
Abstract
In animals, strictly maternal inheritance (SMI) of mitochondria is the rule, but one exception (doubly uniparental inheritance or DUI), marked by the transmission of sex-specific mitogenomes, has been reported in bivalves. Associated with DUI is a frequent modification of the mitochondrial cox2 gene, as well as additional sex-specific mitochondrial genes not involved in oxidative phosphorylation. With the exception of freshwater mussels (for 3 families of the order Unionida), these DUI-associated features have only been shown in few species [within Mytilidae (order Mytilida) and Veneridae (order Venerida)] because of the few complete sex-specific mitogenomes published for these orders. Here, we present the complete sex-specific mtDNAs of two recently-discovered DUI species in two families of the order Venerida, Scrobicularia plana (Semelidae) and Limecola balthica (Tellinidae). These species display the largest differences in genome size between sex-specific mitotypes in DUI species (>10 kb), as well as the highest mtDNA divergences (sometimes reaching >50%). An important in-frame insertion (>3.5 kb) in the male cox2 gene is partly responsible for the differences in genome size. The S. plana cox2 gene is the largest reported so far in the Kingdom Animalia. The mitogenomes may be carrying sex-specific genes, indicating that general mitochondrial features are shared among DUI species.
Similar content being viewed by others

Introduction
Animal mitochondrial DNA (mtDNA) is typically depicted as a strictly maternally inherited (SMI) circular DNA molecule that is relatively small (~16 kb) and genomically streamlined with almost invariant gene content (13 protein-coding genes and 24 structural RNAs)1,2. However, important deviations do occur in the mtDNAs of bivalve molluscs, which not only display dramatic variation in size (<14.7 kb to >67 kb)3,4 and gene arrangement5, but also the presence of additional protein-coding genes not associated with oxidative phosphorylation6,7,8,9. An even more extreme departure from the norm in bivalve mitochondrial genomes is their mode of doubly uniparental inheritance (DUI) — both egg and sperm mitochondria are transmitted from generation to generation in several bivalve species, but only male offspring retain paternally-transmitted mitochondria (with male or M mtDNA) in their gametes10,11,12. Adult females of DUI-exhibiting species usually possess only the female-transmitted mtDNA (F mtDNA) in their soma and gametes whereas males possess F mtDNA in their soma and M mtDNA in their gametes10,11,12,13. The DNA divergence between F and M mtDNAs usually vary from about 8% to 40% depending on the species12,14. Genetic analyses suggested that both F and M mtDNAs in DUI bivalves evolve at a faster rate than typical metazoan mtDNA, and that M mtDNA evolves faster than F mtDNA15,16,17. One factor explaining this observation may be that the M genome is subject to weaker selective pressures than the F genome due to an unequal “division of labor” in the DUI system16. Typical animal mtDNA functions in gonads and somatic tissues of both sexes whereas under DUI, F mtDNA functions in female gonads and the soma of both sexes, while M mtDNAs functions primarily in spermatozoa of male gonads and only partially in the male soma13,16,18. As opposed to SMI that promotes homoplasmy, a state in which all mtDNA copies are typically genetically identical in each cell, thus preventing potentially harmful genomic conflicts, DUI is a naturally heteroplasmic system in which two highly divergent mitochondrial lineages coexist in the same nuclear background, enabling the analysis of the consequences of tissue heteroplamy13.
In addition to a different mode of mitochondrial transmission and evolution rate of mtDNA, two other remarkable differences have been reported between the F and M mtDNAs in DUI bivalves. First, the COX2 protein encoded by M mtDNA (Mcox2 gene) is longer than the FCOX2 protein, which is about the same size as other animal COX2 proteins, although this pattern is not shared by all DUI species for which sex-specific mtDNAs have been completely sequenced (reviewed in Bettinazzi et al.19). For example, male freshwater mussels (order Unionida) have an approximately 550 bp 3′-coding extension to the cox2 gene (Mcox2e), that is absent from other animals mtDNAs20,21,22. This extension proved to be translated and localized in both inner and outer mitochondrial membranes23,24, leading to the hypothesis that it could act as a mitochondrial tag implicated in paternal mitochondria survival in male embryos21. Such a 3′-coding extension of the Mcox2 gene has also been found in the mytilid mussel Musculista senhousia, but in a duplicated version of the cox2 gene25. The extension is apparently absent from M mtDNAs of other mytilid DUI species (e.g., Mytilus spp.)12,26. In the family Veneridae, the M mtDNA of Meretrix lamarckii presents an insertion of 100 codons within the cox2 gene19, whereas a duplicated version of the cox2 gene, similar to that of M. senhousia (i.e., longer at 3′), has been found in Ruditapes philippinarum, but located in the F genome (unpublished GenBank annotation mentioned in Passamonti et al.25). As suggested by Bettinazzi et al.19, non-canonical features of the cox2 gene are often coupled with DUI, but it is difficult to propose a general function because each major lineage of bivalves that possesses the DUI system exhibits some novel features. Clearly further analyses involving additional species are required to better understand the relationship between these structural variations in the cox2 gene and DUI, as well as other general features of DUI-exhibiting mitochondrial genomes.
To date, DUI has been found in over one hundred bivalve species representing four taxonomic orders and twelve bivalve families (reviewed in Gusman et al.27). In these species, complete F and M mtDNAs have been sequenced for ∼25 DUI spp. of the families Mytilidae (order Mytilida), Veneridae (order Venerida), Unionidae, Margaritiferidae and Hyriidae (order Unionida)6,7,22,28,29,30,31,32,33,34,35. To our knowledge, all these species share one DUI-specific feature: they contain additional sex-specific mitochondrial genes without recognizable homologies to other known genes (hereafter called mitochondrial ORFans or mtORFans). These F- and M-specific mtORFans (which have been shown to be expressed) have been respectively called F-orf and M-orf 6,7,8,36,37,38. This discovery is particularly interesting because these mtORFans could be responsible for the different mode of transmission of the mtDNAs and/or the functioning of DUI in bivalves. In unionid freshwater mussels, their discovery established a strong link between DUI and the maintenance of gonochorism (hermaphroditic species possess SMI and usually a highly modified F-orf gene, called H-orf)7,22,39. Otherwise, their predicted functions support their direct involvement in the DUI mechanism: F-ORF proteins are suggested to interact with nucleic acids, adhere to membranes, and have roles in signalling, and M-ORFs are suggested to interact with the cytoskeleton and take part in ubiquitination and apoptosis7,8,40. However, the precise nature of the link between DUI and sex determination and the functions of the F and M mtORFans remains currently unknown. Again, further analyses involving additional species are needed to shed light on this.
Recently, DUI has been discovered in Scrobicularia plana27, a species of the family Semelidae (order Venerida)41, as well as in Limecola balthica42, a species of the family Tellinidae (order Venerida)41, and except for the complete F mtDNA of L. balthica43, their M mtDNAs and F mtDNA, for S. plana, have not previously been sequenced or reported on until now. In this study, we present the complete M and F mitochondrial genomes of these two DUI species. Our main objective was to highlight both unique features and characteristics shared among DUI species of different bivalve families. Obtaining the complete sex-specific mitogenome sequences of the species S. plana was particularly interesting to better understand the hypothesized link between DUI and sex determination since an “intersex” condition, i.e., the appearance of oocytes in male gonads following endocrine disruption, has been reported in this species and was associated with down-regulation of male mitochondrial transcripts in males exhibiting intersex compared to “normal” males (see Gusman et al.27 for details). Overall, besides indicating that the newly sequenced mitogenomes may be carrying sex-specific genes like in other DUI species, our data reveal that the cox2 gene in the M mitogenome of S. plana is the largest reported so far in the Kingdom Animalia. It remains to be demonstrated if such unorthodox features play key roles in DUI and sex determination in bivalves.
Results and Discussion
Main genomic features
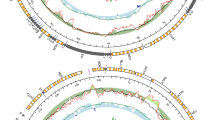
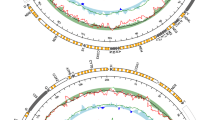
The complete F mtDNAs of Scrobicularia plana and Limecola balthica are 16,170 bp and 17,492 bp in length, respectively, whereas the complete M mtDNAs are respectively 26,270 bp and 24,792 bp long (Fig. 1). To our knowledge, these values represent the biggest differences in genome size between the F and M mtDNAs in species with DUI (Table 1), which usually do not exceed 2 kb. These differences in length are partly explained by the presence of an insertion in the protein-coding gene (PCG) cox2 in the M mtDNA of both species (Fcox2 = 855 bp and Mcox2 = 4,815 bp in L. balthica, whereas Fcox2 = 861 bp and Mcox2 = 5,679 bp in S. plana) (Table 2), which is discussed in more details below. Otherwise, the two sex-specific mtDNAs in each species possess the 13 typical mitochondrial PCGs of metazoans1, all present on the same strand, as is the case in most bivalve species14,19,44. Gene organisation is similar among the four genomes, except for the region between cytb and trnM in the M mtDNA of S. plana. Reorganisation events, such as inversion or transposition, as well as tandem duplication followed by random loss events, are common in animals mtDNAs1.

Gene maps of M and F genomes of Scrobicularia plana and Limecola balthica. All genes are encoded on the heavy strand, total genome lengths are reported inside their corresponding genome. Gene colors correspond to functional groups (OXPHOS gene families, tRNAs and rRNAs).
The initiation and termination codons for the typical 13 PCGs encoded by the four mitogenomes are presented in Table 2. There are differences between sex-specific mtDNAs within species and also between species. For example, in S. plana a similar start codon between the F and M mtDNA for a particular gene is observed in 4 cases out of 13 (only two in L. balthica) whereas 6 PCGs out of 13 have a similar stop codon (4 in L. balthica). Overall, most of the PCGs use the ATD start codon (where D means A, T or G) found in metazoan mtDNAs45: ATA occurs the most (15/52), followed by ATG (14/52) and ATT (11/52). TTG is also found in seven cases, and GTG in five, an observation that has been previously reported in molluscan species46,47. Most of the PCGs are terminated with the TAA (27/52) and TAG (20/52) codons. Some incomplete termination codons are also found, which are assumed to be completed by polyadenylation of their mRNAs48.
The usual 22 tRNAs are found in the four mitochondrial genomes, ranging from 61 to 71 bp, and most of them can be folded into the typical secondary structures (not shown). There is an exception in the M mtDNA of L. balthica, which is lacking trnT, but instead possess a second copy of trnA, which has been identified by MITOS in the same area, between trnY and trnL1 (Fig. S1). This second copy of trnA (trnA_1) does not possess the typical secondary structure and it is not clear if it could be functional. This same mitochondrial genome contains also another copy of trnP (trnP_1) located next to the first trnP (trnP_0), and both possess usual secondary structures (Fig. S1). Additional tRNA copies are common in bivalve mitochondrial genomes19,49,50,51. Concerning the missing trnT, it is possible that the trnA_1 could be a mutated trnT (with a mutation in its anticodon that changed it from trnT to a “secondary” trnA). It is also possible that an unusual secondary structure makes it unidentifiable by the tRNA search programs, and this will require further analyses.
Length variations among rRNA genes range from 765 bp to 896 bp and from 1249 bp to 1556 bp for 12S (rrnS) and 16S (rrnL), respectively (Table 2). Their locations are almost identical in the four genomes; rrnL is found between nad6 and atp6 whereas rrnS is found between trnG and trnM, except for the M genome of S. plana, in which it is located between trnW and trnV (Fig. 1; Tables 2 and 3).
Intraspecific divergences
Genetic distances of individual genes between F and M mtDNAs were analyzed in each species. The results show that the level of conservation is higher for both ribosomal RNA genes (rrnS and rrnL), and also for the PCG cox1, whereas NADH dehydrogenase subunit genes (nad series) and atp8 for L. balthica are generally less conserved (Fig. 2a,c). These results are consistent with the general findings in DUI bivalves32. For S. plana the average nucleotide divergence (measured as p-distance) of combined protein coding and ribosomal RNA genes is 0.43 ± 0.08 SD (based on values in Fig. 2a). For L. balthica the average p-distance of combined protein coding and ribosomal RNA genes is 0.41 ± 0.10 SD (based on values in Fig. 2c). The number of nonsynonymous substitutions per nonsynonymous sites (Ka) relative to the number of synonymous substitutions per synonymous sites (Ks) were also calculated (shown in Fig. 2b,d). This analysis provides an estimate of the degree of selection (either neutral, positive, or purifying) on each PCG. For both species and for all PCGs, all data points fall below the line representing neutrality (expressed as Ka = Ks; see dashed line in Fig. 2b,d), suggesting that the PCGs in S. plana and L. balthica accumulate more synonymous substitutions over evolutionary time relative to nonsynonymous substitutions. Therefore, all Ka and Ks rates supported an initial hypothesis of purifying selection for mitochondrial genes. To statistically test if the genes were indeed under purifying selection, we conducted a Z-test of Selection52. For all PCGs in both species, with the exception of atp8 in L. balthica, there is strong statistical support (at α = 0.05) for rejecting a null hypothesis of neutrality (Hn: Ka = Ks) in support of an alternate hypothesis for purifying selection (Ha: Ka < Ks). This is because all pairwise Z-test p-values were <0.01 for both hypotheses. For atp8 in L. balthica the Z-test of Selection did not support rejecting neutrality (p-value = 0.277 for Hn: Ka = Ks), a result that might be affected by the small size of this gene (129 bp), which in turn altered the statistical power of the test. With regards to the large insertion within the male cox2 gene for both species, these data are of even greater interest as they suggest that this insertion likely does not render the gene functionless. Rather, cox2 (the alignable parts) actually remains among one of the more relatively conserved mt genes (Fig. 2). Overall, our results are in line with what has been observed in other bivalves with DUI, i.e. despite the considerable divergence of DNA and amino acid sequences of PCGs there is evidence of strong purifying selection acting on these mitochondrial genes32.

p-distances and rates of synonymous and nonsynonymous substitutions. Individual M-versus-F gene conservation expressed as p-distance and rates of synonymous and nonsynonymous substitutions within these same mt genes for Scrobicularia plana (A,B, respectively) and Limecola balthica (C,D, respectively). Genes are color-coded by gene family.
For comparative purpose, p-distances of individual genes (PCGs only) between F and M mtDNAs were also calculated for three species known for having the greatest F versus M DNA divergences in the families Mytilidae, Veneridae and Unionidae: i.e., the mytilid Modiolus modiolus53, the venerid Ruditapes philippinarum17, and the unionid Quadrula quadrula14 (Table 4). Our results show an average p-distance (for all 13 PCGs) of 0.45 for nucleotide (nt) and 0.53 for amino acid (aa) in S. plana and 0.44 (nt) and 0.53 (aa) in L. balthica (Table 4). To our knowledge, the nucleotide and amino acid divergences reported in S. plana and L. balthica are the greatest reported among DUI species, surpassing those in freshwater mussels (Table 4), which were previously thought to exhibit the greatest divergences between their sex-specific mtDNAs14. On the other hand, the average uncorrected nucleotide divergence observed between the F and M PCGs of the marine mussel Mytilus edulis is about 0.2326. This low level of divergence has been proposed to be a consequence of masculinization events, which are characterized by an invasion of the male route of inheritance by an F mtDNA that becomes transmitted through sperm as a standard M mtDNA12,54. These events reset the level of divergence between the F and M mitochondrial genomes to zero12,54. Conversely, the high level of divergence observed in freshwater mussels has been hypothesized to be a consequence of a complete absence of masculinization events for over 200 million years in Unionida14,20. According to these studies, masculinization would be no longer possible in this taxon because of the existence of the M-specific extension of the cox2 gene, a specialized feature of the unionid M mtDNA that would prevent recombination between the F and M mtDNAs, i.e. a step necessary for masculinization to occur12,14,20. We thus propose that the high divergences observed between the F and M mtDNAs in S. plana and L. balthica are also related to an absence of masculinization events in these species because of the insertion in their Mcox2 gene (described below).
Mitochondrial ORFans
Unassigned regions in the coding strand were searched for the presence of supernumerary PCGs and mtORFans with a minimal length of 150 bp. Only ORFs encoding proteins with at least one predicted transmembrane domain were retained, because all sex-specific mtORFans characterized to date in DUI species (i.e. those encoding F-ORF in females and M-ORF in males), except for one case, possess at least one transmembrane domain or helix (TMH)7,8,22,40. In both S. plana and L. balthica F mtDNAs, only two unassigned regions were susceptible to contain supernumerary ORFs of >150 bp, i.e. between rrnS-trnM and trnF-cox1 (Table 3). However, the region between trnF and cox1 was discarded because of the presence of a possible 5′ extension of the cox1 gene in all four genomes, an issue that will need to be assessed by looking at expression data. Otherwise, no ORFs corresponding to the expected profile were found in the F mtDNAs of S. plana and L. balthica. However, it is conceivable that smaller ORFs or ORFs encoding proteins without predicted transmembrane domain could be involved in DUI in these distantly-related species. The smallest F-ORF identified to date in a DUI species, i.e. in the unionid Venustaconcha ellipsiformis, is encoding an 89aa-long protein with one predicted TMH7,40, whereas the smallest M-ORF is potentially encoding a 30aa-long protein without TMH in Mytilus californianus8, although the functionality of this latter ORF remains to be demonstrated. Additional F mt sequences and expression data from S. plana and L. balthica will be necessary to clearly demonstrate the presence (or absence) of the F-orf gene in these species.
In both male mitochondrial DNAs, four unassigned regions contained supernumerary ORFs of >150 bp, i.e. between cob-trnW, trnW-rrnS, trnG-cox2 and rrnL-atp6 for S. plana and between cob-cox2, cox2-trnW, trnG-rrnS and rrnS-trnM for L. balthica (Table 3). Five ORFs corresponding to the expected profile were found in S. plana, two between trnW-rrnS and three between trnG-cox2, whereas four ORFs with the expected profile were found in L. balthica, one between trnG-rrnS and three between rrnS-trnM (Fig. S2). Sequence similarity searches using PSI-BLAST55 against non-redundant protein sequences and SWISSPROT databases failed to detect significant sequence similarity with known proteins for all these ORFs except one (i.e. mtORFan1 in the unassigned region trnG-rrnS of L. balthica M mtDNA; Fig. S2). For this sequence, our results revealed moderately significant hits (E-values 2e-07) with microtubule-associated proteins. This result is interesting since previous in silico analyses of M-ORF sequences in other DUI species also indicated connections with cytoskeleton proteins involved in microtubule-binding and actin-binding (e.g. ankyrin)8,40. These observations led to the hypothesis that M-ORF, with its predicted transmembrane domains, may target sites outside sperm mitochondria and be responsible for their cellular positioning in developing embryos8. Indeed, studies have shown that, after fertilization, only in male DUI embryos sperm mitochondria remain grouped together, and are eventually sequestered in the germ line, whereas they are dispersed and/or destroyed in female embryos (reviewed in Zouros12). M-ORFs are thus considered as ideal candidates for M mtDNA-derived masculinizing factors in DUI species8,40.
At this moment, however, we cannot confirm nor disprove the presence of a M-orf gene in the M mtDNAs of S. plana and L. balthica. Pairwise alignments between the ORFs found in both species were performed but it was not possible to obtain satisfactory alignments that could provide support for identifying a conserved ORF (data not shown). Again, additional M mt sequences and expression data from S. plana and L. balthica will be necessary to test for the presence of a M-orf gene in these species.
Insertion in the male cox2 gene
Annotations with MITOS revealed an insertion of >4.5 kb and >3.5 kb in the Mcox2 gene of S. plana and L. balthica, respectively, which is absent in the Fcox2 genes of both species (Figs. 1 and 3). In S. plana, this in-frame insertion, if translated, means that the cox2 gene would be 5,679bp-long and, to our knowledge, would therefore encode for the longest COX2 protein in the animal kingdom (i.e. 1,893 amino acids). The sequencing of additional S. plana male individuals revealed that this insertion is conserved among different individuals from different populations (Tassé et al. unpublished), indicating that it is most probably functional. A multiple sequence alignment of Homo sapiens COX2 and S. plana FCOX2 and MCOX2 amino acids indicates that the insertion is situated between the “heme-patch” region, containing an important residue Trp that functions as the point of electron entry from Cytochrome C, and the first Cua-binding center (Figs. 3 and S3), and in silico analyses suggest that it does not contain any additional transmembrane domain (Tassé et al. unpublished). In-frame insertions resulting in enlarged cox2 genes have also been reported in hymenoptera, ciliates, brown algae and microflagellates56. As mentioned above, modifications to the cox2 gene is a particular feature often found in DUI species. In unionid mussels, the longest cox2 gene is found in the M mtDNA of Hyridella menziesii (1,380 bp)22 and is the result of a 3′ extension, as in other DUI unionids21. Previous studies indicated an extra-mitochondrial localization of MCOX2 as well as a possible involvement of the protein in reproduction in freshwater mussels23,24, further supporting the link between gender and mtDNA transmission patterns. In the order Venerida, i.e. the order to which S. plana and L. balthica belong, an in-frame insertion of 300 nt has been reported in the species Meretrix lamarckii (family Veneridae)19. However, such modifications of the cox2 gene have not been reported in marine mytilid mussels, i.e. Mytilus spp., and in the venerid clam Ruditapes philippinrum, a duplicated copy of the cox2 gene with a 3′ extension has been reported, but in the F mtDNA12,19.

Fcox2 and Mcox2 structural features in Scrobicularia plana and Limecola balthica. For the sequence alignments of the “heme-patch” regions and Cua centers, identical amino acids are indicated by an *.
The situation in L. balthica is quite different than in S. plana regarding the Mcox2 gene, which is split in two by an insertion. Specifically, this insertion divides Mcox2 into Mcox2a, encoding the two transmembrane helices and the “heme-patch” region followed by a complete stop codon (TAA), and Mcox2b, encoding an enlarged intermembrane space and the Cua centers (Figs. 3 and S4). This situation is confirmed by transcriptomic data (Illumina RNAseq reads from sperm cells, used to polish our nanopore reference mitogenomes), which indicate that both regions are transcribed (i.e. with discrete, non-overlapping Mcox2a and Mcox2b transcripts). Although it remains to be determined whether Mcox2a and Mcox2b are translated, it is worth noting that similar cases have been described in the freshwater mussel Anodonta cygnea33 and the Hymenoptera Campsomeris spp56. For example, a translocation of a portion of the nad5 gene has been reported in A. cygnea, and since the translocated portion and the rest of the gene both possessed their own start codon, the authors proposed that they could be transcribed and translated separately33. In the genus Campsomeris, cox2 is also split into two genes and this likely occurred through intragenic insertion of a cluster of several ORFs, one of which encodes a putative endonuclease that might have been directly involved in the process of cox2 fission56. However, no ORFs with similarity to an endonuclease were found in the Mcox2 insertion of L. balthica (nor in S. plana). According to Szafranski et al.56, COXIIA and COXIIB polypeptides in Campsomeris spp. apparently assemble into a functional COXII heterodimer. Further studies will be needed to determine if this is also the case in L. balthica, or to clearly verify if a process of mitochondrial RNA splicing, because a group II intron has already been reported in the cox1 gene of Annelida57, could be involved (i.e. Mcox2 RNA splicing or Mcox2a and Mcox2b RNA trans-splicing into a single mRNA).
It also remains to be determined if L. balthica possesses an enlarged MCOX2 or a MCOX2 protein of a typical length. For example, the stop codon in Mcox2a could be read as a sense codon by a suppressor tRNA, i.e. a mutated (usually) tRNA that would insert an amino acid instead of initiating termination, as seen in several eukaryotes58. The presence of duplicated trn genes in the M mtDNA of L. balthica might be a clue for such scenario. Modification or duplication of tRNAs have already been proven to allow codon reassignment by changing codons from stop-to-sense or sense-to-sense in yeast, green algae and metazoan mitochondria59,60. Alternatively, termination can be avoided by ribosomal frame-shifting, a process that allows for protein merging from two or more overlapped ORFs61. This has been reported for the mitochondrial cox1 gene of the diatom Phaeodactylum tricornutum: to avoid a stop codon, translational frameshift skips a nucleotide (called + 1)62. Similar strategies (+1 or −1 frameshifts) have also been recorded in metazoan mitochondrial genomes including ants, turtles and humans63,64,65. Further transcriptomic and proteomic analyses, and a closest look in mitochondrial tRNAs, will be required to validate any machinery involved in Mcox2 transcription and translation in both L. balthica and S. plana. The results presented in this study clearly indicate that the relationship between cox2 variations and DUI deserves much greater attention.
Conclusion
In summary, the description and comprehensive analysis of complete F and M mtDNAs of two newly discovered bivalve species with DUI from two additional families (Semelidae and Tellinidae), have led to new insights into DUI mitogenomics. Our results revealed uncorrected amino acid p-distance of ~53% for PCGs between the M and F genomes of both species: this is the highest divergences reported in DUI species thus far. Hence the way (or ways) in which males of bivalves with DUI can tolerate heteroplasmy characterized by such high variability remains an important unanswered question (e.g. see Bettinazzi et al.66). Our results also highlighted an extremely unusual feature of the M genomes of S. plana and L. balthica compared to their female-transmitted counterparts. This feature is the presence of an important in-frame insertion (>3.5 to 4.5 kb) in the Mcox2 gene. This is the longest insertion reported in the Kingdom Animalia, which remains to be functionally characterized. The reported data further indicate that the newly-sequenced M mitogenomes may be carrying lineage-specific genes (mtORFans) possibly involved in the DUI process. Analyses of complete mtDNAs from additional bivalve species and further protein-based studies are needed to elucidate the number, taxonomic distribution, evolution, and function of mtORFans and atypical cox2 genes in this group, as well as the molecular mechanisms underlying DUI.
Methods
Specimen collection and sample preparation
Adult specimens of Limecola balthica were collected at low tide on the Aytré sandy mudflat (France; 46.1259 N, 1.1284 W) in April 2016 and May 2017 (these are periods during which gonads are known to be near sexual maturation42. Individuals were carefully opened and gonad tissue nicked with a micro-scalpel. Gametes were then sampled with a micropipette and a small amount was used to sex the animal with a light microscope; the remainder of the sample was flash-frozen in liquid nitrogen and stored at −80 °C. Adult specimens of Scrobicularia plana were collected in May 2013 from Concarneau (France; 47.8728°N, 3.9207°W). Individuals were dissected and gonads inspected under the microscope. Female and male mature gonads were conserved in 95% Ethanol and sent to the Université de Montréal.
For S. plana, total genomic DNA was extracted separately from male and female gonad tissues with a Qiagen DNeasy Blood & Tissue Kit (QIAGEN Inc., Valencia, CA) using the kit provided animal tissue protocol. The quality and quantity of DNA were assessed by electrophoresis on a 1% agarose gel and also with a BioDrop µLITE spectrophotometer. One female and one male gonad sample with the highest purity and concentration were chosen for sequencing. For L. balthica, total genomic DNA from male gonadal tissues (majority of gametic cells, and some somatic carry-over) was extracted using the phenol-chloroform protocol described in67, resuspended in molecular grade water, and quantified using a Nanodrop 2000 and a Qubit fluorometer (dsDNA HS assay kit, Molecular Probes). DNA integrity was also checked by electrophoresis on a 1% agarose gel. One sample characterised by the highest purity and concentration was chosen for sequencing.
Mitochondrial genome sequencing
Total DNA extractions from S. plana male and female gonads were respectively used to prepare two DNA libraries that were paired-end sequenced (2 × 100 bp) using an Illumina HiSeq platform (Génome Québec Innovation Centre, Montréal, QC, Canada). Paired reads were trimmed with Trimmomatic 0.3268 and merged with Pear 0.9.669 on the Galaxy online platform70, checking the quality of reads at every step with FastQC 0.11.571. MITObim 1.872 was used to assemble the complete genome sequence, starting from a S. plana partial cox1 sequence (GenBank accession number KX447421 for the male mtDNA and KX447423 for the female mtDNA) as an initial seed.
For L. balthica, the library preparation was done following the SQK-LSK108. Nanopore protocol. A DNA shearing step (using Covaris gTubes) was included to linearize circular mtDNA molecules (1 min at 11k RPM in a 5415 R Eppendorf centrifuge). The resulting library was sequenced on the Minion MIN-101B sequencer using a R9 flowcell. Basecalling was performed using Albacore 2.1.373, followed by quality control using Minion_QC 1.074. Nanopore adapters were removed and chimeric reads were discarded using Porechop 0.2.3 (Wick R. Porechop, available at: https://github.com/rrwick/Porechop). Reads with quality scores <9 were removed using Nanofilt 2.0.075. Cleaned reads were assembled using Canu 1.676 considering an approximate genome size of 2 Gb and disregarding all reads smaller than 500 bp. Resulting unitigs were blasted locally using blastn55, using male and female rrnL sequences (Genbank accession numbers KX831969 and KX831970, respectively)42. Based on these results, we identified tig00000009 (length of 26,120 bp, assembly based on 61 nanopore reads) as the male mitogenome. We did not retrieve the female mitochondrial genome as one contig but as two (tig00000888: 16,790 bp, 34 reads; tig00000885: 6,319 bp, 1 read). Basecalling errors were corrected using signal-level data with Nanopolish 0.9.077. We used these two cleaned contigs as reference genomes to map Illumina 2 × 150 bp HiSeq sequences (data quality control and pre-processing step as in78) produced from a second individual using Pilon 1.2179. HiSeq sequencing of cDNA was performed based on mRNAs purified from sperm and foot tissue (for mapping onto the male and female mitogenomes, respectively) using the Macherey-Nagel Nucleospin RNA kit for NucleoZOL (RNA quality control, cDNA synthesis, library preparation, sequencing, data quality control and pre-processing as in80). By mapping high-quality Illumina reads on our draft nanopore contigs, we were able to further correct sequencing errors. The two F-type contigs (tig00000885 and tig00000888) were then merged. The quality of the mitogenome assembly was checked at each step of the pipeline (initial Canu assembly, Nanopolish polishing, Pilon polishing) by calculating the percent identity between our F-type unitigs and a published F-type mitogenome from the same sampling locality (KM373201)43 using MUMmer 3.2381.
Mitochondrial genome sequences were deposited in GenBank (accession codes MN528026 and MN528027 for the F and M mtDNAs of S. plana, respectively, and accession codes MN528028 and MN528029 for the F and M mtDNAs of L. balthica, respectively).
Characterization of mtDNAs and sequence analyses
The male and female mitogenome sequences obtained were annotated with MITOS282. Protein-coding genes (PCG) were also predicted using NCBI′s Open Reading Frame Finder, choosing the invertebrate mitochondrial genetic code and alternative start codons. Candidate transfer RNA (tRNA) genes were also identified using tRNAScan-SE83. The ends of 16S and 12S rRNA genes were assumed to extend to the boundaries of their flanking genes. The complete mitochondrial genomes, with their annotated PCG, tRNAs and rRNAs, were illustrated with GenomeVx online tool84.
The degree of genetic conservation among the M-versus-F genes in each species was assessed using two approaches: (i) pairwise p-distances, and (ii) the rates of synonymous and non-synonymous substitutions, i.e. the number of synonymous substitutions per synonymous sites [Ks] and number of non-synonymous substitutions per non-synonymous sites [Ka], which were calculated in MEGA v752. Since there were various indels present between the individual M- and F-type genes within both species, pairwise deletion was used to account for gaps/missing data-points and codon-alignments were used to facilitate accurate calculations of genetic distances, as well as Ka and Ks values. Amino acid sequences were determined using EMBOSS Transeq and by assuming an invertebrate mitochondrial code85. Codon-alignments were generated by aligning M and F amino acid sequences for each gene using MUSCLE86 in the online portal found at http://www.ebi.ac.uk/Tools/msa/muscle/84. Using these protein alignments as references, nucleotide sequences for each respective gene were then codon-aligned via PAL2NAL v1487. The exceptionally large indel within the Mcox2 gene was manually excluded from the gene alignments. MEGA was also used to conduct a Z-test of Selection [via the Nei-Gojobori method] for each gene52,88. All plots relating to gene conservation were generated in R version 3.4.1 (R Core Team 2017) using the package ggplot2 v3.2.089.
For comparative purpose, p-distances of individual genes between F and M mtDNAs were also calculated for three species known for having the greatest F versus M DNA divergences in the families Mytilidae, Veneridae and Unionidae: i.e. the mytilid Modiolus modiolus (GenBank accession: F, KX821782; M, KX821783)53, the venerid Ruditapes philippinarum (GenBank accession: F, AB065375; M, AB065374)17, and the unionid Quadrula quadrula (GenBank accession: F, FJ809750; M, FJ809751)14. These comparisons facilitated a better understanding of the selection pressures acting on the PCGs in these species. Finally, the presence of supernumerary mitochondrial protein-coding genes (and mtORFans) in unassigned regions was examined using ORF Finder (NCBI). We retained only ORFs with a minimal length of 150 bp and localized on the coding strand, as mitochondrial genes are only encoded on one strand in our two species. Transmembrane domains were predicted using the TMHMM server v2.0 (http://www.cbs.dtu.dk/services/TMHMM/). Alignments between predicted ORFs were done with MUSCLE86 and the percentage of identity was visualized with MView90, via the EMBL-EBI online web services85 keeping default parameters.
References
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780 (1999).
Gissi, C., Iannelli, F. & Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 101, 301–20 (2008).
Park, H. & Ahn, D. H. Complete mitochondrial genome of the Antarctic soft-shelled clam, Laternula elliptica (Bivalvia; Laternulidae). Mitochondrial DNA 26, 642–3 (2015).
McCartney, M. A. et al. The Genome of the Zebra Mussel, Dreissena polymorpha: A Resource for Invasive Species Research. bioRxiv 696732 (2019).
Plazzi, F., Puccio, G. & Passamonti, M. Comparative large-scale mitogenomics evidences clade-specific evolutionary trends in mitochondrial DNAs of Bivalvia. Genome Biol. Evol. 8, 2544–2564 (2016).
Breton, S. et al. Comparative mitochondrial genomics of freshwater mussels (Bivalvia: Unionoida) with Doubly Uniparental Inheritance of mtDNA: gender-specific Open Reading Frames (ORFs) and putative origins of replication. Genetics 183, 1575–1589 (2009).
Breton, S. et al. Novel protein genes in animal mtDNA: A new sex determination system in freshwater mussels (Bivalvia: Unionoida)? Mol. Biol. Evol. 28, 1645–1659 (2011).
Milani, L., Ghiselli, F., Guerra, D., Breton, S. & Passamonti, M. A comparative analysis of mitochondrial ORFans: New clues on their origin and role in species with Doubly Uniparental Inheritance of mitochondria. Genome Biol. Evol. 5, 1408–1434 (2013).
Breton, S. et al. A resourceful genome: Updating the functional repertoire and evolutionary role of animal mitochondrial DNAs. Trends Genet. 30, 555–564 (2014).
Breton, S., Beaupré, H. D., Stewart, D. T., Hoeh, W. R. & Blier, P. U. The unusual system of doubly uniparental inheritance of mtDNA: isn′t one enough? Trends Genet. 23, 465–474 (2007).
Passamonti, M. & Ghiselli, F. Doubly Uniparental Inheritance: Two mitochondrial genomes, one precious model for organelle DNA inheritance and evolution. DNA Cell Biol. 28, 79–89 (2009).
Zouros, E. Biparental inheritance through uniparental transmission: The Doubly Uniparental Inheritance (DUI) of mitochondrial DNA. Evol. Biol. 40, 1–31 (2013).
Breton, S. et al. The extremely divergent maternally-and paternally-transmitted mitochondrial genomes are co-expressed in somatic tissues of two freshwater mussel species with doubly uniparental inheritance of mtDNA. PLoS ONE 12 (2017).
Doucet-Beaupré, H. et al. Mitochondrial phylogenomics of the Bivalvia (Mollusca): Searching for the origin and mitogenomic correlates of doubly uniparental inheritance of mtDNA. BMC Evol. Biol. 10, 1–19 (2010).
Hoeh, W. R., Stewart, D. T., Sutherland, B. W. & Zouros, E. Cytochrome c oxidase sequence comparisons suggest an unusually high rate of mitochondrial DNA evolution in Mytilus (Mollusca: Bivalvia). Mol. Biol. Evol. 13, 418–21 (1996).
Stewart, D. T., Kenchington, E. R., Singh, R. K. & Zouros, E. Degree of selective constraint as an explanation of the different rates of evolution of gender-specific mitochondrial DNA lineages in the mussel Mytilus. Genetics 143, 1349–57 (1996).
Passamonti, M. & Scali, V. Gender-associated mitochondrial DNA heteroplasmy in the venerid clam Tapes philippinarum (Mollusca Bivalvia). Curr. Genet. 39, 117–124 (2001).
Dalziel, A. C. & Stewart, D. T. Tissue-specific expression of male-transmitted mitochondrial DNA and its implications for rates of molecular evolution in Mytilus mussels (Bivalvia: Mytilidae). Genome 45, 348–355 (2002).
Bettinazzi, S., Plazzi, F. & Passamonti, M. The complete female- and male-transmitted mitochondrial genome of Meretrix lamarckii. PLoS One 11 (2016).
Curole, J. P. & Kocher, T. D. Ancient sex-specific extension of the cytochrome c oxidase II gene in bivalves and the fidelity of doubly-uniparental inheritance. Mol. Biol. Evol. 19, 1323–8 (2002).
Chapman, E. G. et al. Extreme primary and secondary protein structure variability in the chimeric male-transmitted cytochrome c oxidase subunit II protein in freshwater mussels: Evidence for an elevated amino acid substitution rate in the face of domain-specific purifying selection. BMC Evol. Biol. 8, 165 (2008).
Guerra, D. et al. Evolution of sex-dependent mtDNA transmission in freshwater mussels (Bivalvia: Unionida). Sci. Rep. 7 (2017).
Chakrabarti, R. et al. Presence of a unique male-specific extension of C-terminus to the cytochrome c oxidase subunit II protein coded by the male-transmitted mitochondrial genome of Venustaconcha ellipsiformis (Bivalvia: Unionoidea). FEBS Lett. 580, 862–866 (2006).
Chakrabarti, R. et al. Reproductive function for a C-terminus extended, male-transmitted cytochrome c oxidase subunit II protein expressed in both spermatozoa and eggs. FEBS Lett. 581, 5213–5219 (2007).
Passamonti, M., Ricci, A., Milani, L. & Ghiselli, F. Mitochondrial genomes and Doubly Uniparental Inheritance: New insights from Musculista senhousia sex-linked mitochondrial DNAs (Bivalvia Mytilidae). BMC Genomics 12, 442 (2011).
Breton, S., Burger, G., Stewart, D. T. & Blier, P. U. Comparative analysis of gender-associated complete mitochondrial genomes in marine mussels (Mytilus spp.). Genetics 172, 1107–1119 (2006).
Gusman, A., Lecomte, S., Stewart, D. T., Passamonti, M. & Breton, S. Pursuing the quest for better understanding the taxonomic distribution of the system of doubly uniparental inheritance of mtDNA. PeerJ 4, e2760 (2016).
Soroka, M. & Burzyński, A. Complete female mitochondrial genome of Anodonta anatina (Mollusca: Unionidae): Confirmation of a novel protein-coding gene (F ORF). Mitochondrial DNA 26, 267–269 (2015).
Soroka, M. & Burzyński, A. Complete male mitochondrial genome of Anodonta anatina (Mollusca: Unionidae). Mitochondrial DNA 27, 1679–1680 (2016).
Burzyński, A., Soroka, M., Mioduchowska, M., Kaczmarczyk, A. & Sell, J. The complete maternal and paternal mitochondrial genomes of Unio crassus: Mitochondrial molecular clock and the overconfidence of molecular dating. Mol. Phylogenet. Evol. 107, 605–08 (2017).
Burzyński, A. & Soroka, M. Complete paternally inherited mitogenomes of two freshwater mussels Unio pictorum and Sinanodonta woodiana (Bivalvia: Unionidae). PeerJ 6, e5573 (2018).
Plazzi, F. & Passamonti, M. Footprints of unconventional mitochondrial inheritance in bivalve phylogeny: Signatures of positive selection on clades with doubly uniparental inheritance. J. Zool. Syst. Evol. Res. 57, 258–271 (2019).
Chase, E. E., Robicheau, B. M., Veinot, S., Breton, S. & Stewart, D. T. The complete mitochondrial genome of the hermaphroditic freshwater mussel Anodonta cygnea (Bivalvia: Unionidae): In silico analyses of sex-specific ORFs across order Unionoida. BMC Genomics 19 (2018).
Gomes-dos-Santos, A. et al. The male and female complete mitochondrial genomes of the threatened freshwater pearl mussel Margaritifera margaritifera (Linnaeus, 1758) (Bivalvia: Margaritiferidae). Mitochondrial DNA 4, 1417–1420 (2019).
Soroka, M. & Burzynski, A. Doubly uniparental inheritance and highly divergent mitochondrial genomes of the freshwater mussel Unio tumidus (Bivalvia: Unionidae). Hydrobiologia 810, 239–254 (2018).
Milani, L., Ghiselli, F., Maurizii, M. G., Nuzhdin, S. V. & Passamonti, M. Paternally transmitted mitochondria express a new gene of potential viral origin. Genome Biol. Evol. 6, 391–405 (2014).
Minoiu, I., Burzyński, A. & Breton, S. Analysis of the coding potential of the ORF in the control region of the female-transmitted Mytilus mtDNA. Gene 576, 586–588 (2016).
Ouimet, P. et al. The ORF in the control region of the female-transmitted Mytilus mtDNA codes for a protein. Gene (In press).
Soroka, M. & Burzynski, A. Hermaphroditic freshwater mussel Anodonta cygnea does not have supranumerary open reading frames in the mitogenome. Mitochondrial DNA 2, 862–864 (2017).
Mitchell, A., Guerra, D., Stewart, D. T. & Breton, S. In silico analyses of mitochondrial ORFans in freshwater mussels (Bivalvia: Unionoida) provide a framework for future studies of their origin and function. BMC Genomics 17 (2016).
Bieler, R., Carter, J. G. & Coan E. Classification of Bivalve Families. Pp. 113-133, in: Bouchet P & Rocroi J-P (2010), Nomenclator of Bivalve Families. Malacologia 52, 1–184 (2010).
Pante, E., Poitrimol, C., Saunier, A., Becquet, V. & Garcia, P. Putative sex-linked heteroplasmy in the tellinid bivalve Limecola balthica (Linnaeus, 1758). J. Molluscan Stud. 83, 226–228 (2017).
Saunier, A. et al. Mitochondrial genomes of the Baltic clam Macoma balthica (Bivalvia: Tellinidae): Setting the stage for studying mito-nuclear incompatibilities. BMC Evol. Biol. 14 (2014).
Jameson, D., Gibson, A. P., Hudelot, C. & Higgs, P. G. OGRe: A relational database for comparative analysis of mitochondrial genomes. Nucleic Acids Res. 31, 202–6 (2003).
Wolstenholme, D. R. Genetic novelties in mitochondrial genomes of multicellular animals. Curr. Opin. Genet. Dev. 2, 918–925 (1992).
Grande, C., Templado, J. & Zardoya, R. Evolution of gastropod mitochondrial genome arrangements. BMC Evol. Biol. 8, 61 (2008).
Yuan, Y., Li, Q., Yu, H. & Kong, L. The complete mitochondrial genomes of six heterodont bivalves (Tellinoidea and Solenoidea): Variable gene arrangements and phylogenetic implications. PLoS One 7 (2012).
Ojala, D., Montoya, J. & Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474 (1981).
Milbury, C. A. & Gaffney, P. M. Complete mitochondrial DNA sequence of the eastern oyster Crassostrea virginica. Mar. Biotechnol. 7, 697–712 (2005).
Xu, K., Kanno, M., Yu, H., Li, Q. & Kijima, A. Complete mitochondrial DNA sequence and phylogenetic analysis of Zhikong scallop Chlamys farreri (Bivalvia: Pectinidae). Mol. Biol. Rep. 38, 3067–3074 (2011).
Sun, S., Kong, L., Yu, H. & Li, Q. The complete mitochondrial DNA of Tegillarca granosa and comparative mitogenomic analyses of three Arcidae species. Gene 557, 61–70 (2015).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Robicheau, B. M., Breton, S. & Stewart, D. T. Sequence motifs associated with paternal transmission of mitochondrial DNA in the horse mussel, Modiolus modiolus (Bivalvia: Mytilidae). Gene 605, 32–42 (2017).
Zouros, E. The exceptional mitochondrial DNA system of the mussel family Mytilidae. Genes Genet. Syst. 75, 313–318 (2000).
Altschul, S. F. et al. Blast and Psi-Blast: Protein Database Search Programs. Nucleid Acid Res. 25, 2289–4402 (1997).
Szafranski, P. Evolutionarily recent, insertional fission of mitochondrial cox2 into complementary genes in bilaterian Metazoa. BMC Genomics 18, 269 (2017).
Vallès, Y., Halanych, K. M. & Boore, J. L. Group II introns break new boundaries: Presence in a bilaterian’s genome. PLoS One 3 (2008).
Beier, H. & Grimm, M. Misreading of termination codons in eukaryotes by natural nonsense suppressor tRNAs. Nucleic Acids Res. 29, 4767–82 (2001).
Ling, J., O’Donoghue, P. & Söll, D. Genetic code flexibility in microorganisms: Novel mechanisms and impact on physiology. Nat. Rev. Microbiol. 13, 707–721 (2015).
Noutahi, E., Calderon, V., Blanchette, M., El-Mabrouk, N. & Lang, B. F. Rapid genetic code evolution in green algal mitochondrial genomes. Mol. Biol. Evol. 36, 766–783 (2019).
Rampersad, S. & Tennant, P. Replication and expression strategies of viruses. In: Viruses: Molecular biology, host interactions, and applications to Biotechnology. Elsevier. p. 55–82 (2018).
Oudot-Le Secq, M.-P. & Green, B. R. Complex repeat structures and novel features in the mitochondrial genomes of the diatoms Phaeodactylum tricornutum and Thalassiosira pseudonana. Gene 476, 20–6 (2011).
Beckenbach, A. T., Robson, S. K. A. & Crozier, R. H. Single nucleotide +1 frameshifts in an apparently functional mitochondrial cytochrome b gene in ants of the genus Polyrhachis. J. Mol. Evol. 60, 141–52 (2005).
Russell, R. D. & Beckenbach, A. T. Recoding of translation in turtle mitochondrial genomes: Programmed frameshift mutations and evidence of a modified genetic code. J. Mol. Evol. 67, 682–95 (2008).
Temperley, R. et al. Hungry codons promote frameshifting in human mitochondrial ribosome. Science 327, 301 (2010).
Bettinazzi, S., Rodríguez, E., Milani, L., Blier, P. U. & Breton, S. Metabolic remodelling associated with mtDNA: Insights into the adaptive value of doubly uniparental inheritance of mitochondria. Proc. R. Soc. B Biol. Sci. 286 (2019).
Rosel, P. E. & Block, B. A. Mitochondrial control region variability and global population structure in the swordfish, Xiphias gladius. Mar. Biol. 125, 11–22 (1996).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Zhang, J., Kobert, K., Flouri, T. & Stamatakis, A. PEAR: A fast and accurate Illumina paired-end read merger. Bioinformatics 30, 614–620 (2014).
Afgan, E. et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544 (2018).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. Babraham Bioinform. Available from, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010)
Hahn, C., Bachmann, L. & Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads – a baiting and iterative mapping approach. Nucleic Acids Res. 41, e129 (2013).
Pomerantz, A. et al. Real-time DNA barcoding in a rainforest using nanopore sequencing: Opportunities for rapid biodiversity assessments and local capacity building. Gigascience 7 (2018).
Lanfear, R., Schalamun, M., Kainer, D., Wang, W. & Schwessinger, B. MinIONQC: Fast and simple quality control for MinION sequencing data. Bioinformatics 35, 523–525 (2019).
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669 (2018).
Koren, S. et al. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Loman, N. J., Quick, J. & Simpson, J. T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 12, 733–735 (2015).
Viricel, A., Becquet, V., Dubillot, E. & Pante, E. De novo assembly and functional annotation of the transcriptome of Mimachlamys varia, a bioindicator marine bivalve. Mar. Genomics 41, 42–45 (2018).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9 (2014).
Vagner, M. et al. Ocean warming combined with lower omega-3 nutritional availability impairs the cardio-respiratory function of a marine fish. J Exp. Biol. 222 (2019).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Lowe, T. M. & Eddy, S. R. TRNAscan-SE: A program for improved detection of transfer rna genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Conant, G. C. & Wolfe, K. H. GenomeVx: simple web-based creation of editable circular chromosome maps. Bioinformatics 24, 861–862 (2008).
Madeira, F. et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641 (2019).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–7 (2004).
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–12 (2006).
Nei, M. & Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–26 (1986).
Wickham, H. Ggplot2 Elegant Graphics for Data Analysis (2009).
Brown, N. P., Leroy, C. & Sander, C. MView: A web-compatible database search or multiple alignment viewer. Bioinformatics 14, 380–381 (1998).
Acknowledgements
EP thanks the Molecular Core Facility at the University of La Rochelle and the bioinformatics platform Toulouse Midi-Pyrenees (Bioinfo GenoToul). This work was funded by the French Agence Nationale de la Recherche (HySea project ANR-12-BSV7-0011; DRIVE project ANR-18-CE02-0004-01), La Rochelle Université, the CPER/FEDER (ECONAT project), and by funding from the Natural Sciences and Engineering Research Council of Canada (grant no. RGPIN/435656-2013 to SB).
Author information
Authors and Affiliations
Contributions
C.C., E.P. and S.B. conceived and designed the experiments. C.C. analyzed data and drafted the manuscript. K.B., D.G., B.M.R. and D.T.S. participated in the experimental and analytical work. All authors contributed feedback to the writing process and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Capt, C., Bouvet, K., Guerra, D. et al. Unorthodox features in two venerid bivalves with doubly uniparental inheritance of mitochondria. Sci Rep 10, 1087 (2020). https://doi.org/10.1038/s41598-020-57975-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-57975-y
This article is cited by
-
New Dielis species and structural dichotomy of the mitochondrial cox2 gene in Scoliidae wasps
Scientific Reports (2023)
-
Evidence of multiple genome duplication events in Mytilus evolution
BMC Genomics (2022)
-
No evidence of DUI in the Mediterranean alien species Brachidontes pharaonis (P. Fisher, 1870) despite mitochondrial heteroplasmy
Scientific Reports (2022)
-
Presence of male mitochondria in somatic tissues and their functional importance at the whole animal level in the marine bivalve Arctica islandica
Communications Biology (2021)
-
Mitogenomic architecture of the multivalent endemic black clam (Villorita cyprinoides) and its phylogenetic implications
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
