
Abstract
Two genetically different mitochondrial haplogroups of Brachidontes pharaonis (p-distance 6.8%) have been identified in the Mediterranean Sea. This hinted at a possible presence of doubly uniparental inheritance in this species. To ascertain this possibility, we sequenced two complete mitogenomes of Brachidontes pharaonis mussels and performed a qPCR analysis to measure the relative mitogenome copy numbers of both mtDNAs. Despite the presence of two very similar regions composed entirely of repetitive sequences in the two haplogroups, no recombination between mitogenomes was detected. In heteroplasmic individuals, both mitogenomes were present in the generative tissues of both sexes, which argues against the presence of doubly uniparental inheritance in this species.
Similar content being viewed by others


Introduction
Brachidontes pharaonis (P. Fischer, 1870) is a Lessepsian mussel species that invaded the Mediterranean Sea after the opening of the Suez Canal in 1869 connecting the Indian Ocean through the Read Sea1,2. Due to intrinsic plasticity and overlapping morphological traits, this species is often mistaken for B. variabilis, which inhabits regions of the Indian Ocean and the West Pacific Ocean3. It is a gonochoristic species, with a white gonad-bearing mantle in males and brown mantle in females4. There is only one reported case of a clearly hermaphroditic individual5 in this species, and the sex determination system is otherwise quite stable.
Many bivalve species possess a unique system of mitochondrial inheritance called doubly uniparental inheritance (DUI)6,7,8. Under DUI male individuals are heteroplasmic with an additional, divergent mitogenome located mostly in their gonads. Furthermore, this divergent mitogenome is passed through the sperm to the progeny, unlike the male mitogenome of non-DUI animals, which is lost upon fertilization. In normal circumstances, germlines are homoplasmic towards one of the mitogenomes, M-type in males and F-type in females. However, in rare cases this second male-type mitogenome can also be detected in female individuals (for such exceptions, see9,10,11). After fertilization, initially heteroplasmic embryos behave in one of two ways. If the embryo is a male, the M-type mitochondria group together during the first cell divisions, becoming the dominant mitochondrial fraction in gonads of adult male (somatic tissues are mostly dominated by F-type mitochondria). On the other hand, if the embryo develops into a female individual, the M-type mitochondria get dispersed during the first few division cycles and the signal from the M-type mitochondrial DNA disappears. The mechanism of this elimination is unknown12,13,14,15,16,17,18,19,20,21,22.
Genetic studies based on cox1 and 16S rRNA gene markers revealed the presence of two (p-distance 6.8%) different haplogroups (M and L; referred to respectively A and B later on due to possible misinterpretation of the M haplogroup as male-type mtDNA) in B. pharaonis, suggesting the existence of cryptic species. No heteroplasmic individuals were identified within the Mediterranean population, and the presence of a particular mitochondrial haplogroup did not correlate with the sex of the individuals1,3,23. Nevertheless, many studies suggested the presence of DUI in this genus3,16,19,24,25,26. However, there are well-known difficulties with detecting DUI by end-point PCR with universal primers27. The variability of the sequence divergence between mitogenome pairs ranges from around 5% to over 50%16,28,29. This makes the whole approach problematic for the following reasons. In the cases of low divergence, the amplification of the most prevalent mitogenome, or the one with sequence slightly better matching with PCR primers, may mask the presence of the second mitogenome. In the cases of high divergence, universal primers may not be universal enough to pick up the second genome at all. Moreover, regardless of the divergence, there is always a possibility that a mitogenome of another, contaminating, biological entity, would be co-amplified. This is facilitated by high sensitivity of end-point PCR protocols12,21,27. All this prompted us to use a quantitative mitogenomic approach to further corroborate the possibility of DUI in this species.
Results
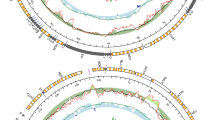
We were able to detect and sequence two slightly divergent mitogenomes in Brachidontes pharaonis samples (p-distance 0.071 for coding genes and 0.086 for the whole mtDNA). The preliminary results of the end-point PCR (Table 1) suggested not only the existence of heteroplasmy, but also showed that it was not linked with sex of individuals in this species. The results between PCR and qPCR differ because not every homoplasmic individual in PCR was checked with qPCR and some of the heteroplasmic individuals were resolved as homoplasmic after the qPCR. This was due to the very low estimated copy number of the second mitogenome, much lower than the copy number of a nuclear gene. Very high Cq values, close to the NTC control, were observed in these samples. Since such results could be either caused by non-specific signal (primer-dimer formation) or the truly very low gene copy number (much lower than that of atpα nuclear gene), these were classified as likely caused by sample contamination and not by true heteroplasmy. In the absence of any sex-bias, instead of naming those mitogenomes as M-type for male and F-type for female, we will refer to them neutrally as type A and type B. The mitogenomes (Fig. 1) are similar in length (type A: 20,066 bp, type B: 20,072 bp), code the typical set of 13 proteins, 2 rRNAs and 23 tRNAs, all on the same strand and in the same order. The striking feature of both mitogenomes is the presence of two long noncoding regions, rich in repetitive sequences, that divide the mitogenomes almost perfectly in half. Fragments of open reading frames in noncoding regions were identified as repeats of the 5′ end of atp6, suggesting the occurrence of multiple tandem duplication random loss events that encompass fragments of the coding sequences, throughout the evolution of these mitogenomes.

Genetic map of Brachidontes pharaonis mitochondrial genomes. The white arrows with orange bands represent protein-coding genes with predicted transmembrane domains. The dark arrows represent the rRNA and tRNA genes; the white boxes indicate the location of repetitive sequences. The figure and compositional indices were generated with MITOCONSTRICTOR as in33,34.
Given the overall relatively small divergence in mitochondrial sequences, the long repetitive sequences, and heteroplasmy, the possibility of widespread mitochondrial recombination at one of the long noncoding regions was considered. Such recombination would impair our ability to detect the type of mitogenome with just one small qPCR fragment. To address this issue, four pairs of specific qPCR primers were used to quantify the two parts of mitogenomes A and B separately (Supplementary data Table S1 and S2). Under the no recombination hypothesis the four quantities, one per part of the mitogenome, should match according to the mitogenome type, whereas if recombination is involved, the four estimates would be mismatched. Analysis of 30 B. pharaonis individuals revealed no noticeable difference between the halves of mitogenome copy number (Fig. 2). We have observed seven cases of heteroplasmy divided more or less evenly (no statistically significant differences for Fisher’s exact test) between sexes. The heteroplasmic individuals were: 4 females, 3 males in qPCR (23%) and 6 females, 7 males in end-point PCR analysis (33%). Both A and B mitogenomes were detectable in males and females (Supplementary data Table S5–S10 and Fig. S1).

The ratio of mtDNA to nDNA in seven heteroplasmic individuals of B. pharaonis. F represents female individuals; M represents male individuals. The yellow and green colored bars represent sequence copy number per nuclear DNA for type A mtDNA; The pink and blue bars represent sequence copy number per nuclear DNA for type B mtDNA. The target genes atp6 and nd4 are located on the opposite sites of the respective mitogenomes separated by repetitive sequences. Samples described as “Mantle” contained mainly gonadal tissues, not just the somatic mantle.
The newly reported mitogenomes of B. pharaonis form a monophyletic clade within the Brachidontes genus clade (Fig. 3). Brachidontes is a large genus composed of around 50 individual species (WoRMS database). However, due to the small number of available complete mitogenomes, the analysis of taxonomic affinities of the mitogenomes, including possible mitochondrial introgression events within this genus can not be conclusive. As a side note, phylogenetic analysis revealed the following inconsistency: genus Brachidontes seems to be paraphyletic, due to the presence of M. solisianus mitogenome within the clade. This hints that the taxonomy of M. solisianus may require reconsideration. Nevertheless, the phylogenetic distance between the two B. pharaonis mitogenomes is similar to the intraspecies distance between F-type mitogenomes form north and south populations of the Chilean mussels Perumytilus purpuratus which were separated from each other in Pleistocene30,31,32. It is also greater than the distance between several interspecies pairs of mitogenomes: Mytilus edulis and M. galloprovincialis, Bathmodiolus marisindicus and B. septemdierum, as well as all available mitogenomes form genus Gigantidas.

The phylogenetic position of Brachidontes pharaonis mitogenomes among mitogenomes of other mytilid mussels. The left tree represents Bayesian phylogenetic analysis done in BEAST2, and the right tree represents maximum likelihood analysis done in IQ-TREE. The nodes support values equaled 1 (Posterior probabilities, left) and 100% (bootstrap values, right), unless indicated differently.
Discussion
We reported two mitogenomes (p-distance 8.6%) in B. pharaonis. These two mitogenomes are quite frequently present in the same individuals (Table 1). Is it possible that the observed heteroplasmy was not true but the result of a recent numt which is segregated in the population? We dismiss this possibility for the following reasons. Both mitogenomes were highly expressed at RNA level (31% of all NGS RNAseq sequencing reads mapped onto the A-type mtDNA and 4% onto the B-type mtDNA sequence), which is not a typical feature of a numt. Moreover, numts are usually much smaller than the complete mitogenomes (but look here35), and their sequence is degenerated36. Here both very large mitogenomes have all their genes intact and seemingly functional. Finally, the ratios of copy numbers of mitogenomes to the nuclear genome are not constant. A tight correlation between a numt and nuclear copy number is expected, which is not the case here. What can be the reason for the existence of two such haplotypes within this species?
Since DUI was postulated for some Brachidontes species based solely on sequence divergence, it is reasonable to consider it here3,24. One may ask what sequence divergence threshold (if there is one) can be unequivocally associated with DUI? The situation present in the Baltic Sea Mytilus mussels37,38, where the divergent paternal mitogenome was replaced by the masculinized F-type mitogenome, is exceptional. Consequently, besides the differences in the noncoding control region, the M and F mitogenomes are identical in that case. If such extreme cases are not considered, the lowest divergence of the complete mitochondrial genomes observed and counted as DUI belongs to Arctica islandica28,29, with the M-F divergence (nucleotide p-distance) at 5.1% for coding genes and 6.9% for the whole mtDNA (Fig. S2). Even if a wider species range is considered, for which only cox116 gene fragments are known, the least divergent is also Arctica islandica: 2.5% at the protein level and 5.2% at the nucleotide level. So, can we count the divergence observed in B. variabilis3 3.6% at the nucleotide level as a suggestion of DUI existence, with the lowest known divergence in the cox1 gene? Or is this just a high haplotype diversity within a species, not linked to sex-related heteroplasmy? This rhetorical question also applies to B. pharaonis.
In the context of DUI, divergence alone is just a secondary factor, depending only on the time of separation of the two lineages and their evolutionary rates. Nevertheless, for the emerging mitogenomes to play different physiological roles, there must be functional differences between their proteins, which is problematic for low genetic distances. However, even with a small number of nonsynonymous substitutions in the sequence, the encoded protein may become different enough (like in quickly evolving viruses or targeted enzyme engineering39,40) to differ in activity. Unfortunately, tracing such substitutions in the context of DUI is difficult, but the possibility that a low-divergence mitogenome may still play distinct physiological role can’t be discounted. Are there any other features that could help classify a mitogenome as involved in DUI? Perhaps we should consider additional open reading frames (ORFs) or gene extensions with no homology to the second mitogenome as a DUI marker? Is this a universal feature of DUI mitogenomes? Indeed, this seems to be true for all known pairs of DUI mitogenomes (Table 2): each of them has some gender-specific structural features, but this is not the case for B. pharaonis. Here, both mitogenomes are structurally identical.
Another feature of mtDNA, which has been reported in the context of DUI, is mitochondrial recombination65,66. The existence of very large repetitive elements suggests, that at least intramolecular recombination would be present in B. pharaonis. Yet, we have not found any evidence for the exchange of large parts between A and B mitogenomes. They apparently maintain their integrity, despite the conditions, which should favor the exchange67. The qPCR method would not detect low frequency, short span recombination so this has to be considered cautiously. However, no detectable signature of recombination is present within the A and B mitogenomic sequences, therefore even if such recombination is present, it is limited to somatic cells.
Several studies postulated the existence of cryptic species within the Brachidontes genus (B. pharaonis/variabilis3 and B. puniceus/exustus24,68). These were usually argued by the distinct divergence (p-distance) at the nucleotide level (~ 7–20%) between mitochondrial sequences. However, at the protein level, these distances were much lower (p-distance ~ 0–1.5%). These must be interpreted cautiously because only short fragments of one mitochondrial gene are available (cox1: AY621835-AY621860; AY621862-AY621865; AY621909; AY621911; AY825105-AY825108; DQ836012; DQ836013; DQ836019-DQ836021) and the mitogenomic distances may be quite different (Fig. 4). However, all nonsynonymous substitutions in the mentioned cox1 fragments stay within the group of nonpolar, mostly aliphatic amino acids (Ile, Val, Leu, and in two cases also Met and Phe). This suggests no alternation in the overall protein structure/reactivity as is indicated by amino acid substitution matrices69,70 derived from empirical data. The issue of cryptic species identification becomes even more complicated when specimens are erroneously assigned68 to their respective species, as in the case of Brachidontes/Mytilaster genera. This is the example of Mytilaster solisianus (d'Orbigny, 1842), known earlier as Brachidontes solisianus (d'Orbigny, 1842). The current taxonomy classifies this species as Mytilaster, but the mitogenomic phylogenetic tree (Fig. 3) does not support this. Furthermore, the M. solisianus mitogenome deposited in GenBank has been wrongly annotated as Perna perna71. A taxonomic revision of Brachidontes seems warranted but should not be based solely on mitochondrial markers, which are known to evolve under strong selective constraints and are prone to introgression72,73. A good example of potential pitfalls associated with such a simplistic use of mitochondrial markers comes from the case of Baltic Mytilus trossulus, which in fact is a nuclear hybrid of M. edulis and M. trossulus that lost their native mitochondrial genome towards one from M. edulis72,74,75 (also see here for M/F recombination66,76). If only the mitogenomic protein level p-distances are considered, the cryptic species hypothesis is becoming less likely. However, the fact that heteroplasmic individuals were consistently observed suggests that anomalies in the mitochondrial inheritance may be involved.

The divergence between A and B type mitogenome of B. pharaonis. The blue color represents the p-distance for nucleotide sequences, and dark red color represents p-distance for translated protein genes.
In conclusion, B. pharaonis represents a species possessing two slightly different mitogenomes (p-distance 8.6%) in every possible combination between the sexes. Total homoplasmy for mitogenomes A or B, as well as heteroplasmy of both mitogenomes within a single individual are possible. This heteroplasmy is not correlated with sex, which excludes DUI. Did we observe the first stages of emerging DUI in this species or is heteroplasmic B. pharaonis a hybrid of two very similar cryptic species and the observed heterpolasmy represents paternal leakage? Future studies concerning Brachidontes populations are needed to conclusively answer this question. On a more practical level, we advise that the use of somatic tissues77, in phylogenetic studies on bivalves, usually practiced as a precaution to avoid amplification of the potential M mitogenome, may not always work as planned78. In the case of B. pharaonis such an approach would lead to seemingly random amplification of one of the two mitogenomes present in an individual.
Methods
Samples of Brachidontes pharaonis mussels were gathered in June 2014–2015 at the salt pan “infersa” of the Marsala lagoon (northwest of Sicily—Italy). Individuals were sectioned with a sterile scalpel blade, checked for male or female gametes under a light microscope, and stored frozen in − 80 °C until further use. DNA was extracted using the modified CTAB extraction method79,80. Small tissue samples (~ 50 mg) were incubated overnight in the 700 µl of extraction buffer (2% CTAB, 0.1 M Tris–HCl, 1.4 M NaCl, 20 mM EDTA, 1 mg/ml proteinase K and 35 mM 2-mercaptoethanol). The digested samples were then extracted with chloroform (1:1 vol/vol ratio) and centrifuged three times at 20,000 × g for 10 min. Then the DNA remaining in the aqueous phase was precipitated by mixing with cold isopropanol (1:1 vol/vol), incubated for 20 min at − 20 °C, and centrifuged (20,000 × g for 30 min at 4 °C). The recovered DNA pellets were washed twice with 75% ethyl alcohol and dried in a vacuum concentrator. At the final stage, DNA pellets were resuspended in Tris–EDTA buffer, checked for DNA concentration and integrity by gel electrophoresis and Epoch microplate Spectrophotometer. RNA was extracted with the GenElute Mammalian Total RNA miniprep kit (Sigma).
Total RNA from three mantles of male individuals was pooled and sent to Macrogen Inc (Korea) for high throughput NGS sequencing (MiSeq Illumina, TruSeq NGS library 2 × 150 bp). Raw sequencing reads have been submitted to the SRA GenBank database under accession number SRR19141670. The acquired data were processed according to the Oyster river protocol34,81 and assembled into the raw transcriptome. Long-range PCR primers for amplification of overlapping mitogenome fragments have been designed based on assembled contigs containing mitochondrial genes identified with Wise2 software82. PCRs were carried out in a volume of 20 µl, containing 25 ng of DNA, primers at 0.4 µM each, dNTP at 200 µM, 1.5 mM MgCl2, and 0.4 U Phusion High-Fidelity polymerase (Thermo Scientific) suspended in GC buffer for difficult GC-rich templates. The PCR conditions were as follows: initial denaturation at 98 °C for 30 s; 35 cycles of denaturation at 98 °C for 10 s, annealing (Table S3 and S4) for 30 s and extension at 72 °C for 8 min. The final extension at 72 °C lasted 10 min. The amplified products were then assigned to their respective mitogenome (A and B) and sent for another NGS sequencing (MiSeq Illumina, TrueSeq NGS library, 2 × 300 bp). Complete mitochondrial DNA sequences have been recovered with NOVOplasty83 software and validated by mapping NGS reads onto the assembled mitogenomes in CLC Genomics Workbench 9.5 (QIAGEN). The two acquired mitogenomes were then annotated with CRITICA84, Wise282, GLIMMER85, ARWEN86, and nhmmer87. Mitogenome circular diagrams and compositional indices were drawn with MITOCONSTRICTOR33,34 (https://github.com/aburzynski/mitoconstrictor). Annotated mitochondrial genomes for Brachidontes pharaonis were deposited in the GenBank under accession numbers ON464163 and ON464164.
Based on assembled transcriptome and two mitogenome sequences, a set of five qPCR primer pairs spanning: nuclear atpα gene and mitochondrial nad4, atp6 (from both mitogenomes; type A and type B) were designed in Primer388. The specificity of each primer pair was verified, and there was no cross-amplification between A and B mitogenomic fragments. Quantitative PCR efficiency has been verified by running standard curves in nine repetitions with seven dilution points of samples. Reactions quantifying A and B mtDNA, as well as nuclear DNA, were performed on an ECO48 (Illumina/now PCRmax) Real-Time PCR System according to the qPCR kit (EurX) manufacturer instructions, in 10 µl reaction volume containing 1 × SG qPCR Master Mix, 2 µl of DNA (at ~ 15 ng/µl) and 0.5 µM of each primer. The thermal profile was as follows: initial denaturation at 95 °C for 10 min, followed by 35 cycles of 10 s denaturation at 94 °C, annealing at 60 °C for 30 s, elongation at 72 °C for 30 s, and a melting curve 55–95 °C. Primer sequences, reaction efficiency, and tabulated results have been placed in Supplementary data S1 and S2. Statistical Fisher’s exact test (Statistica 7, StatSoft) was used to calculate the significance of the association between sex and heteroplasmy.
Reconstruction of phylogenetic relations within the Mytilidae family was based on 53 mitogenomes (Table 3). All mitogenomes from the Mytilidae family available GenBank database (accessed May 2021) were used. The 12 mitochondrial protein coding genes (atp8 was omitted due to high divergence and in a few cases annotation problems89) were used. Each gene was aligned separately, at protein level in MEGA790 software, with the ClustalW algorithm, to ensure that the codon structure of each gene is retained at the alignment level. Gap Extension and Gap Opening costs were set at 5, and the final alignments were visually inspected. No obvious alignment problems were encountered. Phylogenetic reconstruction was done using two approaches: Bayesian inference (BI) and Maximum Likelihool (ML). The same set of individual gene alignments (as separate data partitions) was used in both analyses.
BI was performed in BEAST291. The parameters were as follows: nucleotide substitution model GTR + I + 4G, based on the results from the bModelTest package for BEAST2 software, relaxed log-normal clock, due to the varying evolution rates between F and M-type mtDNA and Yule prior to the common tree. The Markov chain was run in four replicates for 107 generations and sampled every 10,000th step. The convergence of samples was checked with Tracer92, the effective sample size for estimated parameters was greater than 200. ML was performed in IQ-TREE93, under the default parameters with ultrafast bootstrap approximation parameter set to 10,000 replicates. Substitution models for every partition were chosen with build-in ModelFinder tool. Model GTR + F + I + 4G was the best fitting for all protein genes except nd2, nd4, nd5 for which GTR + F + R5 was chosen.
References
Sirna Terranova, M, Lo Brutto, S., Arculeo, M. & Mitton, J. B. Population structure of Brachidontes pharaonis (P. Fisher, 1870) (Bivalvia, Mytilidae) in the Mediterranean Sea, and evolution of a novel mtDNA polymorphism. Mar. Biol. 150, 89–101 (2006).
Antit, M., Amor, N., Urra, J., Alagaili, A. N. & Farjallah, S. Genetic variability of the Lessepsian migrant mussel Brachidontes pharaonis (Bivalvia: Mytilidae) in Tunisia. Afr. J. Mar. Sci. 40, 211–217 (2018).
Sirna Terranova, M., Lo Brutto, S., Arculeo, M. & Mitton, J. B. A mitochondrial phylogeography of Brachidontes variabilis (Bivalvia: Mytilidae) reveals three cryptic species. J. Zool. Syst. 45, 289–298 (2007).
El-Deeb, R. S., Abdel Razek, F. A., Omar, H. A., Khafage, A. R. & Abdul-Aziz, K. K. The gametogenic cycle and spawning of the mussel Brachidontes pharaonis (Fischer, 1876) (Bivalvia: Mytilidae) from Alexandria Coast, Egypt. Egypt. J. Aquat. Res. 44, 353–359 (2018).
Razek, F. A. A., El-Deeb, R. S., Abdul-Aziz, K. K., Omar, H. A. & Khafage, A. R. Hermaphroditism in Brachidontes pharaonis (Fischer, 1876) (Bivalvia: Mytilidae) from the Alexandria Coast, Egypt. Egypt. J. Aquat. Res. 43, 265–268 (2017).
Zouros, E., Oberhauser Ball, A., Saavedra, C. & Freeman, K. R. An unusual type of mitochondrial DNA inheritance in the blue mussel Mytilus. Proc. Natl. Acad. Sci. U S A 91, 7463–7467 (1994).
Skibinski, D. O., Gallagher, C. & Beynon, C. M. Sex-limited mitochondrial DNA transmission in the marine mussel Mytilus edulis. Genetics 138, 801–809 (1994).
Hoeh, W. R., Stewart, D. T., Sutherland, B. W. & Zouros, E. Multiple origins of gender-associated mitochondrial DNA lineages in bivalves (Mollusca: Bivalvia). Evolution 50, 2276–2286 (1996).
Obata, M., Sano, N., Kawamura, K. & Komaru, A. Inheritance of two M type mitochondrial DNA from sperm and unfertilized eggs to offspring in Mytilus galloprovincialis. Dev. Growth Differ. 49, 335–344 (2007).
Obata, M., Sano, N. & Komaru, A. Different transcriptional ratios of male and female transmitted mitochondrial DNA and tissue-specific expression patterns in the blue mussel, Mytilus galloprovincialis. Dev. Growth Differ. 53, 878–886 (2011).
Obata, M., Kamiya, C., Kawamura, K. & Komaru, A. Sperm mitochondrial DNA transmission to both male and female offspring in the blue mussel Mytilus galloprovincialis. Dev. Growth Differ. 48, 253–261 (2006).
Passamonti, M. & Plazzi, F. Doubly uniparental inheritance and beyond: The contribution of the Manila clam Ruditapes philippinarum. J. Zool. Syst. Evol. Res. 58, 529–540 (2020).
Plazzi, F. & Passamonti, M. Footprints of unconventional mitochondrial inheritance in bivalve phylogeny: Signatures of positive selection on clades with doubly uniparental inheritance. J. Zool. Syst. Evol. Res. 57, 258–271 (2019).
Zouros, E. & Rodakis, G. C. Doubly uniparental inheritance of mtDNA: An unappreciated defiance of a general rule. Adv Anat Embryol Cell Biol 231, 25–49 (2019).
Guerra, D. et al. Evolution of sex-dependent mtDNA transmission in freshwater mussels (Bivalvia: Unionida). Sci. Rep. 7, 1551 (2017).
Gusman, A., Lecomte, S., Stewart, D. T., Passamonti, M. & Breton, S. Pursuing the quest for better understanding the taxonomic distribution of the system of doubly uniparental inheritance of mtDNA. PeerJ 4, e2760 (2016).
Ghiselli, F. et al. Structure, transcription, and variability of metazoan mitochondrial genome: Perspectives from an unusual mitochondrial inheritance system. Genome Biol. Evol. 5, 1535–1554 (2013).
Milani, L., Ghiselli, F., Guerra, D., Breton, S. & Passamonti, M. A comparative analysis of mitochondrial ORFans: New clues on their origin and role in species with doubly uniparental inheritance of mitochondria. Genome Biol. Evol. 5, 1408–1434 (2013).
Zouros, E. Biparental inheritance through uniparental transmission: The doubly uniparental inheritance (DUI) of mitochondrial DNA. Evol. Biol. 40, 1–31 (2013).
Passamonti, M. & Ghiselli, F. Doubly uniparental inheritance: Two mitochondrial genomes, one precious model for organelle DNA inheritance and evolution. DNA Cell Biol. 28, 79–89 (2009).
Theologidis, I., Fodelianakis, S., Gaspar, M. B. & Zouros, E. Doubly uniparental inheritance (DUI) of mitochondrial DNA in Donax Trunculus (bivalvia: Donacidae) and the problem of its sporadic detection in bivalvia. Evolution 62, 959–970 (2008).
Breton, S., Beaupré, H. D., Stewart, D. T., Hoeh, W. R. & Blier, P. U. The unusual system of doubly uniparental inheritance of mtDNA: Isn’t one enough?. Trends Genet. 23, 465–474 (2007).
Shefer, S., Abelson, A., Mokady, O. & Geffen, E. Red to Mediterranean Sea bioinvasion: Natural drift through the Suez Canal, or anthropogenic transport?. Mol. Ecol. 13, 2333–2343 (2004).
Lee, T. & Foighil, D. O. Hidden Floridian biodiversity: Mitochondrial and nuclear gene trees reveal four cryptic species within the scorched mussel, Brachidontes exustus, species complex. Mol. Ecol. 13, 3527–3542 (2004).
Alves, F. A. et al. Detection of mitochondrial DNA heteroplasmy suggests a doubly uniparental inheritance pattern in the mussel Mytella charruana. Rev. Bras. Biociênc. 10, 176 (2012).
Guerra, D., Ghiselli, F. & Passamonti, M. The largest unassigned regions of the male- and female-transmitted mitochondrial DNAs in Musculista senhousia (Bivalvia Mytilidae). Gene 536, 316–325 (2014).
Lucentini, L. et al. Additional taxonomic coverage of the doubly uniparental inheritance in bivalves: Evidence of sex-linked heteroplasmy in the razor clam Solen marginatus Pulteney, 1799, but not in the lagoon cockle Cerastoderma glaucum (Bruguière, 1789). J. Zool. Syst. Evol. Res. 58, 561–570 (2020).
Dégletagne, C., Abele, D. & Held, C. A distinct mitochondrial genome with DUI-like inheritance in the ocean Quahog Arctica islandica. Mol. Biol. Evol. 33, 375–383 (2016).
Glöckner, G., Heinze, I., Platzer, M., Held, C. & Abele, D. The mitochondrial genome of Arctica islandica; Phylogeny and variation. PLoS ONE 8, e82857 (2013).
Śmietanka, B., Lubośny, M., Przyłucka, A., Gérard, K. & Burzyński, A. Mitogenomics of Perumytilus purpuratus (Bivalvia: Mytilidae) and its implications for doubly uniparental inheritance of mitochondria. PeerJ 6, e5593 (2018).
Trovant, B., Orensanz, J. M., Ruzzante, D. E., Stotz, W. & Basso, N. G. Scorched mussels (BIVALVIA: MYTILIDAE: BRACHIDONTINAE) from the temperate coasts of South America: Phylogenetic relationships, trans-Pacific connections and the footprints of Quaternary glaciations. Mol. Phylogenet. Evolut. 82, 60–74 (2015).
Guiñez, R. et al. Present-day connectivity of historical stocks of the ecosystem engineer Perumytilus purpuratus along 4500km of the Chilean Coast. Fish. Res. 179, 322–332 (2016).
Lubośny, M., Śmietanka, B., Przyłucka, A. & Burzyński, A. Highly divergent mitogenomes of Geukensia demissa (Bivalvia, Mytilidae) with extreme AT content. J. Zool. Syst. Evol. Res. 58, 571–580 (2020).
Lubośny, M., Przyłucka, A., Śmietanka, B. & Burzyński, A. Semimytilus algosus: First known hermaphroditic mussel with doubly uniparental inheritance of mitochondrial DNA. Sci. Rep. 10, 11256 (2020).
Marshall, C. & Parson, W. Interpreting NUMTs in forensic genetics: Seeing the forest for the trees. Forens. Sci. Int. Genet. 53, 102497 (2021).
Sorenson, M. D. & Quinn, T. W. Numts: A challenge for Avian systematics and population biology. Auk 115, 214–221 (1998).
Sańko, T. J. & Burzyński, A. Co-expressed mitochondrial genomes: Recently masculinized, recombinant mitochondrial genome is co-expressed with the female – transmitted mtDNA genome in a male Mytilus trossulus mussel from the Baltic Sea. BMC Genet. 15, 28 (2014).
Hoeh, W. R., Stewart, D. T., Saavedra, C., Sutherland, B. W. & Zouros, E. Phylogenetic evidence for role-reversals of gender-associated mitochondrial DNA in Mytilus (Bivalvia: Mytilidae). Mol. Biol. Evol. 14, 959–967 (1997).
Duffy, S. Why are RNA virus mutation rates so damn high?. PLoS Biol. 16, e3000003 (2018).
Bezie, Y., Tilahun, T., Atnaf, M. & Taye, M. The potential applications of site-directed mutagenesis for crop improvement: A review. J. Crop Sci. Biotechnol. 24, 229–244 (2021).
Passamonti, M., Ricci, A., Milani, L. & Ghiselli, F. Mitochondrial genomes and Doubly Uniparental Inheritance: New insights from Musculista senhousia sex-linked mitochondrial DNAs (Bivalvia Mytilidae). BMC Genomics 12, 442 (2011).
Ort, B. S. & Pogson, G. H. Molecular Population Genetics of the Male and Female Mitochondrial DNA Molecules of the California Sea Mussel, Mytilus californianus. Genetics 177, 1087–1099 (2007).
Burzyński, A. & Śmietanka, B. Is interlineage recombination responsible for low divergence of mitochondrial nad3 genes in Mytilus galloprovincialis?. Mol. Biol. Evol. 26, 1441–1445 (2009).
Śmietanka, B., Wenne, R. & Burzyński, A. Complete male mitochondrial genomes of European Mytilus edulis mussels. Mitochondrial DNA A 27, 1634–1635 (2016).
Śmietanka, B., Burzyński, A. & Wenne, R. Comparative genomics of marine mussels (Mytilus spp.) gender associated mtDNA: Rapidly evolving atp8. J. Mol. Evol. 71, 385–400 (2010).
Iannello, M., Bettinazzi, S., Breton, S., Ghiselli, F. & Milani, L. A naturally heteroplasmic clam provides clues about the effects of genetic bottleneck on paternal mtDNA. Genome Biol. Evol. 13, eva022 (2021).
Bettinazzi, S., Plazzi, F. & Passamonti, M. The complete female- and male-transmitted mitochondrial genome of Meretrix lamarckii. PLoS ONE 11, e0153631 (2016).
Capt, C. et al. Unorthodox features in two venerid bivalves with doubly uniparental inheritance of mitochondria. Sci. Rep. 10, 1087 (2020).
Gomes-dos-Santos, A. et al. The male and female complete mitochondrial genomes of the threatened freshwater pearl mussel Margaritifera margaritifera (Linnaeus, 1758) (Bivalvia: Margaritiferidae). Mitochondrial DNA B 4, 1417–1420 (2019).
Lopes-Lima, M. et al. The first Margaritiferidae male (M-type) mitogenome: Mitochondrial gene order as a potential character for determining higher-order phylogeny within Unionida (Bivalvia). J. Molluscan Stud. 83, 249–252 (2017).
Breton, S. et al. Comparative mitochondrial genomics of freshwater mussels (Bivalvia: Unionoida) with doubly uniparental inheritance of mtDNA: Gender-specific open reading frames and putative origins of replication. Genetics 183, 1575–1589 (2009).
Breton, S. et al. Novel protein genes in animal mtDNA: A new sex determination system in freshwater mussels (Bivalvia: Unionoida)?. Mol. Biol. Evol. 28, 1645–1659 (2011).
Soroka, M. & Burzyński, A. Doubly uniparental inheritance and highly divergent mitochondrial genomes of the freshwater mussel Unio tumidus (Bivalvia: Unionidae). Hydrobiologia 810, 239–254 (2018).
Soroka, M. & Burzyński, A. Complete sequences of maternally inherited mitochondrial genomes in mussels Unio pictorum (Bivalvia, Unionidae). J. Appl. Genet. 51, 469–476 (2010).
Fonseca, M. M., Lopes-Lima, M., Eackles, M. S., King, T. L. & Froufe, E. The female and male mitochondrial genomes of Unio delphinus and the phylogeny of freshwater mussels (Bivalvia: Unionida). Mitochondrial DNA B 1, 954–957 (2016).
Burzyński, A., Soroka, M., Mioduchowska, M., Kaczmarczyk, A. & Sell, J. The complete maternal and paternal mitochondrial genomes of Unio crassus: Mitochondrial molecular clock and the overconfidence of molecular dating. Mol. Phylogenet. Evol. 107, 605–608 (2017).
Huang, X.-C. et al. The complete maternally and paternally inherited mitochondrial genomes of the endangered freshwater mussel Solenaia carinatus (Bivalvia: Unionidae) and implications for Unionidae taxonomy. PLoS ONE 8, e84352 (2013).
Wen, H. B. et al. The complete maternally and paternally inherited mitochondrial genomes of a freshwater mussel Potamilus alatus (Bivalvia: Unionidae). PLoS ONE 12, e0169749 (2017).
Chase, E. E. et al. The complete male-type mitochondrial genomes of the Fatmucket, Lampsilis siliquoidea, and the endangered Arkansas Fatmucket, Lampsilis powellii. Mitochondrial DNA B 4, 107–109 (2019).
Wang, G., Cao, X. & Li, J. Complete F-type mitochondrial genome of Chinese freshwater mussel Lamprotula tortuosa. Mitochondrial DNA 24, 513–515 (2013).
Soroka, M. & Burzyński, A. Complete male mitochondrial genome of Anodonta anatina (Mollusca: Unionidae). Mitochondrial DNA A 27, 1679–1680 (2016).
Burzyński, A. & Soroka, M. Complete paternally inherited mitogenomes of two freshwater mussels Unio pictorum and Sinanodonta woodiana (Bivalvia: Unionidae). PeerJ 6, e5573 (2018).
Wang, G., Guo, L. & Li, J. The F-type complete mitochondrial genome of Arconaia lanceolata. Mitochondrial DNA A 27, 322–323 (2016).
Froufe, E. et al. Mesozoic mitogenome rearrangements and freshwater mussel (Bivalvia: Unionoidea) macroevolution. Heredity (Edinb) 124, 182–196 (2020).
Ladoukakis, E. D. & Zouros, E. Direct Evidence for Homologous Recombination in Mussel (Mytilus galloprovincialis) Mitochondrial DNA. Mol. Biol. Evol. 18, 1168–1175 (2001).
Burzyński, A., Zbawicka, M., Skibinski, D. O. F. & Wenne, R. Evidence for recombination of mtDNA in the marine mussel Mytilus trossulus from the Baltic. Mol. Biol. Evol. 20, 388–392 (2003).
Burzyński, A., Zbawicka, M., Skibinski, D. O. F. & Wenne, R. Doubly uniparental inheritance is associated with high polymorphism for rearranged and recombinant control region haplotypes in baltic Mytilus trossulus. Genetics 174, 1081–1094 (2006).
García-Souto, D. et al. Detection of invasive and cryptic species in marine mussels (Bivalvia, Mytilidae): A chromosomal perspective. J. Nat. Conserv. 39, 58–67 (2017).
Yampolsky, L. Y. & Stoltzfus, A. The exchangeability of amino acids in proteins. Genetics 170, 1459–1472 (2005).
Prjibelski, A. D., Korobeynikov, A. I. & Lapidus, A. L. Sequence analysis. In Encyclopedia of Bioinformatics and Computational Biology 292–322 (Elsevier, 2019). https://doi.org/10.1016/B978-0-12-809633-8.20106-4.
Uliano-Silva, M. et al. Complete mitochondrial genome of the brown mussel Perna perna (Bivalve, Mytilidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 3955–3956 (2016).
Wenne, R. et al. Trans-Atlantic distribution and introgression as inferred from single nucleotide polymorphism: Mussels Mytilus and environmental factors. Genes 11, 530 (2020).
Väinölä, R. & Strelkov, P. Mytilus trossulus in Northern Europe. Mar. Biol. 158, 817–833 (2011).
Zbawicka, M., Sańko, T., Strand, J. & Wenne, R. New SNP markers reveal largely concordant clinal variation across the hybrid zone between Mytilus spp. in the Baltic Sea. Aquat. Biol. 21, 25–36 (2014).
Zbawicka, M., Wenne, R. & Skibinski, D. O. F. Mitochondrial DNA variation in populations of the mussel Mytilus trossulus from the Southern Baltic. Hydrobiologia 499, 1–12 (2003).
Filipowicz, M., Burzyński, A., Śmietanka, B. & Wenne, R. Recombination in mitochondrial DNA of European mussels Mytilus. J. Mol. Evol. 67, 377–388 (2008).
Tan, K.-S., Tan, S.H.-M., Sanpanich, K., Duangdee, T. & Ambarwati, R. Taxonomic re-description and relationships of two mat-forming mussels from the indo-pacific region, with a proposed new genus. Phuket Mar. Biol. Cent. Res. Bull. 78, 77–115 (2021).
Ghiselli, F., Milani, L. & Passamonti, M. Strict sex-specific mtDNA segregation in the germ line of the DUI species Venerupis philippinarum (Bivalvia: Veneridae). Mol. Biol. Evol. 28, 949–961 (2011).
Hoarau, G., Rijnsdorp, A. D., Veer, H. W. V. D., Stam, W. T. & Olsen, J. L. Population structure of plaice (Pleuronectes platessa L.) in northern Europe: Microsatellites revealed large-scale spatial and temporal homogeneity. Mol. Ecol. 11, 1165–1176 (2002).
Lubośny, M. et al. Next generation sequencing of gonadal transcriptome suggests standard maternal inheritance of mitochondrial DNA in Eurhomalea rufa (Veneridae). Mar. Genomics 31, 21–23 (2017).
MacManes, M. D. The Oyster River Protocol: A multi-assembler and kmer approach for de novo transcriptome assembly. PeerJ 6, e5428 (2018).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18 (2017).
Badger, J. H. & Olsen, G. J. CRITICA: Coding region identification tool invoking comparative analysis. Mol. Biol. Evol. 16, 512–524 (1999).
Delcher, A. L., Harmon, D., Kasif, S., White, O. & Salzberg, S. L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27, 4636–4641 (1999).
Laslett, D. & Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175 (2008).
Wheeler, T. J. & Eddy, S. R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489 (2013).
Untergasser, A. et al. Primer3—New capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012).
Lubośny, M., Przyłucka, A., Śmietanka, B., Breton, S. & Burzyński, A. Actively transcribed and expressed atp8 gene in Mytilus edulis mussels. PeerJ 6, e4897 (2018).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Bouckaert, R. et al. BEAST 2: A software platform for Bayesian evolutionary analysis. PLOS Comput. Biol. 10, e1003537 (2014).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Lee, Y. et al. A mitochondrial genome phylogeny of Mytilidae (Bivalvia: Mytilida). Mol. Phylogenet. Evol. 139, 106533 (2019).
Sun, J. et al. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 1, 1–7 (2017).
Zhang, K., Sun, J., Xu, T., Qiu, J.-W. & Qian, P.-Y. Phylogenetic relationships and adaptation in deep-sea mussels: Insights from mitochondrial genomes. Int. J. Mol. Sci. 22, 1900 (2021).
Ozawa, G. et al. Updated mitochondrial phylogeny of Pteriomorph and Heterodont Bivalvia, including deep-sea chemosymbiotic Bathymodiolus mussels, vesicomyid clams and the thyasirid clam Conchocele cf. bisecta. Mar. Genomics 31, 43–52 (2017).
Śmietanka, B. & Burzyński, A. Complete female mitochondrial genome of Mytilus chilensis. Mitochondrial DNA B 2, 101–102 (2017).
Lee, Y.-C. & Lee, Y.-H. The F type mitochondrial genome of hard-shelled mussel: Mytilus coruscus (Mytiloida, Mytilidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 624–625 (2016).
Bennett, K. F. et al. The F type mitochondrial genome of the scorched mussel: Brachidontes exustus, (Mytiloida, Mytilidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 1501–1502 (2016).
Ranjard, L. et al. Complete mitochondrial genome of the green-lipped mussel, Perna canaliculus (Mollusca: Mytiloidea), from long nanopore sequencing reads. Mitochondrial DNA B Resour. 3, 175–176 (2018).
Li, X., Wu, X. & Yu, Z. Complete mitochondrial genome of the Asian green mussel Perna viridis (Bivalvia, Mytilidae). Mitochondrial DNA 23, 358–360 (2012).
Uliano-Silva, M. et al. The complete mitochondrial genome of the golden mussel Limnoperna fortunei and comparative mitogenomics of Mytilidae. Gene 577, 202–208 (2016).
Robicheau, B. M., Breton, S. & Stewart, D. T. Sequence motifs associated with paternal transmission of mitochondrial DNA in the horse mussel, Modiolus modiolus (Bivalvia: Mytilidae). Gene 605, 32–42 (2017).
Acknowledgements
This work was supported by the Polish National Science Center (UMO-2015/17/N/NZ3/03538 and UMO-2012/07/B/NZ2/01991). The funders did not have a role in the study design and data collection, or the analysis, decision to publish, and preparation of the manuscript. Computer intensive calculations were run on PLGrid supercomputers (www.plgrid.pl). We thank Dr. A. Occhipinti, owner of the salt pan “infersa” who allowed us to collect the samples.
Author information
Authors and Affiliations
Contributions
M.L., M.A., B.Ś. and A.B. designed and conceived the experiments. M.L. and B.Ś. performed the experiments. M.A. gathered and sampled tissues. M.L. and A.B. performed data analysis. M.L., M.A., B.Ś. and A.B. wrote the manuscript. All authors reviewed drafts of the paper and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lubośny, M., Śmietanka, B., Arculeo, M. et al. No evidence of DUI in the Mediterranean alien species Brachidontes pharaonis (P. Fisher, 1870) despite mitochondrial heteroplasmy. Sci Rep 12, 8569 (2022). https://doi.org/10.1038/s41598-022-12606-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-12606-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
