
Abstract
In this work, a novel series of cyanoacetohydrazide linked to 1,2,3-triazoles (9a–n) were designed and synthesized to be evaluated for their anti-α-glucosidase activity, focusing on the fact that α-glucosidase inhibitors have played a significant role in the management of type 2 diabetes mellitus. All synthesized compounds except 9a exhibited excellent inhibitory potential, with IC50 values ranging from 1.00 ± 0.01 to 271.17 ± 0.30 μM when compared to the standard drug acarbose (IC50 = 754.1 ± 0.5 μM). The kinetic binding study indicated that the most active derivatives 9b (IC50 = 1.50 ± 0.01 μM) and 9e (IC50 = 1.00 ± 0.01 μM) behaved as the uncompetitive inhibitors of α-glucosidase with Ki = 0.43 and 0.24 μM, respectively. Moreover, fluorescence measurements were conducted to show conformational changes of the enzyme after binding of the most potent inhibitor (9e). Calculation of standard enthalpy (ΔHm°) and entropy (ΔSm°) values confirmed the construction of hydrophobic interactions between 9e and the enzyme. Also, docking studies indicated desired interactions with important residues of the enzyme which rationalized the in vitro results.
Similar content being viewed by others
Introduction
Diabetes Mellitus (DM) is a common metabolic disease, characterizing by the hyperglycemia that impairs insulin production in the body. The global prevalence of DM and lacking definite treatment of the disease have become a challenging issue in the world1. Long-term dysfunction or failure of various body organs in patients with DM usually leads to severe complications such as kidney diseases, nervous system diseases, leg amputation, heart diseases, and blindness1,2,3. There are three main diabetes types, among which type 2 diabetes (T2DM) with over 85% of diabetics is known as the major type of DM4,5. The first-line medication in T2DM needs a reduction of hepatic glucose production through controlling the digestive enzyme activities or inhibition of carbohydrate digestive enzymes6.
α-Glucosidase (EC 3.2.1.20) is an exocyclic enzyme located in the epithelium of the human small intestine that hydrolyses the 1,4-α-glycosidic linkages of oligosaccharides and disaccharides to form monosaccharides. α-Glucosidase inhibitors slow down the digestion and absorption of simple carbohydrates in the intestine without direct effects on the secretion of insulin leading to the reduction of postprandial plasma glucose levels7. Noteworthy, α-glucosidase inhibitors are also ideal agents for other medical therapies such as hyperlipoproteinemia, obesity, and cancer8,9,10. Clinically approved α-glucosidase inhibitors to target T2DM named acarbose, miglitol, and voglibose have been used in the management of diabetes and obesity. Hence, α-glucosidase is an ideal target against T2DM and their inhibitors are used to alleviate the disease. However, the non-carbohydrate mimicking α-glucosidase inhibitors are limited. In the last years, a variety of synthetic and natural α-glucosidase inhibitors have been developed11,12,13,14,15,16. Most of the potent inhibitors contain heterocyclic compounds11 and coumarins17, thiadiazoles18, imidazoles and benzimidazoles19,20, pyrazoles-benzofurans21, oxindoles22, and isatins16 are examples of synthetic α-glucosidase inhibitors. Further, 1,2,3-triazole based compounds were recently introduced as potent α-glucosidase inhibitors12,16,23,24. 1,2,3-Triazole and its derivatives can be easily prepared through Click multicomponent reaction. The “click” in click chemistry refers to the rapid and selective reactions of small molecules leading to the formation of a wide range of products25. Among many click reactions described to date, copper (I)-catalyzed alkyne-azide cycloaddition introduced by Sharpless et al.26 has attracted much attention due to the potency of production of a library of bioactive 1,2,3-triazole derivatives through heteroatom links27,28,29,30,31,32. The α-glucosidase inhibitory activity of 1,2,3-triazoles have also been fully investigated in the literature33 as a prime choice for medicinal researchers to develop new anti-DM molecules.
In continuation of our efforts to develop novel and efficient anti-α-glucosidase compounds, cyanoacetohydrazide moiety was found to be an ideal and efficient pharmacophore by providing different interactions within the binding site of α-glucosidase. In this respect, new derivatives of cyanoacetohydrazide linked to 1,2,3-triazoles were designed and a library comprising of fourteen compounds was synthesized and evaluated for their in vitro α-glucosidase inhibitory activity. To investigate the interaction of these compounds with α-glucosidase, kinetic as well as molecular docking studies were also performed. Moreover, fluorescence measurements were recorded to characterize conformational changes of the enzyme after inhibition.
Results and discussion
Designing
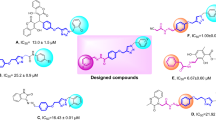
Recently, a large number of research have focused on the physiological and therapeutic properties of benzyl-1,2,3-triazole moiety as a promising pharmacophore to design and develop new potentially useful therapeutic applications23,28,34,35,36,37. Recently, published data revealed that phenoxy-1,2,3-triazole-based scaffolds possessing benzyl substituents are important anti-α-glucosidase agents. For instance, Mahdavi et al. evaluated biscoumarin-phenoxytmethyltriazole derivatives as α-glucosidase inhibitors which exhibited IC50 values in the range of 13–75 μM. Among them, promising compound A (IC50 = 13.0 ± 1.5 µM) showed a competitive mode of inhibition38. The same authors also developed a new series of benzimidazole-1,2,3-triazole hybrid with IC50 values ranging from 25.2 to 176.5 μM. The most potent compound (Fig. 1 compound B) as a competitive inhibitor showed an IC50 = 25.2 ± 0.9 µM. Docking study demonstrated that phenoxy-1,2,3-triazole moiety was stable within the binding site through several π-π, π-cation, and hydrophobic interactions39. Recently, in a study carried out by our group, new hydrazineylideneindolinone derivatives linked to different phenoxymethyl-1,2,3-triazole were designed and the most potent compound (compound C) disclosed 46-fold improvement in the inhibitory activity compared to acarbose with an IC50 value of 750.0 µM. Docking evaluation exhibited H-bonding and π-alkyl interactions between phenoxy ring and Ala284. Also, the 1,2,3-triazole moiety recorded two π-alkyl interactions with leu283 and Ala555 as well as a π-sulfur interaction with Asp28216. As a result, phenoxy-1,2,3-triazole scaffold possessing a 3-benzyl substituent seems to be a good pharmacophore for the inhibition of α-glucosidase and can be further explored to design novel antidiabetic agents.

Design strategy for the SAR studies of current research.
According to our limited literature review, no entry was found for cyanoacetohydrazide moiety as an α-glucosidase inhibitor. Our preliminary docking assessment disclosed that it is a valuable candidate for the exploration of the lead molecule. As depicted in Fig. 1, cyanoacetohydrazide effectively interacted with the critical binding site residues including Trp481, Asp518, Met519, Arg600 and can be considered as an ideal and novel fragment against α-glucosidase.
Pharmacophoric hybridization is known as one of the most efficient strategies in designing novel α-glucosidase inhibitors with improved affinity and efficacy. As a result, the benzyl-1,2,3-triazole moiety which seems to participate in π-stacking and hydrophobic interactions with the enzyme, was linked to the cyanoacetohydrazide pharmacophore. In vitro enzyme inhibition and the mechanism of action as well as docking studies were executed to determine plausible protein–ligand interactions.
Chemistry
Synthesis of the target compounds 9a–n was schematically described in Fig. 2.

Synthesis of compounds 9a–n.
The corresponding derivatives were prepared by the reaction of 1,2,3-triazole-methoxy-benzaldehyde 5 and 2-cyanoacetohydrazide 8 in methanol in the presence of a few drops of acetic acid (HOAc) under microwave irradiation at 700 W for 10–12 min. Aldehyde 5 was prepared by the click reaction of compound 1 and in situ prepared azide derivatives 4 in the presence of triethylamine (NEt3), CuSO4.5H2O, and sodium ascorbate in H2O/tert-BuOH for 24–48 h. It should be mentioned that aldehyde 1 was prepared by the reaction of 4-hydroxy benzaldehyde or 4-hydroxy-3-methoxybenzaldehyde and propargyl bromide in DMF at 80 °C for 4–5 h36. Compound 8 was also obtained by the reaction of excess amount of hydrazine hydrate 6 and ethyl 2-cyanoacetate 7 at room temperature40.
All synthesized compounds were characterized by FTIR, 1H-NMR, 13C-NMR, elemental analysis, and HPLC (Supplementary Information). It should be noted that 1H and 13CNMR spectra of most compounds indicated the presence of two isomers probably due to restricted C-N amide bond rotation41. Also, the presence of two isomers was obvious in HPLC chromatograms.
In vitro α-glucosidase inhibition
Fourteen cyanoacetohydrazide linked to 1,2,3-triazoles 9a–n were synthesized (Table 1). They exhibited varying degrees of α-glucosidase inhibition with IC50 values in the range of 1.00 ± 0.01 to > 750 μM when compared with the standard inhibitor (acarbose: IC50 = 754.1 ± 0.5 μM).
To explain the structure and observed activity correlations, cyanoacetohydrazide-1,2,3-triazole hybrids were divided into three categories based on the presence of methoxy group at X- position (9a–g), the unsubstituted group at X-position (9h–n) along with the substituents at the Y position of benzyl moiety to extract structure–activity relationships (SARs) of α-glucosidase inhibition.
-
(I)
Among the 9a–g bearing OMe at X- position, compound 9e with 4-Cl substituent on the benzyl ring showed the most potent inhibitory activity (IC50 = 1.00 ± 0.01 µM) among all the synthesized compounds. It is worth mentioning that the most active compound 9e recorded 754-fold better potency than the standard drug acarbose (IC50 1.0 Vs 754.1 μM). Changing the chlorine position from para to ortho (9d) led to the decrease of inhibitory activity with an IC50 value of 13.97 ± 0.80 µM. Compound 9b as the second most active analog (Y: 2-F, IC50 = 1.50 μM), showed similar activity compared to the most potent derivatives, 9e (Y: 4-Cl, IC50 = 1.00 μM). Replacing halogen groups with methyl as an electron-donating group in 9f (IC50 = 28.00 μM) and 9g (IC50 = 22.80 μM) caused to decrease of inhibitory activity. Noteworthy, the removal of any substitution from Y position (compounds 9a IC50 > 750 μM) resulted in considerable deterioration of the activity. Overall, it was understood that any substitution at the Y- position improved the inhibitory activity. Also, the electron-donating substituent is less effective compared to electron-withdrawing groups. The presence of halogen groups (2-F and 4-Cl) might play a key role in this inhibition of enzyme due to the high electronegativity, which makes the whole molecule more polar, and the enzyme might have better interaction with it.
-
(II)
Similar to the previous set, among derivatives 9h–n, any substitutions at the Y- position improved the activity significantly as compared with the unsubstituted analog. This trend can easily be seen in compound 9h (Y = H) vs 9i (Y = 2-F), 9j (Y = 4-F), 9k (Y = 2-Cl), 9l (Y = 4-Cl), 9m (Y = 2-Me), 9n (Y = 4-Me). The activity of analogs containing electron-withdrawing group demonstrated that 4-Cl (9l) moiety at Y had good inhibition with IC50 value of 21.66 μM followed by 2-F (9i, IC50 = 45.89 μM) > 4-F (9j, IC50 = 56.64 μM) > 2-Cl (9k, IC50 = 74.68 μM). The minor difference in the activity of the last three analogs may be due to the difference in the position and electron-withdrawing power of the substituents on the benzyl moiety.
By comparing the IC50 values in this set, it can be implied that ortho-methyl group as electron-donating substituent caused a significant improvement in the α-glucosidase inhibition with an IC50 value of 11.28 μM.
-
(III)
Comparison of derivatives bearing the same substitution group at Y while X varies revealed that 9h as an unsubstituted derivative at Y exhibited better potency compared to the 9a counterpart. However, this trend was not followed in the rest of the derivatives as 9i, 9j, 9k, 9l, and 9n were not more potent than their counterparts 9b, 9c, 9d, 9l, and 9g. It can be understood that the SAR was mainly affected by the difference in substituents (Fig. 3).

Summary of SAR studies of the library against α-glucosidase.
Overall, it was perceived that any substitution at the Y position is favorable. Among the first set of compounds bearing OMe at X, it can be found that 4-Cl and 2-F substituents on the benzyl moiety played a substantial role in the anti-α-glucosidase activity. Although the presence of 2-CH3 at the Y-position had destructive effect on the first category, this derivative showed the highest activity in the second category.
To correlate the activity of present molecules with the previously published reports, different interesting SARs were obtained. The comparison of IC50 values of phenoxy derivatives with their corresponding methoxyphenyl analogs of biscoumarin derivatives (Compound A, Fig. 1) revealed that phenoxy analogs of biscoumarin (with 2-chloro and 4-nitro substituents) were more active than 4-methoxyphenoxy counterparts38. These results were supported in other studies on hydrazineylideneindolinone derivatives (Compound C, Fig. 1) so that phenoxy derivatives were more potent than methoxyphenoxy compounds16. Noteworthy, unlike the previous studies, in this work phenoxymethyl-1,2,3-triazole derivatives were more potent inhibitors than phenoxy-1,2,3-triazole counterparts.
Comparison of the benzyl substitutions showed that 2-fluorobenzyl of hydrazineylideneindolinone linked to phenoxymethyl-1,2,3-triazole derivatives (Compound C, Fig. 1) induced better α-glucosidase inhibitory activity than other derivatives16. Also, the same trend was observed by Xie et al., so the 2-fluorobenzyl moiety of isatin-thiazole scaffold disclosed better potency in comparison to different derivatives42. These results are in line with the current study. However, assessments on biscoumarin-1,2,3-triazole hybrids exhibited that 2-Cl substitution on the benzyl pendant recorded better potency than the rest of the derivatives38.
Enzyme kinetic studies
Kinetic studies were conducted for compounds 9b, 9e, 9i, and 9l to identify the type of inhibition. According to Fig. 4, the Lineweaver–Burk plot showed that the Km and Vmax gradually decreased with increasing the inhibitor concentration, indicating an uncompetitive inhibition for compounds 9b and 9e with Ki = 0.43 and 0.24 µM, respectively. However, investigation of their compartments 9i and 9l demonstrated different manner of α-glucosidase inhibition. As can be seen in Figs. 5 and 6, they revealed a competitive inhibition. The Ki value for compound 9i was calculated as 75.0 µM and the corresponding value for compound 9l was obtained as 85.0 µM.

Kinetic study of α-glucosidase inhibition by compounds 9b and 9e. The Lineweaver–Burk plots were obtained in the absence and presence of different concentrations of inhibitors.

Kinetic study of α-glucosidase inhibition by compounds 9i.

Kinetic study of α-glucosidase inhibition by compounds 9l.
Fluorescence spectroscopy measurements
The intrinsic fluorescence property of α-glucosidase is generally due to the presence of tryptophan, tyrosine, and phenylalanine amino acids. α-Glucosidase has 18 tryptophan residues that eight are exposed to the solvent, and four are found in the proposed active site pocket (Trp381, Trp710, Trp715, and Trp789). Therefore, the conformation of the enzyme affected by the local tryptophan environment, can be followed by the change of fluorescence intensity43,44. In fact, fluorescence spectroscopy measurements could be used to predict the tertiary structure of the enzyme. To demonstrate the effect of compound 9e on α-glucosidase activity, fluorescence spectra of the enzyme in the presence of various concentrations of 9e were recorded (Fig. 7). As can be seen in Fig. 7, no shift was observed in the emission maximum (λmax 340 nm) but a significant increase in fluorescence intensity was detected. This effect was directly dependent on the concentration of 9e in the range of 0–1.0 μM.

(Left) Fluorescence spectroscopy of α-glucosidase in the presence of different concentrations of compound 9e (0–1.0 µM) in phosphate buffer (50 mM, pH 6.8). (Right) Inset shows the change in absorbance at 37 °C as a function of compound 9e.
Thermodynamic analysis of binding of compound 9e to α-glucosidase
Noncovalent interactions including hydrogen bonding, hydrophobic, electrostatic, and van der Waals forces are common forces between ligand and protein. To get insight into binding forces in the 9e—α-glucosidase complex, the thermodynamic study was conducted and the thermodynamic parameters of the noncovalent interactions, i.e., standard enthalpy change (ΔH°), standard entropy change (ΔS°), and standard free energy change (ΔG°) were calculated. For this purpose, the stability of α-glucosidase in the presence or absence of compound 9e was investigated by screening the fluorescence intensity at 340 nm at different temperatures (298–338 K) based on the equilibrium model (Native state ↔ Unfolded state). The start and end temperature points were 298 and 338 K, respectively. Denaturation profiles of α-glucosidase were then obtained by thermal scanning in the presence of various concentration of 9e. As shown in Fig. 8, a sigmoidal curve observed by each profile indicated a single denaturant-dependent step based on the two-state theory.

Fraction of unfolded α-glucosidase in various concentrations of compound 9e in phosphate buffer (50 mM, pH 6.8).
The values of ΔH°m and ΔS°m were calculated as reported in Table 2. Tm was estimated to be the lowest for α-glucosidase that incubated in the presence of compound 9e at the concentration of 1.0 µM (311 K), but in the case of concentrations of 0.5 and 0 µM, Tm was estimated to be 315 and 317 K, respectively. These results revealed that the most instability occurred at the higher concentration of compound 9e.
The forces between the protein and ligand can be categorized into I: ΔH° > 0, ΔS° > 0 for hydrophobic interactions; II: ΔH° < 0, ΔS° < 0 for van der Waals forces; III: ΔH° < 0, ΔS° < 0 for hydrogen bond and van der Waals interactions and IV: ΔH° < 0, ΔS° > 0 for electrostatic interactions; as non-covalent interactions. According to our results (Table 2), the presence of compound 9e in aqueous solutions of α-glucosidase indicated the formation of hydrophobic interactions between nonpolar amino acid residues and the enzyme, confirming the unfolded state of the protein.
Docking studies
Molecular docking studies were performed for compounds 9b and 9e to investigate the mode of their interactions with α-glucosidase (PDB ID: 5NN8) using the maestro molecular modeling platform of Schrödinger package. First to validate the in-silico procedure, the acarbose as a crystallographic inhibitor was docked into human lysosomal acid-α-glucosidase. The superimposed structure of acarbose and its crystallographic conformation recorded an RMSD value of 1.69 Å. Next, the docking assessments of the compounds were done based on the same protocol performed on the crystallographic inhibitor.
Figure 9 presented the binding pattern of derivative 9b with the binding site of α-glucosidase (glide score = -− 7.04 kcal/mol). Derivative 9b oriented within the α-glucosidase active site so that phenoxy-cyanoacetohydrazide penetrated the deep gorge of the binding site and the substituted moiety oriented toward the entrance of the active site. In detail, the nitrogen of cyanoacetohydrazide pendant was fixed between the Trp616 (essential residue) and Arg672. Carbonyl and hydrazine moieties of cyanoacetohydrazide group also participated in H-bound interactions with Arg600 (essential residue). The ortho-fluorobenzyl ring was stacked with Phe525 thus stabilizing the molecule at the entrance of the active site to get the suppressed conformation of α-glucosidase.

3D and 2D diagram of compound 9b within the binding pocket of α-glucosidase and potential distribution surface diagram.
According to molecular docking study, 9e recorded the glide score of − 6.89 kcal/mol. As shown in Fig. 10, phenoxy-cyanoacetohydrazide oriented toward the inner core of the binding pocket, while the para-chlorobenzyl part (substituted moiety) of the compound bonded near the active site entrance. Focusing on the cyanoacetohydrazide pendant, confirmed our designing strategy so that nitrogen of the cyano group exhibited two H-bonding interactions with Trp613 (essential residue) and Arg672. Also, NH of hydrazide participated in H-bonding interaction with Asp616 (essential residue). There were π–π stacking and π–cation interactions between Arg600 (essential residue) and the phenoxy linker. Also, 1,2,3-triazole ring recorded a π–π stacking interaction with Phe525.

3D and 2D diagram of compound 9e within the binding pocket of α-glucosidase and potential distribution surface diagram.
ADME-toxicity profiles and physicochemical properties
The pkCSM server45 was used to predict the ADME-toxicity properties of synthesized compounds. As shown in Table 3, all derivatives showed good human intestinal absorption, low clearance values, and low toxicity.
The results of drug-likeness properties were shown in Table 4. All compounds exhibited appropriate molecular properties with no drug-likeness rules violations46.
Conclusion
Novel cyanoacetohydrazide linked to 1,2,3-triazoles were designed, synthesized, and characterized via spectroscopic techniques and evaluated for their α-glucosidase inhibitory potential. These compounds except 9a demonstrated considerable inhibitory activity against α-glucosidase with IC50 value of 1.0 to 271.17 µM compared to acarbose as the positive control (IC50 value of 754.1 µM). Compound 9e (IC50 = 1.00 ± 0.01 μM) with having para chlorobenzyl ring and 9b (IC50 = 1.50 ± 0.01 μM) bearing ortho fluorobenzyl pendant group were found to be the most potent α-glucosidase inhibitors. Kinetic studies revealed that they 9b and 9e behaved uncompetitively against the enzyme with Ki = 0.43 and 0.24 μM, respectively. Also, the binding affinity between compound 9e at different concentrations and α-glucosidase was recorded using fluorescence measurements. It indicated the inhibition of α-glucosidase due to conformational changes of the enzyme. According to the thermodynamic studies, hydrophobic interactions were found to be responsible for the formation of 9e—α-glucosidase complex. The in-silico studies confirmed the designing strategy so that cyanoacetohydrazide group was able to form several important interactions within the cavity which supported the high potency of these compounds and phenoxy-1,2,3-triazole moiety stabilized the derivatives through several hydrophobic and hydrophilic interactions. Interestingly substituted moiety at Y position occupied the entrance of the active site to get the suppressed conformation of α-glucosidase. These results were in accordance with enzymatic assessments that any substitution at the Y position was favorable. As expected, developed pharmacophores used in the design of these hybrids, are involved in the interactions with the enzyme.
Materials and methods
All chemicals and reagents were purchased from Merck and Aldrich. Melting points were determined using Kofler hot stage apparatus and are uncorrected. The IR spectra were obtained on a Nicolet Magna FTIR 550 spectrometer (potassium bromide disks). NMR spectra were recorded on a Varian-INOVA 500 MHz and chemical shifts were expressed as δ (ppm) with tetramethylsilane as internal standard. Analytical HPLC evaluation was performed on a YL9100 HPLC system (Korea) equipped with UV detectors using a RP column (Teknokroma, C18, 5 μm, 150 × 4.6 mm) and solvent: methanol (solvent A) and water, a gradient of 0–100% solvent A in 11 min, 1 min at 0%, to 50% within 3 min, to 100% at 6 min, to 0 within 5 min (total run time 11 min); flow rate, 1 mL/min; detection, 254 nm; injection volume, 20 μL.
Synthesis of compounds 9
The click reaction was conducted by a mixture of aldehyde 1 and in situ prepared azide derivative 4 to obtain compound 516. For this purpose, benzyl chloride/bromide derivative 2 (1.1 mmol) and sodium azide 3 (0.06 g, 0.9 mmol) in the presence of triethylamine (0.13 g, 1.3 mmol) in the mixture of water (4 mL) and tert-butyl alcohol (4 mL) was stirred at room temperature for 30 min. Next, compound 1 (0.5 mmol) and CuSO4·5H2O (7 mol%) were added to the reaction mixture and it was continued for 24–48 h. After completion of the reaction (checked by TLC), the mixture was poured on crushed ice, the precipitates were filtered off and washed with water. Compound 5 was used for further steps with no purification. A mixture of compound 5 (1 mmol) and 2-cyanoacetohydrazide 8 (1 mmol) in methanol (8 mL), in the presence of a few drops of HOAc was irritated under microwave irradiation at 700 W for 10–12 min (1 min interval). After completion of the reaction (checked by TLC), the mixture was poured on crushed ice, the precipitates were filtered off and washed frequently with water (Supplementary Information).
N′-(4-((1-benzyl-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxybenzylidene)-2-cyanoacetohydrazide (9a)
Deep yellow precipitates, Yield: 92%, mp 142–144 °C, IR (KBr, cm−1): 3415, 3222, 2924, 2250, 1690, 1616, 1586. 1H-NMR (500 MHz, DMSO-d6) (two isomers): 11.71 (s, 1H, NH), 11.63 (s, 1H, NH), 8.33 (s, 1H, triazole), 8.31 (s, 1H, triazole), 7.96 (s, 1H, CH), 7.93 (s, 1H, CH), 7.40–7.32 (m, 7H, H5, H6, H2′, H3′, H4′, H5′, H6′), 7.21 (s, 1H, H2), 7.18 (s, 1H, H2), 5.62 (s, 2H, CH2), 5.26 (s, 2H, CH2), 5.17 (s, 2H, CH2), 4.21 (s, 2H, CH2), 3.79 (s, 3H, OMe), 3.77 (s, 3H, OMe) ppm. 13C-NMR (125 MHz, DMSO-d6) (two isomers): 165.1, 153.3, 149.8, 149.7, 144.8, 136.4, 130.4, 129.2, 128.7, 128.5, 127.5, 121.7, 116.6, 115.3, 113.6, 113.1, 110.2, 109.4, 62.1, 55.9, 53.4, 46.3, 24.8 ppm. Calcd for C21H20N6O3: C, 62.37; H, 4.98; N, 20.78. Found: C, 62.50; H, 5.18; N, 20.51.
2-Cyano-N′-(4-((1-(2-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxybenzylidene)acetohydrazide (9b)
Pale yellow precipitates, Yield: 74%, mp 124–126 °C, 3416, 3227, 2936, 2250, 1692, 1600. 1H-NMR (500 MHz, DMSO-d6) (two isomers): 11.70 (s, 1H, NH), 11.60 (s, 1H, NH), 8.27 (s, 1H, triazole), 8.08 (s, 1H, triazole), 7.92 (s, 1H, CH), 7.43–7.18 (m, 14H, H2, H5, H6, H3′, H4′, H5′, H6′), 5.68 (s, 2H, CH2), 5.16 (s, 2H, CH2), 4.21 (s, 2H, CH2), 3.77 (s, 3H, OMe) ppm. 13C-NMR (125 MHz, DMSO-d6): 165.1, 160.6 (d, JC-F = 245.4 Hz), 149.8, 149.7, 144.8, 131.3 (d, JC-F = 3.8 Hz), 131.2, 127.5, 125.6, 125.3 (d, JC-F = 3.7 Hz), 123.3., 123.2, 121.7, 116.7, 116.1 (d, JC-F = 20.6 Hz), 113.6, 109.4, 62.0, 55.9, 47.4, 24.8 ppm. Calcd for C21H19FN6O3: C, 59.71; H, 4.53; N, 19.90. Found: C, 59.62; H, 4.39; N, 19.78.
2-Cyano-N′-(4-((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxybenzylidene)acetohydrazide (9c)
Pale yellow precipitates, Yield: 68%, mp 131–133 °C, IR (KBr, cm−1): 3416, 2963, 2850, 2250, 1690, 1603. 1H-NMR (500 MHz, DMSO-d6) (two isomers): 11.71 (s, 1H, NH), 11.62 (s, 1H, NH), 8.30 (s, 1H, triazole), 8.10 (s, 1H, triazole), 7.93 (s, 1H, CH), 7.88 (s, 1H, CH), 7.42–7.34 (m, 3H, H6, H2′, H6′), 7.21 (t, J = 8.8 Hz, 2H, H3′, H5′), 7.20–7.18 (m, 2H, H2, H5), 5.61 (s, 2H, CH2), 5.16 (s, 2H, CH2), 4.22 (s, 2H, CH2), 3.79 (s, 3H, OMe), 3.77 (s, 3H, OMe) ppm. 13C-NMR (125 MHz, DMSO-d6): 165.1, 162.4 (d, JC-F = 243.3 Hz), 150.0, 149.8, 149.7, 132.7, 130.8 (d, JC-F = 8.4 Hz), 127.5, 125.4, 122.3, 121.7, 116.6, 116.1 (d, JC-F = 21.5 Hz), 113.6, 109.4, 62.1, 55.9, 52.5, 24.8 ppm Calcd for C21H19FN6O3: C, 59.71; H, 4.53; N, 19.90. Found: C, 59.58; H, 4.36; N, 20.21.
N′-(4-((1-(2-chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxybenzylidene)-2-cyanoacetohydrazide (9d)
Yellow precipitates, Yield: 86%, mp 148–150 °C, IR (KBr, cm−1): 3415, 3224, 2922, 2250, 1698, 1662, 1613. 1H-NMR (500 MHz, DMSO-d6) (two isomers): 11.71 (s, 1H, NH), 11.61 (s, 1H, NH), 8.27 (s, 1H, triazole), 8.09 (s, 1H, triazole), 7.92 (s, 1H, CH), 7.88 (s, 1H, CH), 7.52 (d, J = 7.7 Hz, 1H, H3′), 7.40–7.34 (m, 3H, H6, H5′, H6′), 7.24–7.18 (m, 3H, H2, H5, H4′), 5.72 (s, 2H, CH2), 5.17 (s, 2H, CH2), 4.21 (s, 2H, CH2), 3.77 (s, 3H, OMe), 3.67 (s, 3H, OMe) ppm. 13C-NMR (125 MHz, DMSO-d6) (two isomers): 165.1, 151.1, 149.8, 149.7, 149.6, 144.7, 133.7, 133.1, 131.0, 130.7, 130.1, 128.2, 127.5, 125.9, 125.8, 121.7, 116.6, 113.7, 109.4, 62.0, 55.9, 51.1, 24.8 ppm. Calcd for C21H19ClN6O3: C, 57.47; H, 4.36; N, 19.15. Found: C, 57.60; H, 4.54; N, 18.92.
N′-(4-((1-(4-chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxybenzylidene)-2-cyanoacetohydrazide (9e)
Creamy precipitates, Yield: 87%, mp 199–201 °C, IR (KBr, cm−1): 3417, 2928, 2250, 1683, 1616. 1H-NMR (500 MHz, DMSO-d6) (two isomers): 11.71 (s, 1H, NH), 11.63 (s, 1H, NH), 8.34 (s, 1H, triazole), 8.08 (s, 1H, triazole), 7.92 (s, 1H, CH), 7.88 (s, 1H, CH), 7.44 (d, J = 8.2 Hz, 2H, H3′, H5′), 7.35–7.33 (m, 3H, H6, H2′, H6′), 7.20–7.17 (m, 2H, H2, H5), 5.62 (s, 2H, CH2), 5.16 (s, 2H, CH2), 4.21 (s, 2H, CH2), 3.79 (s, 3H, OMe), 3.77 (s, 3H, OMe) ppm. 13C-NMR (125 MHz, DMSO-d6): 165.1, 151.9, 149.8, 149.7, 144.7, 135.4, 133.4, 130.4, 129.2, 127.5, 125.5, 121.7, 116.6, 113.7, 109.4, 62.1, 55.9, 52.5, 24.8 ppm. Calcd for C21H19ClN6O3: C, 57.47; H, 4.36; N, 19.15. Found: C, 57.21; H, 4.20; N, 19.31.
2-Cyano-N′-(3-methoxy-4-((1-(2-methylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzylidene)acetohydrazide (9f)
Pale yellow precipitates, Yield: 94%, mp 105–107 °C, IR (KBr, cm−1): 3415, 2960, 2250, 1688, 1602, 1578. 1H-NMR (500 MHz, DMSO-d6) (two isomers): 11.71 (s, 1H, NH), 11.62 (s, 1H, NH), 8.20 (s, 1H, triazole), 8.10 (s, 1H, triazole), 7.93 (s, 1H, CH), 7.90 (s, 1H, CH), 7.34–7.18 (m, 12H, H2, H5, H6, H3′, H4′, H5′), 7.09 (d, J = 7.6 Hz, 1H, H6′), 5.63 (s, 2H, CH2), 5.17 (s, 2H, CH2), 4.22 (s, 2H, CH2), 3.79 (s, 3H, OMe), 3.77 (s, 3H, OMe), 2.35 (s, 3H, Me) ppm. 13C-NMR (125 MHz, DMSO-d6):165.1, 149.8, 149.7, 144.8, 136.8, 134.5, 113.3, 130.9, 129.1, 128.8, 127.5, 126.7, 125.6, 121.7, 116.6, 113.7, 109.5, 62.1, 55.9, 51.4, 24.8, 19.1 ppm. Calcd for C22H22N6O3: C, 63.15; H, 5.30; N, 20.08. Found: C, 63.37; H, 5.44; N, 19.79.
2-Cyano-N′-(3-methoxy-4-((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzylidene)acetohydrazide (9g)
Off white precipitates, Yield: 84%, mp 98–100 °C, IR (KBr, cm−1): 3415, 3222, 2923, 2250, 1683, 1601. 1H NMR (500 MHz, DMSO-d6) (two isomers): 11.71 (s, 1H, NH), 11.61 (s, 1H, NH), 8.26 (s, 1H, triazole), 8.04 (s, 1H, triazole), 7.92 (s, 1H, CH), 7.88 (s, 1H, CH), 7.33–7.17 (m, 7H, H2, H5, H6, H2′, H3′, H5′, H6′), 5.55 (s, 2H, CH2), 5.15 (s, 2H, CH2), 4.21 (s, 2H, CH2), 3.78 (s, 3H, OMe), 3.77 (s, 3H, OM), 2.28 (s, 3H, Me), 2.24 (s, 3H, Me) ppm. 13C-NMR (125 MHz, DMSO-d6): 165.1, 151.5, 149.8, 149.7, 144.8, 138.0, 133.4, 129.8, 128.5, 127.5, 125.3, 121.7, 116.6, 113.6, 109.4, 62.1, 55.9, 53.1, 24.8, 21.1 ppm. Calcd for C22H22N6O3: C, 63.15; H, 5.30; N, 20.08. Found: C, 63.43; H, 5.18; N, 19.88.
N′-(4-((1-benzyl-1H-1,2,3-triazol-4-yl)methoxy)benzylidene)-2-cyanoacetohydrazide (9h)
Off white precipitates, Yield: 65%, mp 121–123 °C, IR (KBr, cm−1): 3425, 3222, 3100, 22,250, 1680, 1606. 1H NMR (500 MHz, DMSO-d6) (two isomers): 11.68 (s, 1H, NH), 11.58 (s, 1H, NH), 8.31 (s, 1H, triazole), 8.10 (s, 1H, triazole), 7.95 (s, 1H, CH), 7.90 (s, 1H, CH), 7.64 (d, J = 8.4 Hz, 2H, H2, H6), 7.53 (d, J = 8.5 Hz, 2H, H2, H6), 7.39–7.31 (m, 7H, H2′, H3′, H4′, H5′, H6′), 7.09 (d, J = 8.4 Hz, 2H, H3, H5), 5.61 (s, 2H, CH2), 5.19 (s, 2H, CH2), 4.18 (s, 2H, CH2), 4.15 (s, 2H, CH2) ppm. 13C NMR (125 MHz, DMSO-d6) (two isomers): 165.0, 160.1, 144.6, 143.3, 136.4, 129.3, 129.2, 129.1, 128.6, 128.4, 127.2, 125.2, 116.6, 115.5, 61.7, 53.3, 24.7 ppm. Calcd for C20H18N6O2: C, 64.16; H, 4.85; N, 22.45. Found: C, 64.35; H, 4.63; N, 22.60.
2-Cyano-N′-(4-((1-(2-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzylidene)acetohydrazide (9i)
Off white precipitates, Yield: 60%, mp 123–125 °C, IR (KBr, cm−1): 3415, 2963, 2850, 2250, 1681, 1607. 1H NMR (500 MHz, DMSO-d6) (two isomers): 11.68 (s, 1H, NH), 11.60 (s, 1H, NH), 8.30 (s, 1H, triazole), 8.12 (s, 1H, triazole), 7.96 (s, 1H, CH), 7.90 (s, 1H, CH), 7.65 (d, J = 8.5 Hz, 2H, H2, H6), 7.55–7.53 (m, 1H, H4′), 7.44–7.41 (m, 1H, H3′), 7.38–7.35 (m, 2H, H3′, H4′), 7.29–7.22 (m, 4H, 2 × H5′, 2 × H6′), 7.10 (d, J = 8.5 Hz, 2H, H3, H5), 5.69 (s, 2H, CH2), 5.20 (s, 2H, CH2), 4.19 (s, 2H, CH2), 4.16 (s, 2H, CH2) ppm. 13C NMR (125 MHz, DMSO-d6) (two isomers): 165.0, 160.6 (d, JC-F = 245.6 Hz), 160.1, 159.1, 148.1, 144.6, 131.3, 131.2 (d, JC-F = 10.8 Hz), 129.3, 129.1, 127.2, 125.4, 125.3 (d, JC-F = 14.3 Hz), 123.2 (d, JC-F = 14.8 Hz), 116.6, 116.1 (d, JC-F = 20.9 Hz), 115.6, 115.5, 61.6, 47.4, 24.7 ppm. Calcd for C20H17FN6O2: C, 61.22; H, 4.37; N, 21.42. Found: C, 61.40; H, 4.21; N, 21.28.
2-Cyano-N′-(4-((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzylidene)acetohydrazide (9j)
Off white precipitates, Yield: 93%, mp 108–110 °C, IR (KBr, cm−1): 3422, 2961, 2250, 1681, 1605. 1H NMR (500 MHz, DMSO-d6) (two isomers): 11.67 (s, 1H, NH), 11.59 (s, 1H, NH), 8.30 (s, 1H, triazole), 8.10 (s, 1H, triazole), 7.95 (s, 1H, CH), 7.89 (s, 1H, CH), 7.64 (d, J = 8.1 Hz, 2H, H2, H6), 7.53 (d, J = 8.6 Hz, 2H, H2, H6), 7.41–7.38 (m, 2H, H2′, H6′), 7.21 (t, J = 8.7 Hz, 2H, H3′, H5′), 7.08 (d, J = 8.1 Hz, 2H, H3, H5), 7.04 (d, J = 8.8 Hz, 2H, H3, H5), 5.61 (s, 2H, CH2), 5.19 (s, 2H, CH2), 4.17 (s, 2H, CH2), 4.15 (s, 2H, CH2) ppm. 13C NMR (125 MHz, DMSO-d6) (two isomers): 165.0, 162.4 (d, JC-F = 243.3 Hz), 160.0, 148.0, 144.6, 132.7 (d, JC-F = 2.9 Hz), 130.8, 130.7, 130.6, 130.5, 127.2, 125.2, 116.6, 116.1, 115.1 (d, JC-F = 21.4 Hz), 61.7, 52.5, 25.2, 24.7 ppm. Calcd for C20H17FN6O2: C, 61.22; H, 4.37; N, 21.42. Found: C, 61.51; H, 4.14; N, 21.63.
N′-(4-((1-(2-chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzylidene)-2-cyanoacetohydrazide (9k)
White precipitates, Yield: 73%, mp 103–105 °C, IR (KBr, cm−1): 3415, 3217, 3075, 2925, 2250, 1677, 1607. 1H NMR (500 MHz, DMSO-d6) (two isomers): 11.67 (s, 1H, NH), 11.59 (s, 1H, NH), 8.28 (s, 1H, triazole), 8.10 (s, 1H, triazole), 7.95 (s, 1H, CH), 7.64 (d, J = 8.5 Hz, 2H, H2, H6), 7.52 (d, J = 7.7 Hz, 1H, H3′), 7.42–7.35 (m, 2H, H4′, H5′), 7.23 (d, J = 7.7 Hz, 1H, H6′), 7.09 (d, J = 8.5 Hz, 2H, H3, H5), 5.72 (s, 2H, CH2), 5.20 (s, 2H, CH2), 4.18 (s, 2H, CH2) ppm. 13C NMR (125 MHz, DMSO-d6) (two isomers): 165.0, 160.0, 144.6, 143.0, 133.7, 133.1, 131.0, 130.7, 130.1, 129.3, 129.1, 128.2, 127.2, 125.7, 116.6, 115.6, 115.5, 61.6, 51.1, 24.7 ppm. Calcd for C20H17ClN6O2: C, 58.75; H, 4.19; N, 20.56. Found: C, 58.93; H, 3.90; N, 20.38.
N′-(4-((1-(4-chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzylidene)-2-cyanoacetohydrazide (9l)
Off white precipitates, Yield: 89%, mp 123–125 °C, IR (KBr, cm−1): 3414, 3235, 2924, 2250, 1682, 1638. 1H NMR (500 MHz, DMSO-d6) (two isomers): 11.67 (s, 1H, NH), 11.59 (s, 1H, NH), 8.31 (s, 1H, triazole), 8.11 (s, 1H, triazole), 7.95 (s, 1H, CH), 7.89 (s, 1H, CH), 7.64 (d, J = 8.1 Hz, 2H, H2, H6), 7.53 (d, J = 8.6 Hz, 2H, H2, H6), 7.44 (d, J = 8.0 Hz, 2H, H3′, H5′), 7.34 (d, J = 8.0 Hz, 2H, H2′, H6′), 7.28 (d, J = 8.0 Hz, 2H, H2′, H6′), 7.08 (d, J = 8.1 Hz, 2H, H3, H5), 6.98 (d, J = 6.5 Hz, 2H, H3, H5), 5.62 (s, 2H, CH2), 5.19 (s, 2H, CH2), 4.17 (s, 2H, CH2), 4.15 (s, 2H, CH2) ppm. 13C NMR (125 MHz, DMSO-d6) (two isomers): 165.0, 160.0, 144.6, 135.4, 133.4, 130.4, 130.1, 129.4, 129.3, 129.2, 129.1, 127.2, 125.3, 116.6, 115.5, 61.6, 52.5, 24.7 ppm. Calcd for C20H17ClN6O2: C, 58.75; H, 4.19; N, 20.56. Found: C, 58.60; H, 4.37; N, 20.70.
2-Cyano-N′-(4-((1-(2-methylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzylidene)acetohydrazide (9m)
White precipitates, Yield: 490%, mp 116–118 °C, IR (KBr, cm−1): 3415, 3222, 2960, 2250, 1682, 1605. 1H NMR (500 MHz, DMSO-d6) (two isomers): 11.67 (s, 1H, NH), 11.60 (s, 1H, NH), 8.21 (s, 1H, triazole), 8.11 (s, 1H, triazole), 7.95 (s, 1H, CH), 7.90 (s, 1H, CH), 7.26–7.08 (m, 8H, H2, H3, H5, H6, H3′, H4′, H5′, H6′), 5.62 (s, 2H, CH2), 5.20 (s, 2H, CH2), 4.17 (s, 2H, CH2), 2.30 (s, 3H, Me) ppm. 13C NMR (125 MHz, DMSO-d6) (two isomers): 165.0, 160.0, 144.6, 136.8, 134.5, 130.9, 129.3, 129.1, 129.0, 128.8, 127.1, 126.7, 125.5, 116.6, 115.6, 115.5, 61.6, 51.5, 24.7, 19.1 ppm. Calcd for C21H20N6O2: C, 64.94; H, 5.19; N, 21.64. Found: C, 65.15; H, 5.28; N, 21.44.
2-Cyano-N′-(4-((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzylidene)acetohydrazide (9n)
Off white precipitates, Yield: 84%, mp 128–130 °C, IR (KBr, cm−1): 3417, 2924, 2956, 2250, 1682, 1606. 1H NMR (500 MHz, DMSO-d6) (two isomers): 11.67 (s, 1H, NH), 11.58 (s, 1H, NH), 8.26 (s, 1H, triazole), 8.10 (s, 1H, triazole), 7.95 (s, 1H, CH), 7.89 (s, 1H, CH), 7.64 (d, J = 8.1 Hz, 2H, H2, H6), 7.54 (d, J = 8.7 Hz, 2H, H2, H6), 7.22 (d, J = 7.7 Hz, 2H, H2′, H6′), 7.17 (d, J = 7.7 Hz, 2H, H3′, H5′), 7.08 (d, J = 8.1 Hz, 2H, H3, H5), 7.03 (d, J = 7.5 Hz, 2H, H3, H5), 5.55 (s, 2H, CH2), 5.17 (s, 2H, CH2), 4.18 (s, 2H, CH2), 4.15 (s, 2H, CH2), 2.27 (s, 3H, Me), 2.23 (s, 3H, Me) ppm. 13C NMR (125 MHz, DMSO-d6) (two isomers): 165.0, 160.1, 145.1, 144.6, 138.4, 138.0, 133.4, 129.8, 129.3, 129.1, 128.5, 128.2, 127.2, 125.1, 116.6, 115.6, 115.5, 61.7, 53.1, 24.7, 21.1 ppm. Calcd for C21H20N6O2: C, 64.94; H, 5.19; N, 21.64. Found: C, 65.22; H, 5.31; N, 21.80.
In vitro α-glucosidase inhibition assay
α-Glucosidase (Saccharomyces cerevisiae, EC3.2.1.20, 20 U/mg) and the substrate, p-nitrophenyl-β-D-glucopyranoside (p-NPG) were purchased from Sigma-Aldrich and the assay was performed exactly according to our previous report14. In this respect, various concentrations of each synthesized compound dissolved in DMSO, were added to potassium phosphate buffer (50 mM, pH 6.8) including enzyme (at final concentration of 0.1 U/mL), in a 96-well plate. After a 10-min incubation at 37 °C, p-NPG was added to each well to achieve final concentration of 4 mM. Then, the plate was re-incubated at 37 °C for 20 min. It should be noted that the final concentration of DMSO in each enzymatic solution was 10%. Finally, the change in the absorbance was measured at 405 nm using spectrophotometer (Synergy HTX Multi-Mode Microplate Reader–BioTek, Germany). Acarbose, the standard inhibitor of α-glucosidase was used as the positive control and the enzyme activity in the absence of each inhibitor was considered as the negative control. The percentage of inhibition for compounds and control was calculated using Eq. (1):
IC50 values were calculated from the nonlinear regression curve using the Logit method.
Enzyme kinetic studies
The mode of inhibition of compounds 9b, 9e, 9i, and 9l was investigated against α-glucosidase activity with different concentrations of p-nitrophenyl α-D-glucopyranoside (p-NPG) (2–16 mM) as the substrate in the absence and presence of those compounds at different concentrations (9b: 0, 0.38, 0.75, and 1.50 µM; 9e: 0, 0.25, 0.50, and 1.00 µM; 9i: 0, 11.00, 23.00, and 45.00 µM; 9l: 0, 18.70, 37.40, and 74.40 µM). A Lineweaver–Burk plot was generated to identify the type of inhibition and the Michaelis–Menten constant.
Fluorescence spectroscopy measurements
Compound 9e at different concentrations (0–1.0 µM) was added into the 3 mL solution containing a fixed amount of α-glucosidase (0.1 U/mL). All mixtures were held for 10 min to equilibrate before measurements. Then, the fluorescence emission spectra were measured from 300 to 450 nm at the excitation wavelength of 280 nm on a Synergy HTX multi-mode reader (Biotek Instruments, Winooski, VT, USA) equipped with a 1.0 cm quartz cell holder. The fluorescence spectra of the buffer containing compound 9e in the absence of the enzyme were subtracted as the background fluorescence47.
Thermodynamic analysis against α-glucosidase
Thermodynamic analysis was performed as described by Mojtabavi et al., the fluorescent intensity data were plotted as a function of temperature, and the thermodynamic profile was computed48,49. Therefore, the denatured fraction (FD) of protein was calculated from Eq. (2), assuming a two-state mechanism for the protein denaturation:
In Eq. (2), Yobs, YN, and YD are the observed absorbance, the values of absorbance characteristics of a fully native and denatured conformation, respectively. Equation (3) was used to calculate the apparent equilibrium constant (K) for a reversible denaturation process between native and denatured protein states:
The standard Gibbs free energy change (ΔG°) for protein denaturation is given by the Eq. (4):
where T and R are the absolute temperature and the universal gas constant, respectively. The Gibbs free energy (ΔG°) is the most valuable standard of protein conformational stability in thermal denaturation. The integrated Gibbs–Helmholtz equation was utilized for measuring changes in the Gibbs energy of a system as a function of temperature (Eq. (5)):
where ΔCp is the heat capacity of protein denaturation. The ΔCp (11.6 kJ/mol K) of the α-glucosidase denaturation was taken from van der Kamp et al. report48. In thermal denaturation, Tm is the temperature at which the protein is half denatured. ΔH°m and ΔS°m are the standard enthalpy and entropy of denaturation. The standard entropy was calculated from a relation between the standard enthalpy (ΔS) and entropy (ΔH) of denaturation as bellow:
Molecular docking
The molecular docking of compounds 9b and 9e was performed using the maestro molecular modeling platform (version 10.5), Schrödinger suites50. X-ray crystallographic structure of α-glucosidase in complex with acarbose (PDB ID: 5NN8) was obtained from www.rcsb.com44. A protein preparation wizard was used to remove water molecules and co-crystallized atoms from the protein and prepare the receptor. Moreover, heteroatom states were generated at pH: 7.4 by EPIK, and H-bonds were assigned using PROPKA at the same pH. 2D structure of ligands was drawn in Hyperchem and the energies were minimized using molecular mechanics and molecular quantum approaches. Next, the ligand preparation wizard was used to prepare the ligand using the OPLS_2005 force field51. Acarbose, compounds 9b and 9e were docked into the binding sites using glide tasked to report ten poses per ligand with flexible ligand sampling and extra precision52.
In-silico pharmacokinetic properties of synthesized compounds
SwissADME (http://www.swissadme.ch/) and pkCSM (http://biosig.unimelb.edu.au/pkcsm/) servers were used to determine the physicochemical and drug-likeness properties of the derivatives.
References
Nugent, R. A., Fathima, S. F., Feigl, A. B. & Chyung, D. The burden of chronic kidney disease on developing nations: A 21st century challenge in global health. Nephron Clin. Pract. 118, c269–c277 (2011).
Zheng, Y., Ley, S. H. & Hu, F. B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 14, 88–98 (2018).
Sami, W., Ansari, T., Butt, N. S. & Ab Hamid, M. R. Effect of diet on type 2 diabetes mellitus: A review. Int. J. Health Sci. 11, 65 (2017).
Das, T. et al. Alterations in the gut bacterial microbiome in people with type 2 diabetes mellitus and diabetic retinopathy. Sci. Rep. 11, 1–15 (2021).
Dobrică, E.-C. et al. Polypharmacy in type 2 diabetes mellitus: Insights from an internal medicine department. Medicina 55, 436 (2019).
Rines, A. K., Sharabi, K., Tavares, C. D. & Puigserver, P. Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat. Rev. Drug Discov. 15, 786–804 (2016).
Scheen, A. J. Is there a role for α-glucosidase inhibitors in the prevention of type 2 diabetes mellitus? Drugs 63, 933–951 (2003).
Moorthy, N. S. H. N., Ramos, M. J. & Fernandes, P. A. Studies on α-glucosidase inhibitors development: Magic molecules for the treatment of carbohydrate mediated diseases. Mini Rev. Med. Chem. 12, 713–720 (2012).
Tseng, Y.-H., Tsan, Y.-T., Chan, W.-C., Sheu, W.H.-H. & Chen, P.-C. Use of an α-glucosidase inhibitor and the risk of colorectal cancer in patients with diabetes: A nationwide, population-based cohort study. Diabetes Care 38, 2068–2074 (2015).
Hillebrand, I., Boehme, K., Frank, G., Fink, H. & Berchtold, P. The effects of theα-glucosidase inhibitor BAY g 5421 (Acarbose) on meal-stimulated elevations of circulating glucose, insulin, and triglyceride levels in man. Res. Exp. Med. 175, 81–86 (1979).
Saeedi, M., Hadjiakhondi, A., Mohammad Nabavi, S. & Manayi, A. Heterocyclic compounds: Effective α-amylase and α-glucosidase inhibitors. Curr. Top. Med. Chem. 17, 428–440 (2017).
Saeedi, M. et al. Design and synthesis of novel quinazolinone-1, 2, 3-triazole hybrids as new anti-diabetic agents: In vitro α-glucosidase inhibition, kinetic, and docking study. Bioorg. Chem. 83, 161–169 (2019).
Saeedi, M. et al. Design and synthesis of novel 5-arylisoxazole-1, 3, 4-thiadiazole hybrids as α-glucosidase inhibitors. Lett. Drug Des. Discov. 18, 436–444 (2021).
Saeedi, M. et al. Synthesis of 4-alkylaminoimidazo [1, 2-a] pyridines linked to carbamate moiety as potent α-glucosidase inhibitors. Mol. Divers. 25, 1–11 (2020).
Fattaheian-Dehkordi, S., Hojjatifard, R., Saeedi, M. & Khanavi, M. A review on antidiabetic activity of Centaurea spp.: A new approach for developing herbal remedies. Evid. Based Complement. Altern. Med. 2021, 1–23 (2021).
Shareghi-Boroujeni, D. et al. Synthesis, in vitro evaluation, and molecular docking studies of novel hydrazineylideneindolinone linked to phenoxymethyl-1,2,3-triazole derivatives as potential α-glucosidase inhibitors. Bioorg. Chem. 111, 104869. https://doi.org/10.1016/j.bioorg.2021.104869 (2021).
Taha, M. et al. Synthesis, α-glucosidase inhibition and molecular docking study of coumarin based derivatives. Bioorg. Chem. 77, 586–592. https://doi.org/10.1016/j.bioorg.2018.01.033 (2018).
Alomari, M. et al. Synthesis of indole-based-thiadiazole derivatives as a potent inhibitor of α-glucosidase enzyme along with in silico study. Bioorg. Chem. 108, 104638. https://doi.org/10.1016/j.bioorg.2021.104638 (2021).
Zawawi, N. K. et al. Benzimidazole derivatives as new α-glucosidase inhibitors and in silico studies. Bioorg. Chem. 64, 29–36. https://doi.org/10.1016/j.bioorg.2015.11.006 (2016).
Naureen, S. et al. Biological evaluation of new imidazole derivatives tethered with indole moiety as potent α-glucosidase inhibitors. Bioorg. Chem. 76, 365–369. https://doi.org/10.1016/j.bioorg.2017.12.014 (2018).
Azimi, F. et al. Design, synthesis, biological evaluation, and molecular modeling studies of pyrazole-benzofuran hybrids as new α-glucosidase inhibitor. Sci. Rep. 11, 20776. https://doi.org/10.1038/s41598-021-99899-1 (2021).
Taha, M., Imran, S., Rahim, F., Wadood, A. & Khan, K. M. Oxindole based oxadiazole hybrid analogs: Novel α-glucosidase inhibitors. Bioorg. Chem. 76, 273–280. https://doi.org/10.1016/j.bioorg.2017.12.001 (2018).
Nasli Esfahani, A. et al. Design and synthesis of phenoxymethybenzoimidazole incorporating different aryl thiazole-triazole acetamide derivatives as α-glycosidase inhibitors. Mol. Divers. https://doi.org/10.1007/s11030-021-10310-7 (2021).
Saeedi, M. et al. Design, synthesis, in vitro, and in silico studies of novel diarylimidazole-1, 2, 3-triazole hybrids as potent α-glucosidase inhibitors. Bioorg. Med. Chem. 27, 115148 (2019).
Devaraj, N. K. & Finn, M. Introduction: Click Chemistry. Chem. Rev. 121, 6697–6698 (2021).
Kolb, H. C., Finn, M. & Sharpless, K. B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 40, 2004–2021 (2001).
Saeedi, M. et al. Synthesis and bio-evaluation of new multifunctional methylindolinone-1, 2, 3-triazole hybrids as anti-Alzheimer’s agents. J. Mol. Struct. 1229, 129828 (2021).
Askarani, H. K. et al. Design and synthesis of multi-target directed 1, 2, 3-triazole-dimethylaminoacryloyl-chromenone derivatives with potential use in Alzheimer’s disease. BMC Chem. 14, 1–13 (2020).
Safavi, M. et al. Novel quinazolin-4(3H)-one linked to 1,2,3-triazoles: Synthesis and anticancer activity. Chem. Biol. Drug Des. 92, 1373–1381. https://doi.org/10.1111/cbdd.13203 (2018).
Mohammadi-Khanaposhtani, M. et al. Design, synthesis and cytotoxicity of novel coumarin-1, 2, 3-triazole-1, 2, 4-oxadiazole hybrids as potent anti-breast cancer agents. Lett. Drug Des. Discov. 16, 818–824 (2019).
Graciano, I. A., de Carvalho, A. S., de Carvalho da Silva, F. & Ferreira, V. F. 1, 2, 3-Triazole-and quinoline-based hybrids with potent antiplasmodial activity. Med. Chem. 18, 521 (2021).
Alam, M. M. 1, 2, 3-Triazole hybrids as anticancer agents: A review. Arch. der Pharm. 355, 2100158 (2022).
Fallah, Z. et al. A review on synthesis, mechanism of action, and structure-activity relationships of 1, 2, 3-triazole-based α-glucosidase inhibitors as promising anti-diabetic agents. J. Mol. Struct. 1255, 132469 (2022).
Saeedi, M. et al. Synthesis and bio-evaluation of new multifunctional methylindolinone-1,2,3-triazole hybrids as anti-Alzheimer’s agents. J. Mol. Struct. 1229, 129828. https://doi.org/10.1016/j.molstruc.2020.129828 (2021).
Yazdani, M. et al. 5,6-Diphenyl triazine-thio methyl triazole hybrid as a new Alzheimer’s disease modifying agents. Mol. Divers. 24, 641–654. https://doi.org/10.1007/s11030-019-09970-3 (2020).
Mahdavi, M. et al. Synthesis of new benzimidazole-1,2,3-triazole hybrids as tyrosinase inhibitors. Chem. Biodivers. 15, e1800120. https://doi.org/10.1002/cbdv.201800120 (2018).
Iraji, A. et al. Synthesis and structure-activity relationship study of multi-target triazine derivatives as innovative candidates for treatment of Alzheimer’s disease. Bioorg. Chem. 77, 223–235. https://doi.org/10.1016/j.bioorg.2018.01.017 (2018).
Asgari, M. S. et al. Biscoumarin-1,2,3-triazole hybrids as novel anti-diabetic agents: Design, synthesis, in vitro α-glucosidase inhibition, kinetic, and docking studies. Bioorg. Chem. 92, 103206. https://doi.org/10.1016/j.bioorg.2019.103206 (2019).
Asemanipoor, N. et al. Synthesis and biological evaluation of new benzimidazole-1,2,3-triazole hybrids as potential α-glucosidase inhibitors. Bioorg. Chem. 95, 103482. https://doi.org/10.1016/j.bioorg.2019.103482 (2020).
Edraki, N. et al. 2-Imino 2H-chromene and 2-(phenylimino) 2H-chromene 3-aryl carboxamide derivatives as novel cytotoxic agents: Synthesis, biological assay, and molecular docking study. J. Iran. Chem. Soc. 13, 2163–2171. https://doi.org/10.1007/s13738-016-0934-7 (2016).
Saeedi, M. et al. Novel N-benzylpiperidine derivatives of 5-arylisoxazole-3-carboxamides as anti-Alzheimer’s agents. Arch. der Pharm. 354, 2000258. https://doi.org/10.1002/ardp.202000258 (2021).
Xie, Z. et al. Synthesis, biological evaluation, and molecular docking studies of novel isatin-thiazole derivatives as α-glucosidase inhibitors. Molecules 22, 659 (2017).
Jafari, M. et al. Molecular level insight into stability, activity, and structure of Laccase in aqueous ionic liquid and organic solvents: An experimental and computational research. J. Mol. Liq. 317, 113925 (2020).
Karami, M. et al. One-pot multi-component synthesis of novel chromeno[4,3-b]pyrrol-3-yl derivatives as alpha-glucosidase inhibitors. Mol. Divers. https://doi.org/10.1007/s11030-021-10337-w (2021).
Pires, D. E. V., Blundell, T. L. & Ascher, D. B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 58, 4066–4072. https://doi.org/10.1021/acs.jmedchem.5b00104 (2015).
Daina, A., Michielin, O. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Barker, M. K. & Rose, D. R. Specificity of processing α-glucosidase I is guided by the substrate conformation: Crystallographic and in silico studies. J. Biol. Chem. 288, 13563–13574 (2013).
Van Der Kamp, M. W. et al. Dynamical origins of heat capacity changes in enzyme-catalysed reactions. Nat. Commun. 9, 1–7 (2018).
Farhadian, S. et al. Insights into the molecular interaction between sucrose and α-chymotrypsin. Int. J. Biol. Macromol. 114, 950–960 (2018).
Glide, V. 5.8; Schrödinger, LLC (2012).
Release, S. 4: Glide. Schrödinger, LLC (2018).
Friesner, R. A. et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J. Med. Chem. 49, 6177–6196 (2006).
Acknowledgements
This work was supported by Grants from the Research Council of Tehran University of Medical Sciences with project No. 99-2-157-49765. This paper is dedicated to the memory of our unique teacher in Chemistry and Medicinal Chemistry, Professor Abbas Shafiee (1937–2016).
Author information
Authors and Affiliations
Contributions
A.I. wrote the manuscript and performed in-silico study. D.S.-B. synthesized compounds. S.M. performed biological assay. M.A.F. supervised biological assay. T.A. supervised all steps of the project. M.S. designed compounds, characterized final products, and participated in the preparations of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher′s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article′s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article′s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Iraji, A., Shareghi-Brojeni, D., Mojtabavi, S. et al. Cyanoacetohydrazide linked to 1,2,3-triazole derivatives: a new class of α-glucosidase inhibitors. Sci Rep 12, 8647 (2022). https://doi.org/10.1038/s41598-022-11771-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-11771-y
This article is cited by
-
Aryl-quinoline-4-carbonyl hydrazone bearing different 2-methoxyphenoxyacetamides as potent α-glucosidase inhibitors; molecular dynamics, kinetic and structure–activity relationship studies
Scientific Reports (2024)
-
Novel N′-substituted benzylidene benzohydrazides linked to 1,2,3-triazoles: potent α-glucosidase inhibitors
Scientific Reports (2023)
-
Synthesis, in vitro α-glucosidase inhibitory activities, and molecular dynamic simulations of novel 4-hydroxyquinolinone-hydrazones as potential antidiabetic agents
Scientific Reports (2023)
-
Synthesis, in vitro inhibitor screening, structure–activity relationship, and molecular dynamic simulation studies of novel thioquinoline derivatives as potent α-glucosidase inhibitors
Scientific Reports (2023)
-
Synthesis and structure–activity relationship studies of benzimidazole-thioquinoline derivatives as α-glucosidase inhibitors
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
