
Abstract
Establishing microbiome signatures is now recognized as a critical step toward identifying genetic and environmental factors shaping animal-associated microbiomes and informing the health status of a given host. In the present work, we prospectively collected 63 blood samples of the Atlantic cod population of the Southern Gulf of Saint Lawrence (GSL) and characterized their 16S rRNA circulating microbiome signature. Our results revealed that the blood microbiome signature was dominated at the phylum level by Proteobacteria, Bacteroidetes, Acidobacteria and Actinobacteria, a typical signature for fish populations inhabiting the GSL and other marine ecosystems. At the genus level, however, we identified two distinct cod groups. While the microbiome signature of the first group was dominated by Pseudoalteromonas, a genus we previously found in the microbiome signature of Greenland and Atlantic halibut populations of the GSL, the second group had a microbiome signature dominated by Nitrobacter and Sediminibacterium (approximately 75% of the circulating microbiome). Cods harboring a Nitrobacter/Sediminibacterium-rich microbiome signature were localized in the most southern part of the GSL, just along the northern coast of Cape Breton Island. Atlantic cod microbiome signatures did not correlate with the weight, length, relative condition, depth, temperature, sex, and salinity, as previously observed in the halibut populations. Our study provides, for the first time, a unique snapshot of the circulating microbiome signature of Atlantic cod populations and the potential existence of dysbiotic signatures associated with the geographical distribution of the population, probably linked with the presence of nitrite in the environment.
Similar content being viewed by others

In recent years, the marine ecosystems of the Northwest Atlantic have been warming rapidly and are undergoing a rapid transition, jeopardizing the persistence of some once predominant species (e.g.,1). The Atlantic cod (Gadus morhua), a widely distributed groundfish of high economic and cultural importance throughout the region, has remained at relatively low abundance following a massive collapse in the 1990s due to overfishing during a period of low productivity2. Moreover, some cod populations face further declines even without fishing pressure, which is the case for those distributed in the southern Gulf of St. Lawrence (SGSL), which is considered vulnerable to extinction3. Recent data have revealed an extremely high natural mortality rate of 55% in adult SGSL cod, a significant departure from the expected 20%3. Although increased predation by the grey seal (Halichoerus grypus) likely constitutes the primary factor driving this decline4, the health status of Atlantic cod may also be impacted by changes in the environment, leading to concurrent changes in its physiology, immune response and susceptibility to infectious diseases5,6,7,8. Several previous studies have indeed reported the role of environmental factors in limiting Atlantic cod conditions and productivity (e.g.,9,10).
To better understand the role of the environment in Atlantic cod population dynamics, it becomes imperative to set up programs for monitoring the health status of Atlantic cod populations and to develop new sensitive and predictive biomarkers compatible with the sampling logistics of the current monitoring programs. Currently, individual health assessments of wild fish populations remain relatively scarce, mainly relying on calculating the condition factor K with individual length and weight as a measure of “plumpness”11. Even though this method is fast and easy to measure, its sensitivity and predictive value remain relatively low12. Additional methods include data obtained from tissue biopsies, which, unfortunately, are costly and logistically complicated13,14.
In humans, liquid biopsy-based biomarkers have emerged as critical tools for the follow-up of the health status of patients and for detecting signs of the onset of diseases at their earliest stage. Initially developed in oncology to detect mutations in circulating cell-free DNA in the blood of patients with cancer, liquid biopsy-based biomarkers have been extended to the analysis of other omics, including the microbiome. The importance of establishing the host microbiome signature has gained interest in recent years, notably since landmark studies revealed that it is possible to identify specific microbiome signatures that predict the outcome of treating cancer patients with immunotherapy15,16. This approach is now commonly used to compare hosts afflicted with various types of disorders to healthy populations, allowing clinicians to establish a diagnosis, predict future onset of pathologies, and even predict treatment success probability as a function of physical fitness. The approach gained further clinical interest when applied to the blood, leading to the concept of the circulating bacterial microbiome. In this case, the focus is on analyzing bacterial nucleic acids found in the host's blood, allowing the detection of potential pathogens and dysbiosis induced by environmental changes. Dysbiosis, defined here as a change in the abundance or diversity of some groups of microorganisms, is very sensitive to various forms of environmental stress, such as the response to elevated temperatures17. Recent studies have applied this concept to marine species, including invertebrates and fish populations18,19. For example, using a single drop of blood fixed on cellulose paper, we recently reported that the circulating microbiome signature of wild halibut populations is sensitive to physiological and environmental factors20.
In the present work, we prospectively collected blood samples from the SGSL Atlantic cod population in September 2021 and studied the circulating microbiome signature by sequencing the 16S ribosomal RNA (rRNA) gene sequence. This study aimed to characterize the circulating microbiome of wild fish and correlate it with physiological and environmental data, giving us more information on their general health status. Our results suggest the existence of dysbiotic signatures associated with the geographical distribution of the cod population.
Material & methods
Sampling
Blood samples from individual Atlantic cod (n = 63; 15–57 cm) were collected between September 10 and September 26, 2021, during the second leg of the annual Fisheries and Oceans Canada (DFO) bottom trawl survey in the southern sector of the SGSL, Canada (Table 1). Blood samples were taken from the caudal vein immediately upon trawl retrieval, and overall, liquid biopsies were performed at 21 sites for at least one individual. The number of liquid biopsies carried out per station (ranging from 1 to 6) was opportunistic and depended on the presence of Atlantic cod and workload at a given site. Blood samples were collected with a heparin-coated 3 mL sterile syringe and a 22G needle and immediately stored on an FTA™ card (Sigma‒Aldrich®, Oakville, ON, Canada) to minimize contamination. Samples were allowed to air dry in a separate (dry) lab and stored in a plastic bag with a desiccant, as described by Caza et al.18. Drops of blood were collected and immediately stored on an FTA™ card (Sigma‒Aldrich®, Oakville, ON, Canada). Samples were allowed to air dry and stored in a plastic bag with a desiccant, as described by Caza et al.18. The sex of each sampled individual was determined by visual identification of the gonads following the dissection of specimens by the DFO science crew. Scanmar® hydroacoustic sensors attached to the trawl and a conductivity, temperature, and depth (CTD) probe were used to record environmental characteristics. The care and use of field-sampled animals complied with the Government of Canada animal welfare laws, guidelines, and policies approved by Fisheries and Oceans Canada. All methods are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org/).
DNA extraction, amplification and sequencing
All DNA extraction and purification procedures were conducted in a clean room where pressure, temperature, and humidity were controlled to minimize contamination. Individual discs were cut from the FTA™ cards using a sterile 5.0 mm single round hole punch, and total DNA was isolated using the QIAamp DNA Investigator Kit (Qiagen, Toronto, ON, Canada) according to the manufacturer’s protocol. DNA was quantified in duplicate using a Quant-iT™ PicoGreen® dsDNA detection kit (Molecular Probes, Eugene OR, USA). Amplification of the V3–V4 region of the 16S rRNA gene and 16S rRNA gene amplicon sequencing for all DNA samples were performed at Centre d'Expertise et de Services Génome Québec (Montréal, QC, Canada) using the universal primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGGTATCTAATCC-3′). Sequence libraries were prepared by Genome Quebec with the TruSeq® DNA Library Prep Kit (Illumina, San Diego, CA, USA) and quantified using the KAPA Library Quantification Kit for Illumina platforms (Kapa Biosystems). Paired-end sequences were generated on a MiSeq platform PE300 (Illumina Corporation, San Diego, CA, USA) with the MiSeq Reagent Kit v3 600 cycles (Illumina, San Diego, CA, USA). Raw data files are publicly available in the NCBI Sequence Read Archive (PRJNA1015160).
16S rRNA gene data processing and data analysis
Illumina sequence data (FASTQ files) were trimmed using Cutadapt (version 2.8). The 16S rRNA gene (V3–V4) amplicon sequence variants (ASVs) were generated with the DADA2 pipeline (version 1.16.021;) and subsequently within the R environment (R version 4.1.1, Team (2022)). The RDP 16 database was used for the ASV assignment. The software packages phyloseq (1.36.0), microbiomeSeq (0.1), microbiomeMarker (0.99.0) and vegan (2.5.7) were used to characterize the microbial communities22,23,24,25. The maps were created with the packages ggplot2 (3.3.6) and rnaturalearth (0.1.0)26. An ASV was considered part of the core microbiome if it had a minimum prevalence (rate of presence in the group of samples) of 70%, with a detection threshold of 0.01% relative abundance, as described by Palanisamy and colleagues27. A similar decision tree was applied for the core genera (abundance of ASVs of the same genus summed up) but with 90% prevalence and a detection threshold of 0.01%, as described by Fronton and colleagues20.
Bacterial taxonomic α-diversity (intrasample) was estimated using the richness and the Shannon and Simpson indices implemented in the R package microbiome (1.14.0). Variations in bacterial α-diversity and taxon abundances between the two populations were assessed using either the Kruskal‒Wallis test or the Wilcoxon–Mann‒Whitney test since none of the variables had a normal distribution. α-Diversity was also calculated among maturity stages according to the length at maturity (L50), which is estimated at 40 cm for this species3, the sex and the relative condition factor K28. The Kruskal‒Wallis test was followed by a pairwise Wilcoxon–Mann‒Whitney test if the p value (p) was significant (p < 0.05). The β-diversity (intersample) was estimated using phylogenetic weighted UNIFRAC dissimilarities assessed by principal coordinates analysis (PCoA). Differences in community composition were tested using permutational multivariate analysis of variance (PERMANOVA) for weighted UNIFRAC indices with 999 permutations, as implemented in the R vegan package (2.5.7) or the pairwise Adonis package (0.4). A redundancy analysis (RDA) was performed on the standardized (Hellinger transformation) number of reads for each ASV in each matrix and the environmental and physiological variable matrix29. The collinearity between variables was validated with a Pearson correlation, and weight and length were the only variables correlated. Differences were considered statistically significant at p < 0.05. Data analyses were performed in R studio (v4.0.5).
Condition K factor
The relative condition K factor, a broad health index for fish, was calculated based on the length and weight of each individual28,30. A linear regression was performed between log10 (weight) and log10 (length) as follows31:
\({log}_{10}\left(W\right)={log}_{10}\left(a\right)+{b*log}_{10}(L)\)
Where W is the weight, L is the length, and a and b are constant coefficients.
The coefficients a and b were calculated and used to estimate the expected weight We of each individual based on their length with the following equation:
Finally, the Krel of Le Cren was calculated as follows:
The comparison between the individual’s actual and expected weight gives us the fish's plumpness, a glimpse of its health status. If an individual is skinnier than others of the same length within the same species, it is considered to undergo stress, and Krel will be under 1. In contrast, a plump fish is associated with advantageous environmental influence, and Krel will be over 111.
Results
Preliminary characterization of the circulating microbiome
The circulating microbiome signatures were determined by sequencing the V3–V4 hypervariable regions of the 16S rRNA gene. Approximately 2,800,000 raw reads were retrieved after filtering. The number of sequences per sample ranged between 5154 and 111,505. The mean number of reads per individual was 44,620.43 ± 29,34.44. The number of ASVs per sample curve confirmed that the sequencing depth was sufficient to plateau the number of ASVs (Fig. S1). A total of 920 unique ASVs were obtained, with 47.37 ± 1.58 ASVs on average per individual.
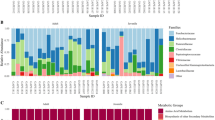
Overall, in the 63 samples we analyzed, we identified 920 ASVs at different taxonomic levels, including 18 phyla, 32 classes, 63 orders, 101 families, and 168 genera. At the phylum level, the blood microbiome signature was dominated by Proteobacteria, Bacteroidetes, Acidobacteria and Actinobacteria, representing an average of 96.89% of the microbiome. The prevalence of Proteobacteria was particularly dominant, with some samples showing almost 100% Proteobacteria in their microbiome (Fig. 1A). However, our analyses at the class, order and family levels revealed two very different signature groups. At the class level, the first group was primarily dominated by the presence of Alphaproteobacteria, while the second group was characterized by the dominance of Gammaproteobacteria (Fig. 1B). This second group also showed substantial heterogeneity at the order, family and gene levels, a notable difference from the first group dominated by Rhizobiales and Bradyrhizobiaceae (Fig. 1C,D).

Relative abundance of the main taxa in the circulating microbiome of Atlantic cod. (A) Mean relative abundance at the phylum level. (B) Relative abundance of individual taxa at the class level. (C) Relative abundance of individual taxa at the order level. (D) Relative abundance of individual taxa at the family level. (E) Relative abundance of individual taxa at the genus level. Data was obtained from a sample size of n = 63. The prevalence of taxa analyzed was 80%, with a detection threshold of 0.01%.
Identification of two groups with distinct microbiome signatures.
Next, we investigated the contrasting microbiome signatures in detail at the genus level (Fig. 1E). We found that the first group (Group A, n = 46) had a relatively homogeneous microbiome signature dominated by the Nitrobacter and Sediminibacterium genera, both of which are involved in the nitrogen cycle32. On average, Nitrobacter accounted for 63.06 ± 2.82%, and Sediminibacterium accounted for 12.27 ± 1.31% of the circulating microbiome of group A (Fig. 2A). Group B (n = 17) was defined as every individual with a low (< 30%) abundance of Nitrobacter and Sediminibacterium, the two genera part of the core microbiome (Fig. 2B and S2). This population was more heterogeneous and dominated by Pseudoalteromonas and Cobetia, which accounted for 48.03 ± 9.58% and 18.69 ± 7.01% of the circulating microbiome, respectively.

Relative abundance of the main genera in the circulating microbiome of Atlantic cod. (A) Main genera found in the circulating microbiome samples of Population A, with mean values. Population A consisted of 46 individuals, where the relative abundance of core microbiome taxa was > 30%. Prevalence of taxa analyzed was 1%, with a detection threshold set at 5%. (B) Main genera found in the circulating microbiome samples of Population B, with mean values. Population B consisted of 17 individuals, where the relative abundance of core microbiome taxa was < 30%. Prevalence of taxa analyzed was 1%, with a detection threshold set at 5%.
The α and β-diversity of the Atlantic cod circulating microbiome.
To further investigate the difference in the microbial composition between the two groups, we carried out a principal coordinate analysis (PCoA). Studies of the community composition by a PERMANOVA revealed a significantly different clustering (p = 0.001), with the separation in two groups explaining 38.4% of the variations (R2 = 0.382) (Fig. 3A)". The Venn diagram of the number of genera in each population showed a more diverse microbiome in group A, with 90 unique genera against 24 in group B. However, both groups shared 54 genera (32.1% of all the genera) (Fig. 3B). When we measured the α-diversity indices of the microbiomes of both populations, including the Shannon diversity index, Simpson’s index, richness, and relative abundance of the most abundant ASV, we found no significant difference (Fig. 3C). Otherwise, there was no difference in the α or the β-diversity when considering sex, relative condition, or maturity classes (Fig. 4).

Diversity indices comparison of the circulating microbiome in two groups of Atlantic cod. (A) Principal Coordinates Analysis (PCoA) plot based on the weighted UniFrac distance, illustrating the differentiation between the two populations of cod circulating microbiomes. (B) Venn diagram depicting the number of genera observed in each population. (C) α-Diversity indices for both cod microbiome groups, represented by solid squares denoting the mean values. Group A consisted of individuals with a relative abundance of core microbiome > 30%, while Group B consisted of individuals with a relative abundance of core microbiome < 30%. The indices analyzed include (i) Simpson's index, (ii) Shannon's diversity index, (iii) Number of ASVs observed, and (iv) Relative abundance of the most abundant ASV. The sample size was n = 63, with Group A consisting of 46 individuals and Group B consisting of 17 individuals.

Diversity indices comparison of physiological variables in the circulating microbiome of Atlantic cod. The diversity indices analyzed include Simpson's index, Shannon's diversity index, the number of ASVs observed, and the relative abundance of the most abundant ASV. Mean values are represented by solid squares. Additionally, a Principal Coordinates Analysis (PCoA) based on the weighted UniFrac distance is shown. The PCoA plot visualizes the differentiation in the circulating microbiome based on the physiological variable being compared. (A) Comparison between sexes: The sample size was n = 61, with 21 individuals classified as males (M) and 40 individuals classified as females (F). (B) Comparison between condition classes: The sample size was n = 63, with 30 individuals classified as having high condition and 33 individuals classified as having low condition. (C) Comparison between maturity classes: The sample size was n = 63, with 38 individuals classified as immature and 25 individuals classified as mature.
Geographical distribution of the cod population with distinct blood microbiomes.
Next, we examined which factors could explain the presence of the two distinct signatures in the blood microbiome. A redundancy analysis (RDA) was thus performed on the normalized number of reads for each ASV in each matrix and the environmental and physiological variable matrix. An ANOVA-like permutation test for RDA revealed that weight, length, relative condition index (Krel), depth, temperature, sex, and salinity did not significantly influence the Atlantic cod circulating microbiome (Table 2). We did find, however, a statistically significant correlation between the blood microbiome signature and sampling sites (p < 0.001). We found that individuals with a Nitrobacter-rich microbiome (group A) were located near Cape Breton Island, whereas group B was closer to the Magdalen Islands (Fig. 5). Of the 21 sampling sites, only one, 165B, contained a mix of populations A and B.

Map indicating the 21 sampling sites. The seafloor depth is labeled on the map, and the number of cod individuals in Group A (yellow) or Group B (purple) is displayed at each site. The lines within the circles denote the number of samples collected at each specific station. N&L represents Newfoundland & Labrador. The figure was generated using ggplot2 sf (1.0–13), rnaturalearthdata (0.1.0), sf(1.0–13), and rnaturalearthdata (0.1.0), ggOceanMaps and ggOceanMapsData (1.3.4), and the scatterpie package (0.1.8) in R (version 3.4.0) and Inkscape.
Discussion
The present work aimed to assess the circulating signature and diversity of the circulating blood microbiome of the Southern Gulf of St. Lawrence Atlantic cod, a population that faces extinction following decades of continuous decline. At the phylum level, the blood microbiome signature was dominated by Proteobacteria, Bacteroidetes, Acidobacteria and Actinobacteria. Apart from a few minor differences, particularly concerning the lower prevalence of Firmicutes, this taxonomic structure at the phylum level was similar to that recently described in the circulating microbiome of the Greenland halibut (Reinhardtius hippoglossoides) and Atlantic halibut (Hippoglossus hippoglossus) in the Gulf of St. Lawrence20. However, we could distinguish two populations with distinct blood microbiome signatures at the lower ranks. The presence of Alphaproteobacteria primarily dominated the first signature. At the same time, the second showed the dominance of Gammaproteobacteria and a very heterogeneous signature at the order and family levels, a notable difference from the first group dominated by Rhizobiales and Bradyrhizobiaceae. Interestingly, the second group had a signature dominated by the Nitrobacter and Sediminibacterium genera, which are involved in the nitrogen cycle32. Finally, we found that the taxonomic and phylogenetic structures of the bacterial community were restricted to specific regions of the SGSL, suggesting that the environment directly impacts the circulating microbiome. The data revealed exciting clinical prospects for using a blood microbiome genetic signature for detecting dysbiosis, risk stratification, and disease surveillance of the cod population in response to environmental changes.
In medicine, the characterization of the peripheral blood-derived microbiome signature, defined as blood microbial DNA, is increasingly used by clinicians to assess an individual's health status, detect dysbiosis and potential pathogens, or as a biomarker to inform disease severity and progression33,34,35,36,37. This concept is also gaining momentum in ecology as the circulating microbiome of dogs, bovine, wild birds and wild fish populations were recently studied, showing that the genetic structure of the blood microbiome, just as in humans, is modulated by genetic and spatiotemporal factors, as well as disease conditions20,38,39,40,41. In the present study, the observed shift toward the dominance of Nitrobacter and Sediminibacterium suggests that environmental factors severely impact the blood microbiome signature. The presence of Sediminibacterium is not uncommon in aquatic species42,43,44. In trout, the presence of this genus is sensitive to seasonal changes45. Its presence is also not uncommon in dysbiotic microbiome profiles. In humans, for example, its presence is associated with lung cancer diagnosis and is in higher abundance in the circulating microbiome of type 2 diabetes mellitus patients46,47. The presence of Nitrobacter is also not uncommon in dysbiotic microbiomes. The Nitrobacter genus plays a role in the nitrogen cycle, as it can oxidize nitrite (NO2−) to nitrate (NO3−). To our knowledge, however, this dominance of Nitrobacter has not been reported in wild fish populations in the past, although bacteria associated with the metabolism of nitro compounds have been found, albeit at lower levels, in the blood microbiome, gut or skin microbiome of various animals, including bovine and fish40,48,49,50. Experimentally, however, exposure of goldfish to nitrite has been shown to induce a shift in the gill, nose and skin microbiome toward bacterial communities involved in the nitrogen cycle and the disappearance of taxa generally found in the microbiome51. Usually, nitrite, an intermediate stage in the nitrogen balance, should not be detectable in a stable environment as it is rapidly transformed into NO3− by biodegrading bacteria such as Nitrobacter or by chemical reaction in the water. It is also quickly consumed by algae at the surface. Yet, high surface nitrite/nitrate concentrations are not uncommon in marine coastal ecosystems, as it is commonly released in seawater because of agricultural activity52,53,54. Whether this dysbiotic microbiome signature is associated with health issues in cod will require future investigation.
The presence of Nitrobacter and Sediminibacterium in population A contrasted with the microbiome profile of population B, where we found DNA derived from Pseudoalteromonas, a genus of marine bacteria commonly found in marine species, such as sponges, shellfish, macroalgae and fish, including wild fish populations of the Greenland halibut and Atlantic halibut of the Gulf of St. Lawrence20,55,56,57,58. Considered a mutualistic bacterium that plays a vital role in the fitness and survival of its host, this genus is known to adapt well to cold environments. It can synthesize bioactive compounds with strong antibacterial and antitumor properties55,59, 60.
Our study revealed the existence of two distinct microbiome signatures in cod populations. We found, however, no significant difference in the α-diversity of both microbiomes. Differences in sex, relative condition or maturity classes were neither associated with a specific signature. Similar conclusions were drawn when we looked for temperature, salinity, or depth differences. Our analysis showed, however, that individuals with a Nitrobacter-rich microbiome (group A) were explicitly found in samples collected near the north coast of Cape Breton Island. Whether this is explained by a specific diet, environmental conditions, or distinct migratory patterns combined with seasonal variations is presently unclear. All these factors have been shown to impact the microbiome of marine fish populations57,61,62,63,64,65,66. Seasonal variations may also explain such variations. For example, during winter, the SGSL contains high concentrations of total nitrate (NO2− + NO3−), which is later consumed in the spring by algal blooms (Fig. S3)67. The presence of Nitrobacter reflects a high concentration of nitrite, but whether the GSL nitrite composition has been impacted by humans (agricultural and aquacultural waste) or by a natural origin in currents remains unclear. Alternatively, both signatures may reflect distinctive migratory behavior.
While our study did not identify potential pathogens, it is important to acknowledge that the analysis of the 16S rRNA gene may not be optimally suited for pathogen detection in a host. One limitation is its limited resolution to distinguish strains or genetic variants within a species, which can hinder the differentiation of pathogenic strains from non-pathogenic ones or tracking the progression of an infection. Additionally, this approach may lack specificity in certain cases. Similar sequences may be found in non-pathogenic bacteria or even in bacteria from other species, leading to potential false positives or difficulties in accurately identifying the target pathogens. To overcome these challenges, more targeted approaches would be necessary to address this issue. It is also important to note that dysbiosis is not solely about shifts in microbial composition but also reflects alterations in microbial functions. Studying dysbiosis at higher taxonomic levels can help assess the functional potential of the microbiota, such as the presence of specific metabolic pathways or the production of bioactive molecules, which can influence host-microbe interactions and disease outcomes.
It is important to remember that our study is based on DNA sequencing analysis of DNA extracted from blood samples. Bacterial DNA found in a blood sample can have diverse origins, including various niches such as the intestines, skin, gills, and oral cavity, among others. It can exist in different forms, such as dormant bacteria, living bacteria originating from the same environment (autochthonous), living bacteria from external sources (allochthonous), degraded bacteria, or solely bacterial DNA without viable cells. It does not necessarily reflect the presence of bacteria in the blood. Although the presence of bacteria in the blood of healthy individuals remains debatable, this does not hold in diseased individuals where damaged epithelial membrane integrity is impaired, leading to dysbiotic profiles in the blood microbiome68. Such changes in the blood microbiome are increasingly used to delineate the onset, progression and treatment of diseases, not only in the case of infectious diseases but also in a variety of other conditions, including cancer, metabolic, neurological and cardiovascular diseases, as well as for detecting behavioral anomalies33,36, 69, 70. The use of the blood microbiome in marine biology is only in its infancy, but it offers a new perspective to better assess the health status of wild fish populations.
Conclusions
In summary, our study provides a snapshot of the circulating microbiome signature in an Atlantic cod population and the existence of clear spatial patterns in dysbiotic signatures. Because this logistically friendly approach is compatible with multi-omics analysis, allowing further measurements of changes at the transcriptomic, epigenomic, metabolomic, and glycomic levels, liquid biopsies hold significant potential for better assessing the impact of climate change on the health of cod populations and other marine resources.
Data availability
The sequence data supporting this study's findings are available on the NCBI website at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1015160. The full dataset is available at https://figshare.com/s/25f476865d094bec1b8f, and the Rstudio code is available at https://figshare.com/s/dde39f44befc0343559b.
Abbreviations
- GSL:
-
Gulf of St. Lawrence
- G. morhua :
-
Gadus morhua
- PCoA:
-
Principal coordinate analysis
- ASV:
-
Amplicon sequence variants
- PERMANOVA:
-
Multivariate analysis of variance with permutation
- RDA:
-
Redundancy analysis
References
Meyer-Gutbrod, E., Greene, C., Davies, K. & Johns, D. Ocean regime shift is driving collapse of the North Atlantic right whale population. Oceanography 34, 22–31 (2021).
Hutchings, J. A. & Rangeley, R. W. Correlates of recovery for Canadian Atlantic cod (Gadus morhua) 1. Can. J. Zool. 89, 386–400 (2011).
Swain, D. P., Ricard, D., Rolland, N. & Aubry, É. Assessment of the southern Gulf of St. Lawrence Atlantic Cod (Gadus morhua) stock of NAFO Div. 4T and 4Vn (November to April), March 2019. DFO Can Sci Advis Sec. Res. Doc. 2019/038:iv + 105 (2019).
Neuenhoff, R. D. et al. Continued decline of a collapsed population of Atlantic cod (Gadus morhua) due to predation-driven Allee effects. Can. J. Fish. Aquat. Sci. 76, 168–184 (2019).
Samuelsen, O. B. et al. Viral and bacterial diseases of Atlantic cod Gadus morhua, their prophylaxis and treatment: A review. Dis .Aquat. Org. 71, 239–254 (2006).
Snow, M., Cunningham, C. & Bricknell, I. Susceptibility of juvenile Atlantic cod Gadus morhua to viral haemorrhagic septicaemia virus isolated from wild-caught Atlantic cod. Dis. Aquat. Org. 41, 225–229 (2000).
Gagné, N., Johnson, S., Cook-Versloot, M., MacKinnon, A. & Olivier, G. Molecular detection and characterization of nodavirus in several marine fish species from the northeastern Atlantic. Dis. Aquat. Org. 62, 181–189 (2004).
Scharsack, J. P. & Franke, F. Temperature effects on teleost immunity in the light of climate change. J. Fish Biol. 101, 780–796 (2022).
Dutil, J.-D. & Lambert, Y. Natural mortality from poor condition in Atlantic cod (Gadus morhua). Can. J. Fish. Aquat. Sci. 57, 826–836 (2000).
Regular, P. M. et al. Indexing starvation mortality to assess its role in the population regulation of Northern cod. Fish. Res. 247, 106180 (2022).
Blackwell, B. G., Brown, M. L. & Willis, D. W. Relative Weight (Wr) Status and Current Use in Fisheries Assessment and Management. Rev Fish Sci. 8, 1–44 (2000).
Wilder, S. M., Raubenheimer, D. & Simpson, S. J. Moving beyond body condition indices as an estimate of fitness in ecological and evolutionary studies. Funct. Ecol. 30, 108–115 (2016).
Bratberg, M. et al. Effects of oil pollution and persistent organic pollutants (POPs) on glycerophospholipids in liver and brain of male Atlantic cod (Gadus morhua). Chemosphere 90, 2157–2171 (2013).
Giannetto, A. et al. Influence of continuous light treatment on expression of stress biomarkers in Atlantic cod. Dev. Comp. Immunol. 44, 30–34 (2014).
Gopalakrishman, V. et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018).
Routy, B. et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 359, 91–97 (2018).
Whittle, E., Leonard, M. O., Harrison, R., Gant, T. W. & Tonge, D. P. Multi-method characterization of the human circulating microbiome. Front. Microbiol. 9, 3266 (2019).
Caza, F., Granger Joly de Boissel, P. G., Villemur, R., Betoulle, S. & St-Pierre, Y. Liquid biopsies for omics-based analysis in sentinel mussels. PLoS ONE 14, 25 (2019).
Ferchiou, S., Caza, F., De Boissel, P. G. J., Villemur, R. & St-Pierre, Y. Applying the concept of liquid biopsy to monitor the microbial biodiversity of marine coastal ecosystems. ISME commun. 2, 61 (2022).
Fronton, F. et al. Insights into the circulating microbiome of Atlantic and Greenland halibut populations: The role of species-specific and environmental factors. Sci. Rep. 13, 5971 (2023).
Callahan, B. J., McMurdie, P. J. & Holmes, S. P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639–2643 (2017).
Cao, Y. microbiomeMarker: Microbiome Biomarker Analysis. R package version 0.0.1.9000 (2020;).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013).
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D. et al. vegan: Community Ecology Package. R package version 2.5-7 (2020).
Ssekagiri, A., Ijaz, U. Z. & Sloan, W.T. microbiomeSeq: An R Package for Analysis of Microbial Communities in an Environmental Context 39 (2017).
South A. rnaturalearth: World Map Data from Natural Earth. 2017;R package version 0.1.0.
Palanisamy, V., Gajendiran, V. & Mani, K. Meta-analysis to identify the core microbiome in diverse wastewater. Int. J. Environ. Sci. Technol. 19, 5079–5096 (2022).
Froese, R. Cube law, condition factor and weight-length relationships: History, meta-analysis and recommendations. J. Appl. Ichthyol. 22, 241–253 (2006).
Legendre, P. & Gallagher, E. D. Ecologically meaningful transformations for ordination of species data. Oecologia 129, 271–280 (2001).
Le Cren, E. D. The length-weight relationship and seasonal cycle in gonad weight and condition in the perch (Perca fluviatilis). J. Anim. Ecol. 20, 201 (1951).
Keys, A. B. The weight-length relation in fishes. Proc. Natl. Acad. Sci. 14, 922–925 (1928).
Ward, B. B. et al. (eds) Nitrification (American Society for Microbiology Press, 2011).
Blauwkamp, T. A. et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat. Microbiol. 4, 663–674 (2019).
Emery, D. C. et al. Comparison of blood bacterial communities in periodontal health and periodontal disease. Front. Cell. Infect. Microbiol. 10, 577485 (2021).
Païssé, S. et al. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 56, 1138–1147 (2016).
Poore, G. D. et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 579, 567–574 (2020).
Tan, C. C. S., Acman, M., Van Dorp, L. & Balloux, F. Metagenomic evidence for a polymicrobial signature of sepsis. Microb. Genom. 7, 000642 (2021).
Ferchiou, S., Caza, F., Villemur, R., Labonne, J. & St-Pierre, Y. Skin and blood microbial signatures of sedentary and migratory trout (Salmo trutta) of the Kerguelen Islands. Fishes 8, 174 (2023).
Herder, E. A., Skeen, H. R., Lutz, H. L. & Hird, S. M. Body size poorly predicts host-associated microbial diversity in wild birds. Microbiol. Spectr. 11, e03749-e3822 (2023).
Mtshali, K. Exploration and Comparison of Bacterial Communities Present in Bovine Milk, Faeces and Blood Using 16S rRNA Metagenomic Sequencing (North-West University, 2022).
Scarsella, E., Meineri, G., Sandri, M., Ganz, H. H. & Stefanon, B. Characterization of the blood microbiome and comparison with the fecal microbiome in healthy dogs and dogs with gastrointestinal disease. Vet. Sci. 10, 277 (2023).
Gonçalves, E. et al. Skin and stinger bacterial communities in two critically endangered rays from the South Atlantic in natural and aquarium settings. MicrobiologyOpen 9, e1141 (2020).
Li, L.-H. et al. Spatial structure of the microbiome in the gut of Pomacea canaliculata. BMC Microbiol. 19, 273 (2019).
Sugden, S., Holert, J., Cardenas, E., Mohn, W. W. & Stein, L. Y. Microbiome of the freshwater sponge Ephydatia muelleri shares compositional and functional similarities with those of marine sponges. ISME J. 16, 2503–2512 (2022).
Savard, P., Fernandes, T., Dao, A., McMeans, B. & Lazar, C. S. Seasons influence the native gut microbiome of lake trout salvelinus namaycush. Appl. Microbiol. 3(1), 276–287. https://doi.org/10.3390/applmicrobiol3010019 (2023).
Cheng, C. et al. Characterization of the lung microbiome and exploration of potential bacterial biomarkers for lung cancer. Transl. Lung Cancer Res. 9, 693–704 (2020).
Qiu, J., Zhou, H., Jing, Y. & Dong, C. Association between blood microbiome and type 2 diabetes mellitus: A nested case–control study. J. Clin. Lab. Anal. 33, e22842 (2019).
Ericsson, A. C. et al. Molecular and culture-based assessment of the microbiome in a zebrafish (Danio rerio) housing system during set-up and equilibration. Anim. Microbiome 3, 55 (2021).
Garbas, K., Zapała, P., Zapała, Ł & Radziszewski, P. The role of microbial factors in prostate cancer development—An Up-to-date review. JCM 10, 4772 (2021).
Minich, J. J. et al. Microbial ecology of Atlantic salmon (Salmo salar) hatcheries: Impacts of the built environment on fish mucosal microbiota. Appl. Environ. Microbiol. 86, 19 (2020).
Ortiz, W. E., Carlos-Shanley, C. & Huertas, M. Impact of sublethal concentrations of nitrite on goldfish (Carassius auratus) microbiomes. Microb. Ecol. https://doi.org/10.1007/s00248-022-02123-4 (2022).
Herbert, R. A. Nitrogen cycling in coastal marine ecosystems. FEMS Microbiol. Rev. 23, 563–590 (1999).
Kieber, R. J., Li, A. & Seaton, P. J. Production of nitrite from the photodegradation of dissolved organic matter in natural waters. Environ. Sci. Technol. 33, 993–998 (1999).
Nixon, S. C., Trent, Z., Marcuello, C. & Lallana, C. Europe’s Water: An Indicator-Based Assessment (European Environment Agency, 2003).
Bowman, J. P. Bioactive compound synthetic capacity and ecological significance of marine bacterial genus Pseudoalteromonas. Mar. Drugs 5, 220–241 (2007).
Bowman, J. P. & McMeekin, T. A. Pseudoalteromonas. In Bergey’s Manual of Systematics of Archaea and Bacteria 1st edn (eds Whitman, W. B. et al. et al.) 1–22 (Wiley, 2015).
Lokesh, J. & Kiron, V. Transition from freshwater to seawater reshapes the skin-associated microbiota of Atlantic salmon. Sci. Rep. 6, 19707 (2016).
Ringø, E., Sperstad, S., Myklebust, R., Refstie, S. & Krogdahl, Å. Characterisation of the microbiota associated with intestine of Atlantic cod (Gadus morhua L.). Aquaculture 261, 829–841 (2006).
Atencio, L. A. et al. Antimicrobial-producing Pseudoalteromonas from the marine environment of Panama shows a high phylogenetic diversity and clonal structure. J. Basic Microbiol. 58, 747–769 (2018).
Bosi, E. et al. The pangenome of (Antarctic) Pseudoalteromonas bacteria: Evolutionary and functional insights. BMC Genom. 18, 93 (2017).
Bagi, A. et al. Gastrointestinal microbial community changes in Atlantic cod (Gadus morhua) exposed to crude oil. BMC Microbiol. 18, 1–14 (2018).
Clements, K. D., Angert, E. R., Montgomery, L. & Choat, J. H. Intestinal microbiota in fishes: What’s known and what’s not. Mol. Ecol. 23, 1891–1898 (2014).
Egerton, S., Culloty, S., Whooley, J., Stanton, C. & Ross, R. P. The gut microbiota of marine fish. Front. Microbiol. 9, 873 (2018).
Ringø, E., Strøm, E. & Tabachek, J.-A. Intestinal microflora of salmonids: A review. Aquac. Res. 26, 773–789 (1995).
Sylvain, F. -É. et al. Fish skin and gut microbiomes show contrasting signatures of host species and habitat. Appl. Environ. Microbiol. 86, e00789-e820 (2020).
Walter, J. M., Bagi, A. & Pampanin, D. M. Insights into the potential of the Atlantic cod gut microbiome as biomarker of oil contamination in the marine environment. Microorganisms 7, 209 (2019).
Blais, M., Galbraith, P. S., Plourde, S., Devred, E., Clay, S., Lehoux, C. et al. Chemical and Biological Oceanographic Conditions in the Estuary and Gulf of St. Lawrence During 2020. DFO Can Sci Advis Sec. 2021;Res. Doc. 2021/060:iv + 67 (2021).
Camilleri, M. Leaky gut: Mechanisms, measurement and clinical implications in humans. Gut 68, 1516–1526 (2019).
Cussotto, S., Sandhu, K. V., Dinan, T. G. & Cryan, J. F. The neuroendocrinology of the microbiota-gut-brain axis: A behavioural perspective. Front. Neuroendocrinol. 51, 80–101 (2018).
Kuti, D. et al. Gastrointestinal (non-systemic) antibiotic rifaximin differentially affects chronic stress-induced changes in colon microbiome and gut permeability without effect on behavior. Brain Behav. Immun. 84, 218–228 (2020).
Acknowledgements
This research was performed with the logistic support of Fisheries and Oceans Canada (DFO). The authors would like to thank all the personnel from the DFO for their help and hospitality during the survey of the Southern Gulf of St. Lawrence. The author would like to thank Sophia Ferchiou and France Caza for their support on the bioinformatical analysis and Marlène Fortier for her excellent technical help.
Funding
This work was funded by the Fonds de Recherche du Québec-Nature et Technologie (YSP, RV and DR). D.R. is supported by the Canada Research Chair Program.
Author information
Authors and Affiliations
Contributions
D.R., R.V., F.F. and Y.S.P. conceived the study. All authors were responsible for the interpretation of data and critical appraisal. All authors executed the experiments and contributed to the experimental design and analyses of the results. F.F. and Y.S.P. drafted the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fronton, F., Villemur, R., Robert, D. et al. Divergent bacterial landscapes: unraveling geographically driven microbiomes in Atlantic cod. Sci Rep 14, 6088 (2024). https://doi.org/10.1038/s41598-024-56616-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-56616-y
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
