
Abstract
Herein, various N′-substituted benzylidene benzohydrazide-1,2,3-triazoles were designed, synthesized, and screened for their inhibitory activity toward α-glucosidase. The structure of derivatives was confirmed using 1H- and 13C-NMR, FTIR, Mass spectrometry, and elemental analysis. All derivatives exhibited good inhibition with IC50 values in the range of 0.01 to 648.90 µM, compared with acarbose as the positive control (IC50 = 752.10 µM). Among them, compounds 7a and 7h showed significant potency with IC50 values of 0.02 and 0.01 µM, respectively. The kinetic study revealed that they are noncompetitive inhibitors toward α-glucosidase. Also, fluorescence quenching was used to investigate the interaction of three inhibitors 7a, 7d, and 7h, with α-glucosidase. Accordingly, the binding constants, the number of binding sites, and values of thermodynamic parameters were determined for the interaction of candidate compounds toward the enzyme. Finally, the in silico cavity detection plus molecular docking was performed to find the allosteric site and key interactions between synthesized compounds and the target enzyme.
Similar content being viewed by others

Introduction
Diabetes mellitus (DM) is one of the major health challenges of the twenty-first century, characterized by chronic hyperglycemia1. The disease is associated with many complications, including neuropathy, nephropathy, retinopathy, cardiovascular disease, kidney disease, and liver disease as well as skin complications which result in a heavy economic and social burden2,3,4. Type-2 diabetes (T2DM) known as the most common type of DM, takes up 90% of all diabetes cases resulting from pancreatic β-cell dysfunction and resistance to insulin action in peripheral tissues (muscle and adipose)5,6. α-Glucosidase (EC 3.2.1.20) is an important hydrolase enzyme present on the surface of the small intestine. It catalyzes the hydrolysis of carbohydrates into absorbable glucose monomers by cleaving the bond between glucosidic oxygen and glucosyl residues of carbohydrates7,8. The inhibition of α-glucosidase delays the production of glucose monomers leading to the reduction of postprandial blood glucose levels, which is an effective approach to the management strategy of T2DM9. To date, a large number of α-glucosidase inhibitors have been introduced from natural origin or chemical routes10,11,12,13,14. Amongst just acarbose, voglibose, and miglitol enter the market, which is associated with side effects, especially gastrointestinal effects and hepatotoxicity15,16. Thus, the design and synthesis of potent α-glucosidase inhibitors is an attractive subject among researchers.
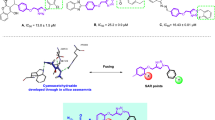
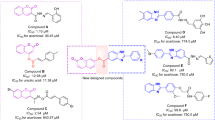
1,2,3-Triazole and its derivatives are the key skeletons possessing various pharmacological effects, including antioxidant, anti-inflammatory17,18, anti-Alzheimer’s19,20, anti-bacterial21, and anti-cancer22,23,24 activity. Recently, various studies have indicated 1,2,3-triazoles as potent anti-α-glucosidase agents13,25,26,27, such as compound A (Fig. 1) which is a potent competitive inhibitor of α-glucosidase. These derivatives exhibited IC50 values in the range of 13.0–75.5 µM28. A series of benzimidazole-1,2,3-triazoles containing phenoxy linker (Compound B) were rationally designed as α-glucosidase inhibitors and exhibited significant inhibitory activity compared with its parental compounds as well as the positive control, acarbose29. Recently, a series of indolinone substituted phenoxy-methyltriazole derivatives were synthesized and the derivative C, the most potent derivative, inhibited the enzyme competitively. In silico assessments confirmed that the phenoxy-1,2,3-triazole moiety makes compounds stable through H-bonding and pi-alkyl interactions30.

Representative inhibitors of α-glucosidase and newly designed compounds.
On the other hand, substituted acetohydrazide derivatives were introduced as the key skeleton anchoring agent that functionality interacts with amino acid residues toward the α-glucosidase. Through systematic chemical optimization, compound D demonstrated improved inhibition (IC50 = 21.9 µg/ml) against α-glucosidase compared with acarbose (IC50 = 34.5 µg/ml)31. Moreover, the moiety is perfectly nestled in the pocket of α-glucosidase32. Also, atranorin, a secondary metabolite of lichen, was conjugated with different hydrazines. Among the synthesized derivatives, compound E afforded high anti-α-glucosidase activity (IC50 = 6.67 µM) without toxicity on the HEK293 cell line. Recently, cyanoacetohydrazide linked to 1,2,3-triazole derivatives were found to be potent α-glucosidase inhibitors, and among them, compound F showed very good activity (IC50 = 1.00 µM) as compared with acarbose (IC50 = 754.1 μM). Also, fluorescence measurements confirmed conformational changes of the enzyme after binding of compound F33 (Fig. 1).
In this work, considering the structural characteristics of substituted acetohydrazide as α-glucosidase inhibitor and the advantages of 1,2,3-triazoles, a new series of N'-substituted benzylidene benzohydrazides linked to 1,2,3-triazoles were synthesized to evaluate their α-glucosidase inhibitory activity. Subsequently, structure−activity relationship studies (SARs) and kinetic studies were performed. Cavity detection was further executed to find a suitable site for noncompetitive inhibitors for in silico assessments.
Results and discussion
Chemistry
The synthetic route for the preparation of compounds 7a-p was schematically described in Fig. 2. It was initiated by the reaction of methyl 4-(prop-2-yn-1-yloxy)benzoate 1 and hydrazine hydrate in refluxing ethanol leading to the formation of 4-(prop-2-yn-1-yloxy)benzohydrazide 2. Compound 1 in turn was prepared from the reaction of 4-hydroxy benzoic acid and propargyl bromide in DMF at 80 °C. Compound 2 reacted with different aromatic aldehydes 3 in ethanol, heated at reflux catalyzed by acetic acid to afford the corresponding Schiff base derivatives 4. Finally, the click reaction of compound 4 and in situ prepared azides 6 in the presence of triethylamine (NEt3), CuSO4.5H2O, and sodium ascorbate in H2O/tert-BuOH for 24−48 h at room temperature, gave the desired products 7.

Synthesis of compounds 7a-p.
In vitro inhibition of α-glucosidase and the structure-activity relationships
Compounds 7a-p were evaluated for their in vitro potential toward α-glucosidase compared with acarbose as the positive reference, and the IC50 values are presented in Table 1. Results indicated that all derivatives are potent α-glucosidase inhibitors compared to acarbose. Amongst, compound 7h showed promising activity with an IC50 value of 0.01 µM regarded as the most potent inhibitor compared to acarbose as a positive control (IC50 = 752.10 μM).
A distinct inhibitory pattern was observed regarding the type of substituent at Y position on the benzyl moiety connected to the 1,2,3-triazole ring. In detail, derivatives 7a-d (X = H) showed IC50 values in the range of 0.02 to 61.70 μM. Noteworthy, the presence of 4-NO2 substituent at X position significantly improved the inhibition and recorded IC50 values in the range of 0.01 to 8.09 μM. A strong electron-withdrawing group at the para position seems to have improved the activity.
Assessments on the 7i-l bearing para-chlorine as halogen group at X position disclosed a considerable reduction in the potency (IC50 = 261.40 to 648.90 μM). However, the methoxy group at the para position (7m-p) increased the inhibitory trend with IC50 = 0.86 to 101.80 μM. The more detailed analysis demonstrated that the presence of NO2 substituent as a strong electron-withdrawing group at X (7e-h), improved the inhibitory potency highlighting the pivotal role of the nitro functional group for inhibition. The other potent entry was unsubstituted derivatives at X (7a-d) followed by 4-OMe (7m-p).
Precise assessments on the 7a-d (X = H) derivatives also indicated that 7a as an unsubstituted derivative (IC50 = 0.02 μM) and 7d bearing 4-methyl group at Y, as a lipophilic and weak electron-donating group (IC50 = 0.04 μM), were more potent in comparison to derivatives bearing 4-F and 2-Cl in this set. The evaluations on 7e-h (X = NO2) as the most potent set of derivatives showed the flowing order of potency 4-Me (7h) ˃ 2-Cl (7g) ˃ H (7e) ˃ 4-F (7f). As can be seen in these two sets of derivatives, 4-Me substituted analog at Y exhibited good potency. A different trend was observed in the 7i-l bearing 4-Cl and 7m-p containing 4-OMe at X, so that 7l (X = 4-Cl, Y = 4-Me) and 7p (X = 4-OMe, X = 4-Me) were not as potent as their counterparts. In the two mentioned sets of compounds, the unsubstituted derivatives in each set, compounds 7i (X = 4-Cl, Y = H, IC50 = 261.40 μM) and 7m (X = 4-OMe, Y = H, IC50 = 0.86 μM) were highly potent.
To sum up, the straightforward SAR was extracted so that the presence of 4-NO2 as a bulk-electron withdrawing group at X, regardless of the type of substituent at Y significantly improved the inhibitory activity of compounds against α-glucosidase. Based on Hammett electronic σ and Hansch π constants, the electronic effect and lipophilicity of the substituents can affect the biological activity34. Herein, the α-glucosidase inhibitory activities of compounds 7e-h were influenced by the NO2 group (σ = 0.78, π = − 0.28), and generally, high activity was obtained by these compounds. However, the right-forward trend cannot be seen in Hammett's electronic properties, so compounds 7i-l (X = Cl, σ = 0.23, π = 0.71) exhibited lower activity in comparison with compounds 7m-p (X = OMe, σ = -− 0.27, π = − 0.02)35 which were supposed to be weak inhibitors due to low σ value. It seems that the effects of Y substituents should not be ignored. Also, it was revealed that the increased lipophilicity at the X position reduced the potency as high lipophilic Cl-derivatives 7i-l gave lower activity against α-glucosidase.
Unsubstituted compounds at the X and Y positions can also be categorized as the second top potent group. Investigation on Y showed different patterns so that if X = 4-Cl or OMe, the unsubstituted derivative at Y is more favorable, and the Me group is inferior to that in the potency. Vice versa, 4-Me moiety seems to improve the inhibitory activity in the X = NO2 set of compounds.
The SAR comparison between the designed compounds and previously reported scaffolds was executed. The benzimidazole-1,2,3-triazole hybrids demonstrated IC50 values ranging from 25.2 to 176.5 μM. SARs exhibited that the presence of ethyl or methyl as lipophilic electron-donating substituent on the benzyl ring improved the potency vs halogen and electron-withdrawing groups. The exception returned to 2-NO2 moiety, which was categorized as the second top potent derivative36.
Hydrazineylideneindolinone linked to phenoxymethyl-1,2,3-triazole derivatives showed IC50 in the range of 16.43 to > 750 µM. Interesting results were obtained with SAR analysis, and it was concluded that the presence of a substituent on the phenoxy linker deteriorated the potency, and fluoride substituent on the terminal benzyl ring induced better α-glucosidase inhibitory activity compared to other derivatives30. Recently, novel series of cyanoacetohydrazide linked to phenoxy-triazole was developed (IC50 = 1.00 to 750 against α-glucosidase), and different results were obtained in this case. The presence of methoxy on the phenoxy linker was favorable, and fluorine and chlorine substituents on the benzyl ring empowered the activity37. Although the backbones of these derivatives are not similar to fully extracting the SARs; it was understood that nitro group at the X position is favorable and in this case, 4-Me moiety at Y seems to increase the activity (Supplementary Information 1).
Enzyme kinetic studies
A kinetic study was achieved to investigate the mode of inhibition of α-glucosidase by compounds 7a and 7h. According to Fig. 3, the Lineweaver−Burk plots showed that the Km gradually increased, and Vmax remained unchanged with increasing the inhibitors concentration, indicating a noncompetitive inhibitory activity, with Ki = 0.01 and 0.02 μM for compounds 7h and 7a, respectively.

Kinetic study of α-glucosidase inhibition by compounds 7h (left) and 7a (right).
Fluorescence spectroscopy
Fluorescence spectrometry was used for studying the interactions between selected α-glucosidase inhibitors (7a, 7d, and 7h) and the enzyme at 25 °C and pH 6.8 to determine the binding constants, number of binding sites, and thermodynamic parameters of the corresponding interactions. Herein, the maximum fluorescence intensity of α-glucosidase was obtained at 340 nm, which can be attributed to tryptophan, tyrosine, and phenylalanine residues that can act as intrinsic fluorescence probes with excitation at 280 nm. As shown in Fig. 4, the fluorescence intensity of α-glucosidase was gradually decreased in the presence of compounds 7a, 7d, and 7h in a concentration dependent manner at 340 nm, indicating that those compounds interacted with α-glucosidase and may change the fluorescence characteristics of the enzyme.

Fluorescence spectra of α-glucosidase in the presence of compounds 7a (a), 7d (b) and 7h (c) at 25 °C.
The fluorescence quenching process was characterized by the Stern−Volmer equation (experimental section, Eq. 1). As shown in Table 2, the quenching model was static as the quenching rate constant values Kq (L mol−1 s−1) for selected inhibitors 7a, 7d, and 7h at 25 and 35 ºC were obtained much higher than 2.0 × 1010 L mol−1 s−1 which is the maximum collision rate constant for all quencher compounds that collide with biological macromolecules.
In the static quenching, it is assumed that there are the same and independent binding sites (n) in the protein (herein α-glucosidase) which all are potent to construct interactions with the inhibitor (experimental section, Eq. 2).
Plots of fluorescence intensity ratio (F0/F) versus [Dt] F0/(F0 − F) were obtained (experimental section, Eqs. 3–5) by keeping the total concentrations of α-glucosidase at 0.1 µM and varying the total concentrations of compounds 7a, 7d, and 7h (Fig. 5) to calculate the binding constants and binding sites (n) at different temperatures (Table 3). Among investigated inhibitors, compound 7d demonstrated the lowest binding constant and the highest value was calculated for compound 7h.

The plots F0/F vs function of [Dt] F0/(F0 − F) at 25 °C for 7a (a), 7d (b) and 7h (c).
Finally, the acting forces between selected inhibitors (7a, 7d, and 7h) and α-glucosidase were determined (Table 4) according to the standard thermodynamic values (experimental section, Eqs. 6 and 7). These forces may include Van der Waals, electrostatic, hydrogen bonding, and hydrophobic interactions. The ΔH values for three inhibitors were less than zero (ΔH < 0) and those of ΔS were all greater than 0 (ΔS > 0). These results indicated that the acting force between these inhibitors and α-glucosidase was mainly electrostatic interactions.
Molecular docking studies
To achieve more information on the molecular mechanisms behind the ability to inhibit the α-glucosidase, designed compounds were docked into the target enzyme. First, to validate docking assessments, self-docking was performed with an actual inhibitor obtained from the PDB (5NN8). The top-ranked docking score of the crystallographic inhibitor was superimposed over the X-ray coordinate of the experimentally derived structure and showed an RMSD of 1.9 Å (RMSD < 2 Å is the acceptable value).
The enzyme kinetic study showed that compounds 7a and 7h acted as noncompetitive inhibitors. In this type of inhibition, the inhibitor binds to the enzyme at a location other than the active site. To find the suitable sites, the enzyme was subjected to mastreo sitemap tool to find the possible cavity of the enzyme. Five possible binding sites were detected on the surface of the enzyme (Fig. 6) which can be suitable for noncompetitive inhibition. Next, compound 7h was docked in all the potential binding sites of the enzyme as the most potent structure.

Potential binding sites of the α-glucosidase detected by cavity detection tools.
Considering the Moledock score and interactions, site 4 appeared to have the maximum affinity in comparison to other identified sites. Next, the same docking procedure was applied to all selected derivatives at site 4. In this respect, derivatives 7h and 7d as the potent inhibitors and 7l as the least active analog as well as 7p were docked within the allosteric site (Y = methyl in all selected compounds). Residues involved in the interaction of these compounds plus MolDock Score were shown in Table 5 and 3D structures of each ligand were presented in Fig. 7.

The proposed binding modes of compounds 7d, 7h, 7l, and 7p with the allosteric site of α-glucosidase.
At first look, it can be seen that the order of IC50 values was consistent within the MolDock Score values so that compounds 7h and 7d as potent inhibitors in the list, IC50 = 0.01 and 0.04 µM, generated the MolDock Score value of − 148.49 and − 120.57 kJ/mol. Compound 7l with IC50 value of 289.20 µM generated a low score value of − 89.89 kJ/mol which showed that the in silico assessments could successfully categorize compounds based on their experimental activities. Also, the least potent compounds recorded some unfavorable interactions and were unable to participate in strong interactions with the proposed site while 7h and 7d effectively participate in H-bonding interactions with the allosteric site.
Physicochemical properties
According to the physicochemical properties predicted from SwissADME web tool, all compounds exhibited desirable molecular properties with no drug-likeness rules violations (Table 6). All inhibitors also showed a desirable bioavailability score.
In the case of molar refractivity values which were related to the overall bulkiness and lipophilicity of the synthesized compounds, the most potent compound 7h, depicted a relatively high value (127.96). However, compound 7a showed a low value of 117.92, indicating no definite relation between the bulkiness and α-glucosidase inhibition, which can be confirmed in the series of compounds 7a−p.
Conclusion
Following our expertise in the rational design of α-glucosidase inhibitors; herein, a series of N'-substituted benzylidene benzohydrazides linked to 1,2,3-triazoles were designed and synthesized.
The chemical structures of all derivatives were characterized using NMR, FTIR, MS spectrometry, and elemental analysis. All compounds exhibited pronounced anti-α-glucosidase activity with IC50 values in the range of 0.01−648.90 μM compared with the reference compound, acarbose (IC50 = 752.10 µM). The SAR data showed that the designed backbone was highly potent against α-glucosidase and the incorporation of a para-nitro moiety on the benzyl moiety connected to 1,2,3-triazole ring significantly improved the potency. Compounds 7a (IC50 = 0.02 μM) and 7h (IC50 = 0.01 μM) as the most potent inhibitors, were subjected to kinetic experiments and revealed the noncompetitive inhibition pattern. In addition, fluorescence spectroscopy demonstrated that the intrinsic fluorescence of α-glucosidase was quenched by three inhibitors 7a, 7d, and 7h, due to the formation of inhibitor-fluorophore complex, resulting in the decrease in enzyme activity. This result was in good agreement with our results obtained from the kinetic studies. It should be noted that the calculation of standard thermodynamic values (ΔG, ΔH, and ΔS) revealed that the acting force between these inhibitors and the α-glucosidase was mainly electrostatic interactions. In the docking study, mastreo sitemap tools were applied to find the allosteric site for noncompetitive inhibitors and an appropriate cavity was proposed. To evaluate the behavior of derivatives within the proposed site, enzyme molecular docking assessments were performed. Derivatives 7d and 7h showed the lowest score and could effectively participate in H-bonding interactions with the proposed site, while derivatives with fewer activities exhibited no strong interactions with the allosteric site. This study has made good progress in anti-T2DM agents and promoted the development of related fields.
Method and materials
Melting points were measured on a Electrothermal digital melting point apparatus IA9300 and are uncorrected. 1H- and 13C-NMR spectra were recorded on a Varian-INOVA 500 MHz, using TMS as an internal standard. IR spectra were obtained on a Nicolet Magna FTIR 550 spectrophotometer (KBr disks). Mass spectra were recorded on an Agilent Technology (HP) mass spectrometer operating at an ionization potential of 70 eV. Elemental analysis was performed on an Elementar Analysensystem GmbH VarioEL CHNS mode.
Synthesis of compounds 7
A mixture of methyl 4-(prop-2-yn-1-yloxy)benzoate 1 (1 mmol) and hydrazine hydrate (4 mmol) in EtOH (10 ml) was heated at reflux overnight. After completion of the reaction, the mixture was poured over ice and water, and the precipitate was filtered off to give 4-(prop-2-yn-1-yloxy)benzohydrazide 2. Next, a mixture of compound 2 (1 mmol) and aldehyde 3 (1 mmol) in EtOH (15 ml), in the presence of a few drops acetic acid was heated at reflux for 24 h. After completion of the reaction, the mixture was poured over ice and water and the precipitate was filtered off to afford compound 4. Finally, the click reaction was conducted by a mixture of compound 4 and in situ prepared azide derivative 630. For this purpose, benzyl chloride/bromide derivative 5 (1.1 mmol) and sodium azide (0.06 g, 0.9 mmol) in the presence of triethylamine (0.13 g, 1.3 mmol) in water (4 ml) and tert-butyl alcohol (4 ml) was stirred at room temperature for 30 min. Next, compound 4 (0.5 mmol) and CuSO4·5H2O (7 mol%) were added to the reaction mixture and it was continued for 24−48 h at the same temperature. After completion of the reaction as checked by TLC, the mixture was poured on the crushed ice, the precipitates were filtered off and washed with water. All products were recrystallized from ethyl acetate and petroleum ether.
4-((1-Benzyl-1H-1,2,3-triazol-4-yl)methoxy)-N′-benzylidenebenzohydrazide (7a)
Gray precipitates, Yield: 78%, mp 248−251 °C. IR (KBr, cm−1): 3417, 3051, 2924, 2850, 1687, 1635, 1589. 1HNMR (500 MHz, DMSO-d6): 11.75 (s, 1H, NH), 8.50 (s, 1H, CH), 8.29 (s, 1H, triazole), 7.91 (d, J = 8.3 Hz, 2H, H3′, H5′), 7.78 (d, J = 7.0 Hz, 2H, H2, H6), 7.50−7.46 (m, 3H, H3, H4, H5), 7.35−7.19 (m, 7H, H2′, H6′, H2′′, H3′′, H4′′, H5′′, H6′′), 5.62 (s, 2H, CH2), 5.21 (s, 2H, CH2) ppm. 13CNMR (125 MHz, DMSO-d6): 163.1, 161.3, 147.5, 142.6, 133.6, 133.2, 131.1, 129.7, 129.1, 128.9, 128.3, 127.5, 125.4, 125.1, 122.8, 114.5, 61.5, 52.3 ppm. MS (m/z, %): 411 (M+, 3), 295 (8), 245 (27), 182 (33), 154 (51), 121 (23), 105 (87), 77 (55), 57 (100). Calcd for C24H21N5O2: C, 70.06; H, 5.14; N, 17.02. Found: C, 70.23; H, 5.05; N, 16.91.
N′-Benzylidene-4-((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzohydrazide (7b)
Gray precipitates, Yield: 78%, mp 160−163 °C. IR (KBr, cm−1): 3415, 3051, 2922, 2851, 1687, 1636, 1590. 1HNMR (500 MHz, DMSO-d6): 11.73 (s, 1H, NH), 8.45 (s, 1H, CH), 8.31(s, 1H, triazole), 7.89 (d, J = 8.5 Hz, 2H, H3′, H5′), 7.71 (d, J = 7.0 Hz, 2H, H2, H6), 7.45−7.38 (m, 5H, H3, H4, H5, H2′′, H6′′), 7.20−7.14 (m, 4H, H2′, H6′, H3′′, H5′′), 5.60 (s, 2H, CH2), 5.22 (s, 2H, CH2) ppm. 13CNMR (125 MHz, DMSO-d6): 163.3, 162.4, 161.3, 147.7, 146.4, 143.1, 134.7, 132.6, 130.8, 129.9, 129.3, 128.6, 127.4, 125.2, 116.0, 114.9, 61.7, 52.5 ppm. Calcd for C24H20FN5O2: C, 67.12; H, 4.69; N, 16.31. Found: C, 66.98; H, 4.53; N, 16.59.
N′-Benzylidene-4-((1-(2-chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzohydrazide (7c)
White precipitates, Yield: 80%, mp 203−207 °C. IR (KBr, cm−1): 3416, 3052, 2927, 2848, 1688, 1631, 1588. 1HNMR (500 MHz, DMSO-d6): 11.72 (s, 1H, NH), 8.44 (s, 1H, CH), 8.28 (s, 1H, triazole), 7.90 (d, J = 8.3 Hz, 2H, H3′, H5′), 7.71 (d, J = 7.0 Hz, 2H, H2, H6), 7.52 (d, J = 7.5 Hz, 1H, H3′′), 7.45−7.35 (m, 5H, H3, H4, H5, H4′′, H5′′), 7.23 (d, J = 7.5 Hz, 1H, H6′′), 7.16 (d, J = 8.3 Hz, 2H, H2′, H6′), 5.72 (s, 2H, CH2), 5.24 (s, 2H, CH2) ppm. 13CNMR (125 MHz, DMSO-d6): 163.1, 162.6, 147.6, 142.9, 137.8, 134.9, 133.6, 133.1, 131.0, 130.7, 130.4, 130.1, 129.9, 128.7, 128.2, 127.4, 125.1, 114.9, 61.6, 51.1 ppm. Calcd for C24H20ClN5O2: C, 64.65; H, 4.52; N, 15.71. Found: C, 65.02; H, 4.41; N, 15.45.
N′-Benzylidene-4-((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzohydrazide (7d)
Gray precipitates, Yield: 75%, mp 154−156 °C. IR (KBr, cm−1): 3417, 3050, 2924, 2853, 1690, 1637, 1587. 1HNMR (500 MHz, DMSO-d6): 11.76 (s, 1H, NH), 8.47 (s, 1H, CH), 8.29 (s, 1H, triazole), 7.90−7.72 (m, 4H, H2, H6, H3′, H5′), 7.51−7.47 (m, 3H, H3, H4, H5), 7.22−7.14 (m, 6H, H2′, H6′, H2′′, H3′′, H5′′, H6′′), 5.55 (s, 2H, CH2), 5.19 (s, 2H, CH2), 2.27 (s, 3H, CH3) ppm. 13CNMR (125 MHz, DMSO-d6): 163.1, 161.4, 146.1, 143.8, 138.0, 135.1, 134.8, 133.4, 132.00, 129.7, 129.4, 129.4, 129.3, 128.5, 127.5, 114.9, 60.8, 53.4, 21.5 ppm. Calcd for C25H23N5O2: C, 70.57; H, 5.45; N, 16.46. Found: C, 70.88; H, 5.22; N, 16.13.
4-((1-Benzyl-1H-1,2,3-triazol-4-yl)methoxy)-N′-(4-nitrobenzylidene)benzohydrazide (7e)
Yellow precipitates, yield: 48%, mp 193−196 °C. IR (KBr, cm−1): 3415, 3052, 2926, 2851, 1687, 1630, 1586, 1533, 1375. 1HNMR (500 MHz, DMSO-d6): 12.04 (s, 1H, NH), 8.54 (s, 1H, CH), 8.33 (s, 1H, triazole), 8.30−7.97 (m, 6H, H2, H3, H5, H6, H3′, H5′), 7.35−7.22 (m, 5H, H2′′, H3′′, H4′′, H5′′, H6′′), 7.18 (d, J = 8.3 Hz, 2H, H2′, H6′), 5.62 (s, 2H, CH2), 5.24 (s, 2H, CH2). 13CNMR (125 MHz, DMSO-d6): 162.0, 160.1, 147.7, 144.6, 140.8, 136.0, 129.7, 128.8, 128.2, 128.0, 127.9, 125.1, 124.9, 124.1, 121.0, 114.5, 61.3, 52.9 ppm. Calcd for C24H20N6O4: C, 63.15; H, 4.42; N, 18.41. Found: C, 63.29; H, 4.73; N, 18.57.
4-((1-(4-Fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-N′-(4-nitrobenzylidene)benzohydrazide (7f)
Light green precipitates, Yield: 75%, mp 202−205 °C. IR (KBr, cm−1): 3415, 3050, 2925, 2850, 1684, 1635, 1590, 1535, 1375. 1HNMR (500 MHz, DMSO-d6): 12.03 (s, 1H, NH), 8.53 (s, 1H, CH), 8.31−8.29 (m, 3H, H3, H5, triazole), 7.97−7.92 (m, 4H, H2, H6, H3′, H5′), 7.39−7.38 (m, 2H, H2′′, H6′′), 7.22−7.17 (m, 4H, H2′, H6′, H3′′, H5′′), 5.60 (s, 2H, CH2), 5.23 (s, 2H, CH2) ppm. 13CNMR (125 MHz, DMSO-d6): 163.3, 161.4, 160.9 (d, JC−F = 256.9 Hz), 148.1, 145.1, 143.1, 141.2, 132.7, 130.8 (d, JC−F = 8.1 Hz), 129.6, 128.3, 125.7, 125.2, 124.5, 116.1 (d, JC−F = 21.5 Hz), 114.9, 61.7, 52.5 ppm. Calcd for C24H19FN6O4: C, 60.76; H, 4.04; N, 17.71. Found: C, 60.88; H, 3.87; N, 17.42.
4-((1-(2-Chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-N′-(4-nitrobenzylidene)benzohydrazide (7g)
Yellow precipitates, Yield: 83%, mp 204−206 °C. IR (KBr, cm−1): 3417, 3051, 2922, 2853, 1686, 1634, 1587, 1535, 1375.1HNMR (500 MHz, DMSO-d6): 12.03 (s, 1H, NH), 8.53 (s, 1H, CH), 8.29−8.27 (m, 3H, H3, H5, triazole), 7.96 (d, J = 8.2 Hz, 2H, H2, H6), 7.91 (d, J = 8.3 Hz, 2H, H3′, H5′) 7.51 (d, J = 7.5 Hz, 1H, H3′′), 7.41−7.35 (m, 2H, H4′′, H5′′), 7.23 (d, J = 7.5 Hz, 1H, H6′′), 7.17 (d, J = 8.3 Hz, 2H, H2′, H6′,), 5.72 (s, 2H, CH2), 5.25 (s, 2H, CH2) ppm. 13CNMR (125 MHz, DMSO-d6): 162.4, 161.2, 148.2, 145.1, 142.9, 141.2, 133.6, 133.1, 131.0, 130.7, 130.1, 129.3, 128.3, 128.2, 125.8, 125.7, 124.5, 115.0, 61.7, 51.1 ppm. Calcd for C24H19ClN6O4: C, 58.72; H, 3.90; N, 17.12. Found: C, 58.41; H, 4.14; N, 17.29.
4-((1-(4-Methylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-N′-(4-nitrobenzylidene)benzohydrazide (7h)
Brown precipitates, yield: 38%, mp 247−250 °C. IR (KBr, cm−1): 3416, 3052, 2925, 2851, 1690, 1634, 1585, 1536, 1372. 1HNMR (500 MHz, DMSO-d6): 12.04 (s, 1H, NH), 8.54 (s, 1H, CH), 8.29- 7.93 (m, 7H, H2, H3, H5, H6, H3′, H5′, triazole), 7.22−7.18 (m, 6H, H2′, H6′, H2′′, H3′′, H5′′, H6′′), 5.56 (s, 2H, CH2), 5.23 (s, 2H, CH2), 2.27 (s, 3H, CH3). 13CNMR (125 MHz, DMSO-d6): 161.8, 160.1, 147.7, 145.8, 140.8, 137.5, 132.9, 129.6, 129.2, 128.0, 127.5, 127.3, 124.7, 124.0, 120.0, 114.5, 61.2, 52.7, 20.6 ppm. MS (m/z, %): 470 (M+, 2), 207 (7), 182 (15), 161 (100), 105 (72), 77 (41), 57 (58). Calcd for C25H22N6O4: C, 63.82; H, 4.71; N, 17.86. Found: C, 64.17; H, 4.82; N, 17.61.
4-((1-Benzyl-1H-1,2,3-triazol-4-yl)methoxy)-N′-(4-chlorobenzylidene)benzohydrazide (7i)
Light green precipitates, yield: 51%, mp 211−214 °C. IR (KBr, cm−1): 3415, 3048, 2924, 2853, 1690, 1627, 1588. 1HNMR (500 MHz, DMSO-d6): 11.72 (s, 1H, NH), 8.44 (s, 1H, CH), 8.33 (s, 1H, triazole), 7.90 (d, J = 8.3 Hz, 2H, H3′, H5′), 7.76−7.15 (m, 11H, H2, H3, H4, H5, H2′, H6′, H2′′, H3′′, H4′′, H5′′, H6′′), 5.62 (s, 2H, CH2), 5.24 (s, 2H, CH2). 13CNMR (125 MHz, DMSO-d6): 162.0, 160.5, 147.5, 141.5, 135.9, 134.1, 132.0, 129.5, 128.9, 128.7, 128.2, 128.0, 127.0, 124.9, 120.5, 114.4, 61.2, 52.9 ppm. Calcd for C24H20ClN5O2: C, 64.65; H, 4.52; N, 15.71. Found: C, 64.92; H, 4.46; N, 15.88.
N′-(4-Chlorobenzylidene)-4-((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzohydrazide (7j)
Orange precipitates, yield: 49%, mp 236−238 °C. IR (KBr, cm−1): 3417, 3051, 2925, 2852, 1687, 1632, 1586. 1HNMR (500 MHz, DMSO-d6): 11.86 (s, 1H, NH), 8.33−8.31 (m, 2H, triazole, CH), 7.95 (d, J = 8.2 Hz, 2H, H3′, H5′), 7.79−7.50 (m, 4H, H2, H3, H5, H6), 7.23−7.11 (m, 6H, H2′, H6′, H2′′, H3′′, H5′′, H6′′), 5.59 (s, 2H, CH2), 5.21 (s, 2H, CH2). 13CNMR (125 MHz, DMSO-d6): 164.1, 161.7 (d, JC−F = 242.9 Hz), 159.9, 145.3, 142.0, 135.1, 133.0, 132.0, 130.1 (d, JC−F = 7.8 Hz), 129.5, 128.8, 128.6, 124.8, 121.0, 115.4 (d, JC−F = 22.9 Hz), 114.8, 61.1, 51.9 ppm. Calcd for C24H19ClFN5O2: C, 62.14; H, 4.13; N, 15.10. Found: C, 62.25; H, 4.09; N, 14.76.
4-((1-(2-Chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-N′-(4-chlorobenzylidene)benzohydrazide (7k)
Yellow precipitates, yield: 43%, mp 246−248 °C. IR (KBr, cm−1): 3416, 3049, 2923, 2852, 1688, 1631, 1591. 1HNMR (500 MHz, DMSO-d6): 11.82 (s, 1H, NH), 8.45−8.31 (m, 2H, CH, triazole), 8.22−7.91 (m, 2H, H3′, H5′), 7.76−7.51 (m, 5H, H2, H3, H5, H6, H3′′), 7.39- 7.17 (m, 5H, H2′, H6′, H4′′, H5′′, H6′′), 5.72 (s, 2H, CH2), 5.25 (s, 2H, CH2). 13CNMR (125 MHz, DMSO-d6): 162.2, 160.7, 147.4, 142.0, 134.5, 133.1, 132.6, 130.5, 130.2, 129.6, 129.2, 128.9, 128.8, 128.6, 127.7, 125.3, 120.5, 114.4, 61.1, 50.6 ppm. Calcd for C24H19Cl2N5O2: C, 60.01; H, 3.99; N, 14.58. Found: C, 60.19; H, 3.76; N, 14.45.
N′-(4-Chlorobenzylidene)-4-((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzohydrazide (7l)
Red precipitates, yield: 40%, mp 216−218 °C. IR (KBr, cm-1): 3417, 3051, 2922, 2850, 1689, 1625, 1589. 1HNMR (500 MHz, DMSO-d6): 11.81 (s, 1H, NH), 8.60 (s, 1H, CH), 8.28 (s, 1H, triazole), 7.96- 7.54 (m, 6H, H2, H3, H5, H6, H3′, H5′), 7.21- 7.09 (m, 6H, H2′, H6′, H2′′, H3′′, H5′′, H6′′), 5.55 (s, 2H, CH2), 5.22 (s, 2H, CH2), 2.28 (s, 3H, CH3). 13CNMR (125 MHz, DMSO-d6): 161.8, 160.5, 147.5, 140.9, 137.4, 132.8, 132.0, 129.5, 129.2, 128.8, 128.5, 127.9, 125.8, 123.7, 120.9, 114.8, 62.0, 52.6, 20.6 ppm. Calcd for C25H22ClN5O2: C, 65.29; H, 4.82; N, 15.23. Found: C, 65.63; H, 4.63; N, 15.19.
4-((1-Benzyl-1H-1,2,3-triazol-4-yl)methoxy)-N′-(4-methoxybenzylidene)benzohydrazide (7m)
Yellow precipitates, yield: 53%, mp 237−240 °C. IR (KBr, cm−1): 3415, 3053, 2921, 2852, 1686, 1633, 1589. 1HNMR (500 MHz, DMSO-d6): 11.86 (s, 1H, NH), 8.31−8.30 (m, 2H, triazole, CH), 7.88−7.71 (m, 4H, H2, H6, H3′, H5′), 7.36−7.32 (m, 5H, H2′′, H3′′ H4′′, H5′′, H6′′), 7.15−7.01 (m, 4H, H3, H5, H2′, H6′), 5.56 (s, 2H, CH2), 5.21 (s, 2H, CH2), 3.78 (s, 3H, OCH3). 13CNMR (125 MHz, DMSO-d6): 162.0, 161.5, 158.8, 147.5, 142.8, 135.9, 130.3, 128.7, 128.1, 127.9, 127.6, 126.4, 125.5, 120.2, 114.4, 113.8, 61.2, 55.2, 52.8 ppm. Calcd for C25H23N5O3: C, 68.01; H, 5.25; N, 15.86. Found: C, 68.1; H, 5.42; N, 15.81.
4-((1-(4-Fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-N′-(4-ethoxybenzylidene)benzohydrazide (7n)
Light green precipitates, yield: 58%, mp 244−247 °C. IR (KBr, cm−1): 3416, 3051, 2926, 2852, 1685, 1633, 1591. 1HNMR (500 MHz, DMSO-d6): 11.63 (s, 1H, NH), 8.40 (s, 1H, CH), 8.32 (s, 1H, triazole), 7.89 (d, J = 8.3 Hz, 2H, H3′, H5′), 7.41−7.20 (m, 6H, H2, H6, H2′′, H3′′, H5′′, H6′′), 7.15 (d, J = 8.3 Hz, 2H, H2′, H6′), 7.02 (d, J = 8.2 Hz, 2H, H3, H5), 5.62 (s, 2H, CH2), 5.23 (s, 2H, CH2), 3.81 (s, 3H, OCH3). 13CNMR (125 MHz, DMSO-d6): 162.8, 161.3, 161.2 (d, JC−F = 245.0 Hz), 157.0, 147.1, 142.0, 132.2, 130.3 (d, JC−F = 8.2 Hz), 129.8, 129.4, 128.7, 124.8, 122.5, 115.6 (d, JC−F = 21.6 Hz), 114.4, 114.3, 61.2, 55.3, 52.1 ppm. Calcd for C25H22FN5O3: C, 65.35; H, 4.83; N, 15.24. Found: C, 65.57; H, 5.21; N, 15.13.
4-((1-(2-Chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-N′-(4-methoxybenzylidene)benzohydrazide (7o)
Green precipitates, yield: 43%, mp 222−225 °C. IR (KBr, cm−1): 3417, 3050, 2926, 2850, 1688, 1637, 1591. 1HNMR (500 MHz, DMSO-d6): 11.61 (s, 1H, NH), 8.30−8.29 (m, 2H, triazole, CH), 7.94−7.78 (m, 5H, H2, H6, H3′, H5′, H3′′), 7.24−7.16 (m, 5H, H2′, H6′, H4′′, H5′′, H6′′), 7.07 (d, J = 8.1 Hz, 2H, H3, H5), 5.73 (s, 2H, CH2), 5.24 (s, 2H, CH2), 3.81 (s, 3H, OCH3). 13CNMR (125 MHz, DMSO-d6): 163.0, 160.6, 159.0, 144.9, 142.5, 133.1, 132.6, 130.5, 130.2, 129.6, 129.4, 128.6, 127.7, 126.8, 126.2, 121.1, 114.3, 113.5, 61.2, 55.2, 50.6 ppm. Calcd for C25H22ClN5O3: C, 63.09; H, 4.66; N, 14.72. Found: C, 63.22; H, 4.39; N, 14.87.
N′-(4-Methoxybenzylidene)-4-((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzohydrazide (7p)
Light brown precipitates, yield: 57%, mp 224−228 °C. IR (KBr, cm−1): 3415, 3052, 2927, 2850, 1687, 1630, 1587. 1HNMR (500 MHz, DMSO-d6): 11.68 (s, 1H, NH), 8.26−8.25 (m, 2H, triazole, CH), 7.88−7.69 (m, 4H, H2, H6, H3′, H5′), 7.21−7.02 (m, 8H, H3, H5, H2′, H6′, H2′′, H3′′, H5′′, H6′′), 5.54 (s, 2H, CH2), 5.19 (s, 2H, CH2), 3.79 (s, 3H, OCH3), 2.28 (s, 3H, CH3). 13CNMR (125 MHz, DMSO-d6): 161.9, 160.8, 159.3, 146.0, 144.0, 137.5, 132.9, 130.0, 129.2, 128.9, 128.5, 128.0, 125.5, 121.0, 114.1, 113.9, 61.2, 54.8, 52.6, 20.6 ppm. Calcd for C26H25N5O3: C, 68.56; H, 5.53; N, 15.37. Found: C, 68.35; H, 5.61; N, 15.53.
In vitro α-glucosidase inhibition assay
The assay was performed exactly according to our previous report30,38.
Enzyme kinetic studies
The mode of inhibition of the most active compounds 7a and 7h (identified with the lowest IC50) was investigated against α-glucosidase activity with different concentrations of p-nitrophenyl α-D-glucopyranoside (1−10 mM) as substrate in the absence and presence of inhibitors at different concentrations. 7h: 0, 2, 4, and 8 nM and 7a: 0, 0.2, 0.5, and 1 µM.
Fluorescence spectroscopy
The fluorescence measurements were performed on a Synergy HTX multi-mode reader (Biotek Instruments, Winooski, VT, USA) equipped with a quartz cuvette of 10 mm. The excitation wavelength was 280 nm and the emission spectra were measured in the range from 300 to 440 nm with 10 accumulations for each collection point. The emission spectrum was corrected for the background fluorescence from the buffer solution and for the inner filter effect promoted by the inhibitors. The protein-inhibitors interaction measurements were performed by collecting the fluorescence.
The fluorescence quenching of the protein (P) by a drug or inhibitor (D) was analyzed by Stern−Volmer equation (Eq. 1), where F0 and F are the fluorescence intensity in the absence and presence of a quencher (inhibitor), respectively. [D] is the concentration of inhibitor, Kq is the quenching rate constant, τ0 is the average life-time of the molecule without the quencher (10−8 s), and KSV is the Stern−Volmer dynamic quenching constant.
The reaction of the protein (P) and a drug molecule or inhibitor (D) was considered as Eq. 2, where P is the protein, D is the drug molecule, and DnP is the new complex molecule with the binding constant of KA.
As the number of the binding site of protein and drug is respectively n and 1, the equivalent concentration of the complex DnP is n[DnP]; the equivalent concentration of the protein is n[P] and the equivalent concentration of the drug is [D]. In this respect, the KA value is calculated based on the Eq. (3).
If the total concentration of protein is [Pt] and the total concentration of the drug is [Dt]: [Pt] = [Pf] + [DnP] and [Df] = [Dt] − n[DnP].
If protein (P) is the only fluorescence in the reaction system, thus:
where F and F0 are the fluorescence intensity of protein in the presence and absence of drug D, respectively. Therefore, the relationship between the fluorescence intensity and the drug total concentration can be deduced from Eq. (5).
Keeping the total concentration of protein at a definite value and varying the total concentration of the drug leads to obtaining the plot of F0/F as a function of [Dt] F0/(F0 − F) following with the calculation of KA.
The standard thermodynamic values were calculated according to Eqs. (6), (7), where ΔH, ΔG, and ΔS are enthalpy, free energy, and entropy changes, respectively. In the case of small changes in temperature, the enthalpy change can be considered as a constant and ΔH is calculated from Eq. (6) following with the calculation of ΔG and ΔS from Eq. (7).
Molecular docking procedure
To prepare the ligands, the 2D structures of the ligands were drawn in ChemDraw (ver. 16), converted into SDF and software, and energy was minimized by the MM1 force field. The docking simulation was performed on the crystal structures of human lysosomal acid-alpha-glucosidase (PDB ID: 5NN8) to figure out the binding modes of the selected compounds. The crystal structures of acarbose were retrieved from the PDB and docked using the Molegro Virtual Docker software. The site map was tasked to report up to 5 potential binding sites with at least 15 site points per each reported site by a more restrictive definition of hydrophobicity. Flexible ligand dockings were accomplished for the selected compounds with the scoring function of MolDock Score, Grid resolution of 0.30, Radios 10, search algorithm of Moldock Se, Max population size of 100, and max iteration 1500. The best pose of the selected compound in the target protein was chosen by analyzing the interactions between the enzyme and inhibitors. The best-scoring positions, as achieved by the docking score, were then selected, and visualized using Discovery Studio Client 2017.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Giri, B. et al. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 107, 306–328. https://doi.org/10.1016/j.biopha.2018.07.157 (2018).
Toender, A. et al. Risk of diabetic complications and subsequent mortality among individuals with schizophrenia and diabetes—A population-based register study. Schizophr. Res. https://doi.org/10.1016/j.schres.2020.01.024 (2020).
Paul, S., Ali, A. & Katare, R. Molecular complexities underlying the vascular complications of diabetes mellitus—A comprehensive review. J. Diabetes Complicat. https://doi.org/10.1016/j.jdiacomp.2020.107613 (2020).
Gan, Q. et al. The role of diosgenin in diabetes and diabetic complications. J. Steroid Biochem. 198, 105575. https://doi.org/10.1016/j.jsbmb.2019.105575 (2020).
Kumawat, V. S. & Kaur, G. Therapeutic potential of cannabinoid receptor 2 in the treatment of diabetes mellitus and its complications. Eur. J. Pharmacol. 862, 172628. https://doi.org/10.1016/j.ejphar.2019.172628 (2019).
Rivera-Mancía, S., Lozada-García, M. C. & Pedraza-Chaverri, J. Experimental evidence for curcumin and its analogs for management of diabetes mellitus and its associated complications. Eur. J. Pharmacol. 756, 30–37. https://doi.org/10.1016/j.ejphar.2015.02.045 (2015).
Moghaddam, F. M. et al. Synthesis and characterization of 1-amidino-O-alkylureas metal complexes as α- glucosidase Inhibitors: Structure-activity relationship, molecular docking, and kinetic studies. J. Mol. Struct. 1250, 131726. https://doi.org/10.1016/j.molstruc.2021.131726 (2022).
Karami, M. et al. One-pot multi-component synthesis of novel chromeno[4,3-b]pyrrol-3-yl derivatives as alpha-glucosidase inhibitors. Mol. Divers. https://doi.org/10.1007/s11030-021-10337-w (2021).
Fattaheian-Dehkordi, S., Hojjatifard, R., Saeedi, M. & Khanavi, M. A review on antidiabetic activity of Centaurea spp.: A new approach for developing herbal remedies. Evid.-Based Complement. Altern. Med. 2021, 5587938. https://doi.org/10.1155/2021/5587938 (2021).
Dhameja, M. & Gupta, P. Synthetic heterocyclic candidates as promising α-glucosidase inhibitors: An overview. Eur. J. Med. Chem. 176, 343–377. https://doi.org/10.1016/j.ejmech.2019.04.025 (2019).
Santos, C. M. M., Freitas, M. & Fernandes, E. A comprehensive review on xanthone derivatives as α-glucosidase inhibitors. Eur. J. Med. Chem. 157, 1460–1479. https://doi.org/10.1016/j.ejmech.2018.07.073 (2018).
Guo, J.-C. et al. Triterpenoids and meroterpenoids with α-glucosidase inhibitory activities from the fruiting bodies of Ganoderma australe. Bioorganic Chem. 117, 105448. https://doi.org/10.1016/j.bioorg.2021.105448 (2021).
Saeedi, M., Hadjiakhondi, A., Mohammad Nabavi, S. & Manayi, A. Heterocyclic compounds: effective α-amylase and α-glucosidase inhibitors. Curr. Top. Med. Chem. 17, 428–440 (2017).
Saeedi, M. et al. Synthesis of 4-alkylaminoimidazo[1,2-a]pyridines linked to carbamate moiety as potent α-glucosidase inhibitors. Mol. Divers. 25, 2399–2409. https://doi.org/10.1007/s11030-020-10137-8 (2021).
De Marzio, D. & Navarro, V. Drug-Induced Liver Disease Vol. 523 (Academic Press, 2013).
Khan, S. et al. Probing 2-acetylbenzofuran hydrazones and their metal complexes as α-glucosidase inhibitors. Bioorganic Chem. 102, 104082. https://doi.org/10.1016/j.bioorg.2020.104082 (2020).
Saroha, B., Kumar, G., Kumar, R., Kumari, M. & Kumar, S. A minireview of 1,2,3-triazole hybrids with O-heterocycles as leads in medicinal chemistry. Chem. Biol. Drug Des. 100, 843–869 (2022).
Bozorov, K., Zhao, J. & Aisa, H. A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 27, 3511–3531 (2019).
Karimi Askarani, H. et al. Design and synthesis of multi-target directed 1,2,3-triazole-dimethylaminoacryloyl-chromenone derivatives with potential use in Alzheimer’s disease. BMC Chem. 14, 64. https://doi.org/10.1186/s13065-020-00715-0 (2020).
Saeedi, M. et al. Synthesis and bio-evaluation of new multifunctional methylindolinone-1,2,3-triazole hybrids as anti-Alzheimer’s agents. J. Mol. Struct. 1229, 129828 (2021).
Li, J. & Zhang, J. The antibacterial activity of 1,2,3-triazole-and 1,2,4-triazole-containing hybrids against Staphylococcus aureus: An updated review (2020-present). Curr. Top. Med. Chem. 22, 41–63 (2022).
Safavi, M. et al. Novel quinazolin-4 (3H)-one linked to 1,2,3-triazoles: Synthesis and anticancer activity. Chem. Biol. Drug Des. 92, 1373–1381 (2018).
Mohammadi-Khanaposhtani, M. et al. Design, synthesis and cytotoxicity of novel coumarin-1, 2, 3-triazole-1, 2, 4-oxadiazole hybrids as potent anti-breast cancer agents. Lett. Drug Des. Discovery 16, 818–824 (2019).
Mahdavi, M. et al. Synthesis and anticancer activity of N-substituted 2-arylquinazolinones bearing trans-stilbene scaffold. Eur. J. Med. Chem. 95, 492–499 (2015).
Saeedi, M. et al. Design and synthesis of novel quinazolinone-1, 2, 3-triazole hybrids as new anti-diabetic agents: In vitro α-glucosidase inhibition, kinetic, and docking study. Bioorg. Chem. 83, 161–169 (2019).
Saeedi, M. et al. Design, synthesis, in vitro, and in silico studies of novel diarylimidazole-1,2,3-triazole hybrids as potent α-glucosidase inhibitors. Bioorg. Med. Chem. 27, 115148 (2019).
Farid, S. M. et al. Quinazolinone-1,2,3-triazole-acetamide conjugates as potent α-glucosidase inhibitors: Synthesis, enzyme inhibition, kinetic analysis, and molecular docking study. RSC Med. Chem. (2023).
Asgari, M. S. et al. Biscoumarin-1,2,3-triazole hybrids as novel anti-diabetic agents: Design, synthesis, in vitro α-glucosidase inhibition, kinetic, and docking studies. Bioorganic Chem. 92, 103206. https://doi.org/10.1016/j.bioorg.2019.103206 (2019).
Asemanipoor, N. et al. Synthesis and biological evaluation of new benzimidazole-1,2,3-triazole hybrids as potential α-glucosidase inhibitors. Bioorganic Chem. 95, 103482. https://doi.org/10.1016/j.bioorg.2019.103482 (2020).
Shareghi-Boroujeni, D. et al. Synthesis, in vitro evaluation, and molecular docking studies of novel hydrazineylideneindolinone linked to phenoxymethyl-1,2,3-triazole derivatives as potential α-glucosidase inhibitors. Bioorganic Chem. 111, 104869. https://doi.org/10.1016/j.bioorg.2021.104869 (2021).
Settypalli, T. et al. Design, synthesis, in silico docking studies and biological evaluation of novel quinoxaline-hydrazide hydrazone-1,2,3-triazole hybrids as α-glucosidase inhibitors and antioxidants. New J. Chem. 43, 15435–15452. https://doi.org/10.1039/C9NJ02580D (2019).
Abbasi, I. et al. Isatin-hydrazide conjugates as potent α-amylase and α-glucosidase inhibitors: Synthesis, structure and invitro evaluations. Bioorganic Chem. 116, 105385. https://doi.org/10.1016/j.bioorg.2021.105385 (2021).
Iraji, A. et al. Cyanoacetohydrazide linked to 1,2,3-triazole derivatives: A new class of α-glucosidase inhibitors. Sci. Rep. 12, 1–15 (2022).
Hammett, L. P. The effect of structure upon the reactions of organic compounds. Benzene Deriv. J. Am. Chem. Soc. 59, 96–103 (1937).
Hansch, C. et al. Aromatic substituent constants for structure-activity correlations. J. Med. Chem. 16, 1207–1216. https://doi.org/10.1021/ja01280a022 (1973).
Asemanipoor, N. et al. Synthesis and biological evaluation of new benzimidazole-1,2,3-triazole hybrids as potential α-glucosidase inhibitors. Bioorg Chem 95, 103482. https://doi.org/10.1016/j.bioorg.2019.103482 (2020).
Iraji, A. et al. Cyanoacetohydrazide linked to 1,2,3-triazole derivatives: A new class of α-glucosidase inhibitors. Sci. Rep. 12, 8647. https://doi.org/10.1038/s41598-022-11771-y (2022).
Saeedi, M. et al. Synthesis of 4-alkylaminoimidazo [1,2-a] pyridines linked to carbamate moiety as potent α-glucosidase inhibitors. Mol. Diversity 25, 2399–2409 (2021).
Acknowledgements
This work was supported by grants from the Research Council of Tehran University of Medical Sciences with project No. 1400-2-104-53319. This paper is dedicated to the memory of our unique teacher in Chemistry and Medicinal Chemistry, Professor Abbas Shafiee (1937−2016).
Author information
Authors and Affiliations
Contributions
M.S. designed, and characterized the target compounds and wrote the manuscript. R.H. contributed to the synthesis of compounds. A.I. wrote the manuscript and performed in silico study. A.A. synthesized compounds. S.M. and S.G. performed biological assays. M.A.F. supervised biological assays. T.A. supervised all steps of the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saeedi, M., Hariri, R., Iraji, A. et al. Novel N′-substituted benzylidene benzohydrazides linked to 1,2,3-triazoles: potent α-glucosidase inhibitors. Sci Rep 13, 8960 (2023). https://doi.org/10.1038/s41598-023-36046-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-36046-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
