doi.org/10.1016/j.marpolbul.2004.02.033 (2004).
Storhaug, E. et al. Seasonal and spatial variations in biomarker baseline levels within Arctic populations of mussels (Mytilus spp.). Sci. Total Environ. 656, 921–936. https://doi.org/10.1016/j.scitotenv.2018.11.397 (2019).
Caza, F. et al. Liquid biopsies for omics-based analysis in sentinel mussels. PLoS ONE 14, e0223525. https://doi.org/10.1371/journal.pone.0225359 (2019).
Ignatiadis, M., Sledge, G. W. & Jeffrey, S. S. Liquid biopsy enters the clinic - implementation issues and future challenges. Nat. Rev. Clin. Oncol. 18, 297–312. https://doi.org/10.1038/s41571-020-00457-x (2021).
Kowarsky, M. et al. Numerous uncharacterized and highly divergent microbes which colonize humans are revealed by circulating cell-free DNA. Proc. Natl. Acad. Sci. USA 114, 9623–9628. https://doi.org/10.1073/pnas.1707009114 (2017).
Chen, H. et al. Circulating microbiome DNA: An emerging paradigm for cancer liquid biopsy. Cancer Lett. 521, 82–87. https://doi.org/10.1016/j.canlet.2021.08.036 (2021).
Lokmer, A. et al. Spatial and temporal dynamics of Pacific oyster hemolymph microbiota across multiple scales. Front. Microbiol. 7, 1367. https://doi.org/10.3389/fmicb.2016.01367 (2016).
Lokmer, A. & Wegner, M. K. Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress and infection. ISME J. 9, 670–682. https://doi.org/10.1038/ismej.2014.160 (2015).
Auguste, M. et al. Exposure to TiO2 nanoparticles induces shifts in the microbiota composition of Mytilus galloprovincialis hemolymph. Sci. Total Environ. 670, 129–137. https://doi.org/10.1016/j.scitotenv.2019.03.133 (2019).
Vezzulli, L. et al. Climate influence on Vibrio and associated human diseases during the past half-century in the coastal North Atlantic. Proc. Natl. Acad. Sci. USA 113, E5062–E5071. https://doi.org/10.1073/pnas.1609157113 (2016).
Musella, M. et al. Tissue-scale microbiota of the Mediterranean mussel (Mytilus galloprovincialis) and its relationship with the environment. Sci. Total Environ. 717, 137209. https://doi.org/10.1016/j.scitotenv.2020.137209 (2020).
Féral, J.-P. et al. PROTEKER: Implementation of a submarine observatory at the Kerguelen islands (Southern Ocean). Underw. Technol. 34, 3–10. https://doi.org/10.3723/ut.34.003 (2016).
Spain, E. A. et al. Shallow seafloor gas emissions near Heard and McDonald Islands on the Kerguelen Plateau, southern Indian Ocean. Earth Space Sci. 7, e2019EA000695. https://doi.org/10.1029/2019EA000695 (2020).
Cao, S. et al. Structure and function of the Arctic and Antarctic marine microbiota as revealed by metagenomics. Microbiome. 8, 47. https://doi.org/10.1186/s40168-020-00826-9 (2020).
Wang, L.-Y. et al. Comparison of bacterial community in aqueous and oil phases of water-flooded petroleum reservoirs using pyrosequencing and clone library approaches. Appl. Microbiol. Biotechnol. 98, 4209–4221. https://doi.org/10.1007/s00253-013-5472-y (2014).
Gutierrez, T., Berry, D., Teske, A. & Aitken, M. D. Enrichment of Fusobacteria in sea surface oil slicks from the Deepwater Horizon oil spill. Microorganisms. 4, 24. https://doi.org/10.3390/microorganisms4030024 (2016).
Michelou, V. K., Caporaso, J. G., Knight, R. & Palumbi, S. R. The ecology of microbial communities associated with Macrocystis pyrifera. PLoS ONE 8, e67480. https://doi.org/10.1371/annotation/48e29578-a073-42e7-bca4-2f96a5998374 (2013).
Florez, J. Z. et al. Structure of the epiphytic bacterial communities of Macrocystis pyrifera in localities with contrasting nitrogen concentrations and temperature. Algal Res. 44, 101706. https://doi.org/10.1016/j.algal.2019.101706 (2019).
Minich, J. J. et al. Elevated temperature drives kelp microbiome dysbiosis, while elevated carbon dioxide induces water microbiome disruption. PLoS ONE 13, e0192772. https://doi.org/10.1371/journal.pone.0192772 (2018).
Lin, J. D., Lemay, M. A. & Parfrey, L. W. Diverse bacteria utilize alginate within the microbiome of the giant kelp Macrocystis pyrifera. Front. Microbiol. 9, 1914. https://doi.org/10.3389/fmicb.2018.01914 (2018).
Pierce, M. L. & Ward, J. E. Microbial ecology of the Bivalvia, with an emphasis on the family Ostreidae. J. Shellfish Res. 37, 793–806. https://doi.org/10.2983/035.037.0410 (2018).
Pierce, M. L. & Ward, J. E. Gut Microbiomes of the Eastern Oyster (Crassostrea virginica) and the Blue Mussel (Mytilus edulis): Temporal variation and the influence of marine aggregate-associated microbial communities. mSphere. 4, e00730-19. https://doi.org/10.1128/mSphere.00730-19 (2019).
Delille, D. & Gleizon, F. Distribution of enteric bacteria in Antarctic seawater surrounding the Port-aux-Francais permanent station (Kerguelen Island). Mar. Pollut. Bull. 46, 1179–1183. https://doi.org/10.1016/S0025-326X(03)00164-4 (2003).
Nguyen, T. V. & Alfaro, A. C. Metabolomics investigation of summer mortality in New Zealand Greenshell mussels (Perna canaliculus). Fish Shellfish Immunol. 106, 783–791. https://doi.org/10.1016/j.fsi.2020.08.022 (2020).
Vezzulli, L. et al. Comparative 16SrDNA gene-based microbiota profiles of the Pacific oyster (Crassostrea gigas) and the Mediterranean Mussel (Mytilus galloprovincialis) from a shellfish farm (Ligurian Sea, Italy). Microb. Ecol. 75, 495–504. https://doi.org/10.1007/s00248-017-1051-6 (2018).
Romalde, J. L., Diéguez, A. L., Lasa, A. & Balboa, S. New Vibrio species associated to molluscan microbiota: A review. Front. Microbiol. 4, 413. https://doi.org/10.3389/fmicb.2013.00413 (2014).
Narayan, N. R. et al. Piphillin predicts metagenomic composition and dynamics from DADA2-corrected 16S rDNA sequences. BMC Genom. 21, 56. https://doi.org/10.1186/s12864-019-6427-1 (2020).
Peng, W. et al. Integrated 16S rRNA sequencing, metagenomics, and metabolomics to characterize gut microbial composition, function, and fecal metabolic phenotype in non-obese type 2 diabetic Goto-Kakizaki rats. Front. Microbiol. 10, 3141. https://doi.org/10.3389/fmicb.2019.03141 (2020).
Koner, S. et al. Assessment of carbon substrate catabolism pattern and functional metabolic pathway for microbiota of limestone caves. Microorganisms 9, 1789. https://doi.org/10.21203/rs.3.rs-549787/v1 (2021).
Li, Y. F. et al. Temperature elevation and Vibrio cyclitrophicus infection reduce the diversity of haemolymph microbiome of the mussel Mytilus coruscus. Sci. Rep. 9, 16391. https://doi.org/10.1038/s41598-019-52752-y (2019).
Scanes, E. et al. Climate change alters the haemolymph microbiome of oysters. Mar. Pollut. Bull. 164, 111991. https://doi.org/10.1016/j.marpolbul.2021.111991 (2021).
Hylander, B. L. & Repasky, E. A. Temperature as a modulator of the gut microbiome: What are the implications and opportunities for thermal medicine?. Int. J. Hyperth. 36, 83–89. https://doi.org/10.1080/02656736.2019.1647356 (2019).
Lo Giudice, A. et al. Marine bacterioplankton diversity and community composition in an antarctic coastal environment. Microb. Ecol. 63, 210–223. https://doi.org/10.1007/s00248-011-9904-x (2012).
Yumoto, I. et al. Temperature and nutrient availability control growth rate and fatty acid composition of facultatively psychrophilic Cobetia marina strain L-2. Arch. Microbiol. 181, 345–351. https://doi.org/10.1007/s00203-004-0662-8 (2004).
Weingarten, E. A., Atkinson, C. L. & Jackson, C. R. The gut microbiome of freshwater Unionidae mussels is determined by host species and is selectively retained from filtered seston. PLoS ONE 14, e0224796. https://doi.org/10.1371/journal.pone.0224796 (2019).
Rosa, M., Ward, J. E. & Shumway, S. E. Selective capture and ingestion of particles by suspension-feeding bivalve molluscs: A review. J. Shellfish Res. 37, 727–746. https://doi.org/10.2983/035.037.0405 (2018).
Griffiths, C. L. & King, J. A. Some relationships between size, food availability and energy balance in the ribbed mussel Aulacomya ater. Mar. Biol. 51, 141–149. https://doi.org/10.1007/BF00555193 (1979).
Riisgård, H. U. Filtration rate and growth in the blue mussel, Mytilus edulis Linneaus, 1758: Dependence on algal concentration. J. Shellfish Res. 10, 29–36 (1991).
Sonier, R. et al. Picophytoplankton contribution to Mytilus edulis growth in an intensive culture environment. Mar. Biol. 163, 73. https://doi.org/10.1007/s00227-016-2845-7 (2016).
Jacobs, P., Troost, K., Riegman, R. & Van der Meer, J. Length-and weight-dependent clearance rates of juvenile mussels (Mytilus edulis) on various planktonic prey items. Helgol. Mar. Res. 69, 101–112. https://doi.org/10.1007/s10152-014-0419-y (2015).
Ward, J. E. & Shumway, S. E. Separating the grain from the chaff: Particle selection in suspension- and deposit-feeding bivalves. J. Exp. Mar. 300, 83–130. https://doi.org/10.1016/j.jembe.2004.03.002 (2004).
Waite, A. M., Safi, K. A., Hall, J. A. & Nodder, S. D. Mass sedimentation of picoplankton embedded in organic aggregates. Limnol. Oceanogr. 45, 87–97. https://doi.org/10.4319/lo.2000.45.1.0087 (2000).
Ward, J. E. & Kach, D. J. Marine aggregates facilitate ingestion of nanoparticles by suspension-feeding bivalves. Mar. Environ. Res. 68, 137–142. https://doi.org/10.1016/j.marenvres.2009.05.002 (2009).
Ward, J. E. Biodynamics of suspension-feeding in adult bivalve molluscs: Particle capture, processing, and fate. Invertebr. Biol. 115, 218–231. https://doi.org/10.2307/3226932 (1996).
Rosa, M. et al. Physicochemical surface properties of microalgae and their combined effects on particle selection by suspension-feeding bivalve molluscs. J. Exp. Mar. 486, 59–68. https://doi.org/10.1016/j.jembe.2016.09.007 (2017).
Allam, B. & Espinosa, E. P. Bivalve immunity and response to infections: Are we looking at the right place?. Fish Shellfish Immunol. 53, 4–12. https://doi.org/10.1016/j.fsi.2016.03.037 (2016).
Barr, J. J. et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. USA 110, 10771–10776. https://doi.org/10.1073/pnas.1305923110 (2013).
Allam, B. & Espinosa, E. P. Mucosal immunity in mollusks. In Mucosal Health in Aquaculture (eds Beck, B. H. & Peatman, E.) 325–370 (Academic Press, 2015).
Huang, J. et al. Hemocytes in the extrapallial space of Pinctada fucata are involved in immunity and biomineralization. Sci. Rep. 8, 4657. https://doi.org/10.1038/s41598-018-22961-y (2018).
Kim, H. J. et al. Isolation and characterization of two bacteriophages and their preventive effects against pathogenic Vibrio coralliilyticus causing mortality of Pacific oyster (Crassostrea gigas) larvae. Microorganisms. 8, 926. https://doi.org/10.3390/microorganisms8060926 (2020).
Ihara, H. et al. Sulfur-oxidizing bacteria mediate microbial community succession and element cycling in launched marine sediment. Front. Microbiol. 8, 152. https://doi.org/10.3389/fmicb.2017.00152 (2017).
Jørgensen, B. B. & Nelson, D. C. Sulfide oxidation in marine sediments: Geochemistry meets microbiology. Geol. S. Am. S. 379, 63–81. https://doi.org/10.1130/0-8137-2379-5.63 (2004).
Zhou, M. et al. Surface currents and upwelling in Kerguelen Plateau regions. Biogeosci. Discuss. 11, 6845–6876. https://doi.org/10.5194/bgd-11-6845-2014 (2014).
Gille, S. T., Carranza, M. M., Cambra, R. & Morrow, R. Wind-induced upwelling in the Kerguelen Plateau region. Biogeosciences 11, 6389–6400. https://doi.org/10.5194/bg-11-6389-2014 (2014).
Park, Y. H., Roquet, F., Durand, I. & Fuda, J. L. Large-scale circulation over and around the Northern Kerguelen Plateau. Deep Sea Res. II(55), 566–581. https://doi.org/10.1016/j.dsr2.2007.12.030 (2008).
Renac, C. et al. Hydrothermal fluid interaction in basaltic lava units, Kerguelen Archipelago (SW Indian Ocean). Eur. J. 22, 215–234. https://doi.org/10.1127/0935-1221/2009/0022-1993 (2010).
Vancanneyt, M. et al. Sphingomonas alaskensis sp. nov., a dominant bacterium from a marine oligotrophic environment. Int. J. Syst. Evol. 51, 73–79. https://doi.org/10.1099/00207713-51-1-73 (2001).
Helmuth, B. S. & Hofmann, G. E. Microhabitats, thermal heterogeneity, and patterns of physiological stress in the rocky intertidal zone. Biol. Bull. 201, 374–384. https://doi.org/10.2307/1543615 (2001).
Testut, L., Wöppelmann, G., Simon, B. & Téchiné, P. The sea level at Port-aux-Français, Kerguelen Island, from 1949 to the present. Ocean Dyn. 56, 464–472. https://doi.org/10.1007/s10236-005-0056-8 (2006).
Pohl, B. et al. Recent climate variability around the Kerguelen Islands (Southern Ocean) seen through weather regimes. J. Appl. Meteorol. Climatol. 60, 711–731. https://doi.org/10.1175/JAMC-D-20-0255.1 (2021).
PROTEKER. Ilôt Channer (Passe Royale)—Sea water temperature at 5 and 13 m depth (T°C) daily average 2014–2019. https://www.proteker.net/swt-ilot-channer-passe-royale/ (2021).
Caza, F. et al. Comparative analysis of hemocyte properties from Mytilus edulis desolationis and Aulacomya ater in the Kerguelen Islands. Mar. Environ. Res. 110, 174–182. https://doi.org/10.1016/j.marenvres.2015.09.003 (2015).
Caza, F., Cledon, M. & St-Pierre, Y. Biomonitoring climate change and pollution in marine ecosystems: A review on Aulacomya ater. J. Mar. Biol. 2016, 7183813. https://doi.org/10.1155/2016/7183813 (2016).
Rey-Campos, M. et al. High individual variability in the transcriptomic response of Mediterranean mussels to Vibrio reveals the involvement of myticins in tissue injury. Sci. Rep. 9, 3569. https://doi.org/10.1038/s41598-019-39870-3 (2019).
Caza, F. et al. Hemocytes released in seawater act as Trojan horses for spreading of bacterial infections in mussels. Sci. Rep. 10, 19696. https://doi.org/10.1038/s41598-020-76677-z (2020).
Yao, C. L. & Somero, G. N. Thermal stress and cellular signaling processes in hemocytes of native (Mytilus californianus) and invasive (M. galloprovincialis) mussels: Cell cycle regulation and DNA repair. Comp. Biochem. Physiol. 165, 159–168. https://doi.org/10.1016/j.cbpa.2013.02.024 (2013).
Lockwood, B. L., Sanders, J. G. & Somero, G. N. Transcriptomic responses to heat stress in invasive and native blue mussels (genus Mytilus): Molecular correlates of invasive success. J. Exp. Biol. 213, 3548–3558. https://doi.org/10.1242/jeb.046094 (2010).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1. https://doi.org/10.1093/nar/gks808 (2013).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 13, 581–583. https://doi.org/10.1038/nmeth.3869 (2016).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2021).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217. https://doi.org/10.1371/journal.pone.0061217 (2013).
Oksanen, J. & Blanchet, F. G. Vegan: Community Ecology Package. 2. 3-0 (2015).
Ssekagiri, A., Sloan, W. & Ijaz, U. Z. microbiomeSeq: an R package for analysis of microbial communities in an environmental context, In ISCB Africa ASBCB Conference (Kumasi, Ghana, 2017).
Cao, Y. Microbiome marker: Microbiome Biomarker Analysis Toolkit. R package version 0.99.0 (2020). https://github.com/yiluheihei/microbiomeMarker. Accessed March 2022.
Kanehisa, M. et al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114. https://doi.org/10.1093/nar/gkr988 (2012).
Iwai, S. et al. Piphillin: Improved prediction of metagenomic content by direct inference from human microbiomes. PLoS ONE 11, e0166104. https://doi.org/10.1371/journal.pone.0166104 (2016).
Dhariwal, A. et al. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 45, W180–W188. https://doi.org/10.1093/nar/gkx295 (2017).
Acknowledgements
This research was performed at Kerguelen with the logistic support of the French Polar Institute Paul-Emile-Victor (IPEV). The authors would like to thank all the personnel from the IPEV and the Terres Australes et Antarctiques Françaises (TAAF) for their help and hospitality during our stays. The authors would also like to thank Jean-Pierre Féral and Thomas Saucède and the PROTEKER team for their expert advice and technical support. The author would also like to thank Ms. Marlène Fortier for her excellent technical help. This work was funded in part by the National Science and Engineering Research Council of Canada (YSP) (Grant No. RGPIN-2019-06607) and the IPEV (YSP and SB).
Author information
Authors and Affiliations
Contributions
F.C., S.F., S.B. and Y.S.P. conceived the study. All authors were responsible for interpretation of data and critical appraisal. All authors executed experiments and/or contributed to the experimental design and/or analyses of the results. F.C., S.F, and Y.S.P. drafted the manuscript with input from all authors at all stages.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ferchiou, S., Caza, F., Villemur, R. et al. Species- and site-specific circulating bacterial DNA in Subantarctic sentinel mussels Aulacomya atra and Mytilus platensis. Sci Rep 12, 9547 (2022). https://doi.org/10.1038/s41598-022-13774-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-13774-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
