
Abstract
A first clue to gene function can be obtained by examining whether a gene is required for life in certain standard conditions, that is, whether a gene is essential. In bacteria, essential genes are usually identified by high-density transposon mutagenesis followed by sequencing of insertion sites (Tn-seq). These studies assign the term “essential” to whole genes rather than the protein domain sequences that encode the essential functions. However, genes can code for multiple protein domains that evolve their functions independently. Therefore, when essential genes code for more than one protein domain, only one of them could be essential. In this study, we defined this subset of genes as “essential domain-containing” (EDC) genes. Using a Tn-seq data set built-in Burkholderia cenocepacia K56-2, we developed an in silico pipeline to identify EDC genes and the essential protein domains they encode. We found forty candidate EDC genes and demonstrated growth defect phenotypes using CRISPR interference (CRISPRi). This analysis included two knockdowns of genes encoding the protein domains of unknown function DUF2213 and DUF4148. These putative essential domains are conserved in more than two hundred bacterial species, including human and plant pathogens. Together, our study suggests that essentiality should be assigned to individual protein domains rather than genes, contributing to a first functional characterization of protein domains of unknown function.
Similar content being viewed by others

Introduction
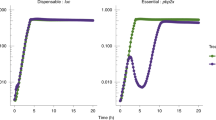
A first step when characterizing gene function should be asking whether a given gene encodes an essential cellular function, whether the gene is necessary for the survival of the organism. A widely accepted method to identify essential genes in bacteria is high-density transposon mutagenesis, followed by Illumina-sequencing of the transposon insertion junctions (Tn-seq)1. During Tn-seq, transposon mutant cells are pooled and grown in optimal conditions, allowing cells with a transposon insertion located in a non-essential element to survive. Cells with a transposon insertion in an essential element should be lost or depleted from the population. When transposon insertions are identified by Illumina sequencing, read counts per gene in the central 70–90% of the open reading frame (disruptive insertions) are normalized by gene length and used to predict essentiality. 5–15% sequences from the 3′ and 5′ ends are usually removed from the analysis, as insertions within the terminal regions are likely non-disruptive2,3,4,5. While disrupted genes are regarded as “non-essential,” the method yields a list of putative essential genes as those with zero or very few mapped reads (Fig. 1a, b)3.

Schematics of Tn-Seq reads mapped to the insertion sites in non-essential (a), essential (b), and essential domain-containing (EDC) genes (c–d). The number of transposon insertions related to the length of the gene (minus the non-informative 10% towards the 5′ and 3′ ends) is quantified and used to classify genes as non-essential (a), or essential (b) according to the relative number of reads mapped to that gene. Tn-seq analysis may miss EDC genes which are essential genes that contain an essential domain not spanning throughout the whole length of the gene (c–d). Genes are represented by arrows. Tn-seq reads that map to regions of those genes are represented by black boxes. Essential and non-essential regions are colored in red and green, respectively.
Another step towards identifying gene function is the annotation of the protein domains encoded by genes. Protein domains are functional or structural units that can fold, evolve, and function independently. Homology-based protein domain prediction and function assignment are effective starting points for understanding protein function, even when diverse protein architectures add complexity to functional annotations6,7. While domain databases such as Pfam8 and InterPro9 aim to provide maximum sequence coverage to predict protein domain identity, approximately 30% of all domains listed in these databases (Pfam 33.1 and InterPro 81.0) are ‘domains of unknown function (DUFs).’ Single DUFs are usually predicted to span through functionally uncharacterized proteins. However, studies suggest that at least some of these proteins may contain more than one domain10,11.
While robust and comprehensive, very few Tn-seq studies12,13,14 consider that genes may encode for more than one protein domain. Tn-seq analysis may classify a gene as “non-essential” due to the presence of transposon insertions in a non-essential coding region, despite the gene coding for a second domain not spanning through the whole gene length that might be essential3,15,16. We operationally defined this subclass of essential genes as “essential domain-containing” (EDC) genes (Fig. 1c, d) and present a computational pipeline to identify them in a Tn-seq dataset built-in Burkholderia cenocepacia K56-217. Unlike the previously reported methods, our method does not require in-depth understanding of computational platforms and generates a list of candidate EDC genes. By analyzing biases in transposon density in genes previously identified as “non-essential”, we found 40 genes where the encoded proteins contained putative essential and non-essential domains. Using a CRISPR Interference (CRISPRi)18 platform we developed for Burkholderia19, we experimentally confirmed growth defects, representing the loss of a putative essential function, in 27 EDC gene knockdowns. The identified EDC genes include ten encoding known multidomain proteins and two entirely uncharacterized genes encoding different N-terminal DUFs, demonstrating the utility of the approach. This study highlights that gene essentiality depends on the function of individual protein domains rather than entire proteins.
Results
Identification of EDC genes from Tn-seq data
To identify EDC genes in B. cenocepacia K56-2, we built a custom script that used our previous Tn-seq data17 to select genes that (i) were not previously found to be essential in B. cenocepacia K56-217, and (ii) had an asymmetric distribution of transposon insertions (Fig. 2). The script split each gene into two equal parts and selected genes with reads in only one region to identify genes with transposon insertion biases. We worked under the assumption that (i) each half could represent one functional domain and (ii) one of the domains may be essential while the other may not. We arbitrarily set the parameters “min ratio” and “min reads” to 0 and 0.14, respectively (see Material and Methods and Supplementary Fig. 1). These settings looked for genes that had zero reads at one end, while the number of reads in the non-empty end was at least 14% of that region's length. For example, if a section of a gene was 100 bp in length, it would require at least 14 reads mapped to that section to be considered non-essential. With these settings, the script produced an extensive list of 178 candidate EDC genes (Supplementary Table 1).

Identification of putative essential domain-containing (EDC) genes from a Tn-seq dataset. Tn-Seq reads are first mapped to the reference genome. A custom-built script identifies genes with biased location of transposon insertions towards one half of the gene. The script parameters “min ratio’ and “min reads” were set such that genes were selected when (i) a half-region of that gene (at least 40% of the total gene length) showed no insertions (min ratio = 0), and (ii) the other half contained mapped reads in at least 14% of that gene-half length (min reads = 0.14) (see Supplementary Fig. 1and Material and Methods for details). Reads mapping to each 5′ or 3′ 10% end of the gene were discarded from the analysis.
Bioinformatic analysis of the candidate EDC genes
We reasoned that if EDC genes contained essential protein domains, then the essential protein domains may be encoded by essential genes in at least some other bacteria. We then searched for essential ortholog genes of the 178 candidate EDC genes by BLASTx searches against the ‘Database of Essential Genes (DEG)20 using 50% sequence alignment and 30% sequence identity as the cut-off. We found that 40 of the 178 genes had orthologs annotated as ‘essential” in other bacterial species. We wished to interrogate the domains encoded by these 40 genes using UniProt21 based on InterPro domains9. InterPro predicts the domain information by matching the protein or nucleic acid sequences against the member databases (collectively known as InterPro consortium) to identify ‘signatures’ associated with known domains. Thus, the InterPro prediction relies on the availability of sequence characterization and annotation. This analysis showed that from the 40 candidate EDC genes predicted to be essential by homology with other essential genes, 10 genes encoded multidomain proteins, and 7 of them were well-characterized, such as the N-terminal domain of DnaK and NusA (Fig. 3a). The remaining genes were predicted to have one single annotated domain (19 genes) that did not span the whole gene-length or encoded uncharacterized proteins (11 genes) (Supplementary Table 2). All 40 genes had transposon insertions located in one half of the gene, showing that the script was able to identify genes with biased transposon insertions (Supplementary Fig. 2). Taken together, these results suggest that the identified genes could be essential due to the presence of essential protein domain orthologues. Notably, 17 DNA regions were identified as coding for new putative essential protein domains (Table 1).

Biased transposon insertion identifies putative essential domains of uncharacterized hypothetical proteins. Tn-seq reads from17 were mapped to the B. cenocepacia K56-2 genome and predicted to contain essential domains. (a) The script identified the well characterized essential N-terminal domains of DnaK (BCAL3270) and NusA (BCAL1506). Their respective CRISPRi mutants demonstrated a conditional growth defect. (b) Two uncharacterized genes BCAM1066 (WQ49_RS16145) and BCAS0158 (WQ49_RS10495) contain the Pfam domains DUF2213 (PF09979) and DUF4148 (PF13663), respectively, at the N-terminal end. The Tn-seq reads map to the C-terminal end of these genes, demonstrating the essentiality of DUF2213 and DUF4148. Putative essential domains are highlighted in blue. Black triangles represent the transposon insertion sites. Numbers on top of the domains denote amino acid sequence positions. Blue and red lines in the growth curves (a and b) represent growth in the absence and presence of rhamnose, respectively. Growth curves are shown for the most efficient sgRNAs. Growth curves values are the average of three independent biological replicates. Error bars indicate mean ± SD.
CRISPRi knockdowns of EDC genes show growth defects
To phenotypically characterize the effect of knocking down EDC genes, we used CRISPR interference or CRISPRi19 to create knockdown mutants of the genes of interest. CRISPRi comprises a chromosomally integrated dcas9 under the control of a rhamnose-inducible promoter and plasmid-borne sgRNA driven by a constitutively active synthetic promoter, PJ2311919. Simultaneous expression of dcas9 and a target-specific sgRNA allows the dCas9 to bind the target DNA region and, thus, sterically interfere with transcription by RNA polymerase18,19. To inhibit the expression of the candidate genes, we designed two sgRNAs against each of the candidate genes targeting the start codon and adjacent region on the non-template strand (Supplementary Fig. 3a,c). For phenotypic characterization, we grew the cells in LB with and without rhamnose. Upon induction of dCas9 with rhamnose, 27 out of the 40 candidate genes showed at least 25% growth inhibition relative to the uninduced condition (Supplementary Fig. 3b,d).
DUF2213 and DUF4148 appear to be essential domains
The presence of DUFs is a common feature of hypothetical or uncharacterized proteins. To initiate functional characterization of DUFs, we focused on two genes containing DUF-coding sequences, which their respective CRISPRi mutants demonstrated a conditional growth defect (Fig. 3b). WQ49_RS16145 (BCAM1066) and WQ49_RS10495 (BCAS0158) contain DUF2213 (Pfam accession PF09979) and DUF4148 (Pfam accession PF13663), respectively at the N-terminal end of the proteins (Fig. 3b). BLAST searches of BCAM1066 and BCAS0158 genes as a query against the DEG20 showed that BCAM1066 (WQ49_RS16145) had 30% sequence similarity with lysK (B8GXH3) from Caulobacter crescentus, and BCAS0158 (WQ49_RS10495) had a 52% sequence identity with a predicted amino acid permease (BPSS1112) from Burkholderia pseudomallei K96243 (data not shown). Mining of the Pfam database (https://pfam.xfam.org/) showed that these DUFs are well conserved across the bacterial species: DUF2213 is present in 209 bacterial species, including bacterial pathogens (Acinetobacter baumannii, Enterobacter cloacae, Haemophilus influenzae, Burkholderia cepacia, Shigella flexneri), plant pathogens (Agrobacterium tumefaciens), and biotechnologically relevant species (Pseudomonas putida) (Fig. 4a and Supplementary Table 5). DUF4148 is found in 204 bacterial species, primarily in Burkholderia species (i. e Burkholderia cepacia, Burkholderia mallei, Burkholderia vietnamiensis) and plant pathogens such as Ralstonia solanacearum (Fig. 4b; Supplementary Table 5). DUF2213 is also present in many phage-related proteins (Fig. 4a). Eight unique domain architectures were observed for proteins containing DUF2213 and five for DUF4148 (Fig. 4c, d). DUF2213 is associated with another essential domain PF00293, a NUDIX hydrolase (Fig. 4c). In other proteins, DUF2213 is associated with the LPD3 domain (PF18798) and DUF1073 (PF06381) which is also conserved across bacterial species11 (Fig. 4c). On the other hand, Pfam analysis of DUF4148 shows that DUF4148 differs in domain length among species and is associated with the Pfam domain PF00144, known to confer resistance against β-lactams (Fig. 4d)22. Nonetheless, the encoded N-terminus was highly conserved, suggesting that it is functionally significant. The Pfam-based analysis of species distribution also revealed that DUF2213 is present in six eukaryotic species (five metazoans and one fungal species), whereas DUF4148 is present in five eukaryotic species (three viridiplantae species and two metazoan species). The widespread distribution of these DUFs indicates the functional importance of these putative essential domains, creating an impetus for further characterization.

Phylogenetic trees with taxonomic information of DUF2213 (PF09979) and DUF4148 (PF13663) and domain architectures of proteins containing these domains. (a–b) Phylogenetic trees of DUF2213 (a) and DUF4148 (b) across the species with taxonomic annotations. DUF2213 is widely distributed within bacterial, archaeal, phage and eukaryotic species, whereas DUF4148 is mostly distributed in bacteria (primarily in Proteobacterial species). Trees shown here are the majority rule consensus trees. Taxonomic annotations were labelled based on NCBI taxonomy database. Representative bacterial, archaeal, phage and eukaryotic species are highlighted in lilac, yellow, grey and green, respectively. The orange circles on the branches represent the bootstraps values. (c)–(d) Domain architectures of proteins containing DUF2213 (c) and DUF4148 (d) across species. Numbers on top of the domains in (c) and (d) represent amino acid sequence positions.
Discussion
A first step in the functional characterization of genes is performed through gene deletion or gene silencing and growth phenotype characterization. For genes that encode multidomain proteins performing multiple functions driven by the activity of their individual domains23, the function assigned to a gene could indeed correspond to one of its encoded protein domains and not to the whole protein. That is the case of essential genes identified by Tn-seq1. In standard Tn-seq analysis the condition of essentiality is assigned to genes and not to encoded domains, resulting in incorrect classification of many essential genes as non-essential. Rather, the essentiality assignment pipeline should be revised to analyze the essentiality of encoded individual protein domains24. Indeed, essentiality can be assigned to individual domains of a multidomain protein rather than the entire protein15,16. In this work, we defined as essential-domain-containing (EDC) genes those genes that encode more than one protein domain, with one of the domains coding for an essential function. By analyzing a Tn-seq dataset17 for transposon insertion biases, we show that standard Tn-seq analysis pipelines may miss EDC genes, whose detection often requires either manual curation or additional considerations25.
We validated our approach by identifying genes encoding previously characterized multidomain essential proteins in which the essential function is assigned to one single domain. For instance, our analysis of biases in the Tn-seq dataset showed that the gene region coding for the N-terminal domain of NusA26 is sufficient to mediate the essential function, in agreement with previous work27. Similarly, the B. cenocepacia K56-2 dnaK gene was previously defined as non-essential17; however, we found that the Tn-seq reads mapped onto dnaK were biased toward the C-terminal domain (CTD), suggesting that only the NTD is necessary for its essential function. (Fig. 3b; Supplementary Fig. 2). DnaK is a multidomain protein and a master regulator of the chaperone network28. DnaK comprises an N-terminal ATPase domain (NTD) and a C-terminal substrate-binding domain (CTD)28. Perturbations either within the NTD that leads to the abrogation of the ATPase activity or within the conserved linker peptide that impairs the interdomain mechanistic interaction abrogate the in vivo activity of DnaK29,30.
While 14 EDC genes that demonstrated a growth defect when knocked down code for proteins annotated to have a single domain, none of these domains span the entire gene, and transposon insertions are only mapped to the annotated domain (Supplementary Fig. 2). Thus, it is possible that the remaining regions code for novel domains that perform the essential biological functions independently of the adjacent sequences. Indeed, multidomain proteins that are involved in direct protein–protein interactions are more often detected as essential than proteins with a single domain15, hinting towards the functional contribution of individual domains within a protein complex. However, it should be noted that the presence of multiple domains in an essential protein does not necessarily mean that the protein is composed by essential and non-essential domains. An example is the Bacillus subtillis SMC, a multidomain essential protein involved in chromosomal segregation31,32.
We demonstrated a conditional growth defect in 27 out of 40 CRISPRi mutants of EDC genes. It remains a possibility that the sgRNAs designed for CRISPRi-mediated gene silencing of the remaining 13 genes were not efficient in target binding, thus yielding no growth defect. CRISPRi is more effective in blocking transcription initiation than elongation, and is the most efficient in silencing gene expression when promoter regions are targeted with gRNAs18,33,34,35. However, as promoter regions for B. cenocepacia genomes remained largely unannotated we targeted translation start sites. It remains to be investigated whether targeting the promoter region to block the transcription initiation rather than elongation might yield conditional a growth phenotype in the remaining 13 genes.
Eighteen of the 27 EDC genes CRISPRi mutants that demonstrated a conditional growth defect are in an operon (Supplementary Fig. 3). It is possible then that due to the polar effect of CRISPRi, the observed growth defect could result from the transcriptional silencing of any other gene(s) in the same operon. However, we consider this possibility unlikely. These genes (other than the candidate gene in the operon) had transposon insertions greater than the defined threshold in the script across the entire genes (data not shown), suggesting that they are dispensable. The only exceptions are BCAL0245 and BCAL0250, where both genes are located in the same operon (Supplementary Fig. 3). Thus, it remains a possibility that observed growth defect could be due to transcriptional silencing of either or both the genes. A large portion of the protein domains that lack functional assignment can be grouped within the DUF category. DUFs are members of ever-increasing uncharacterized protein families; they are the object of experimental and computational efforts towards their functional characterization10,36,37,38. Determining if a DUF is essential is among the first steps in functional characterization. In this study, we focused on two EDC genes that encode putative essential DUFs: DUF2213 and DUF4148. Both domains have a high degree of conservation across diverse phyla, which highlights their biological relevance. DUF2213, a phage-associated domain (PF09979), is well distributed across bacteria and phages. Interestingly, we found that DUF4148 (PF13663) is putatively essential and associated with β-lactamase (PF00144) (Fig. 4).
In summary, our study identified 27 EDC genes whose knockdown produced a growth defect, suggesting the essential nature of one of their protein domains. By leveraging a Tn-Seq dataset in B. cenocepacia K56-217, we demonstrate that the essential nature of protein-coding genes is a function of the individual protein domains they encode. The utility of our work lies in the identification of gene regions encoding essential and conserved protein domains, which will help de-orphan the many remaining proteins of unknown function. Therefore, we propose that determining essentiality of a domain of unknown function should be the first step in the process to define their function.
Methods
Bacterial strains and growth conditions
The list of bacterial strains and plasmids used in this study is provided in Supplementary Table 3. Bacterial strains were grown in LB-Lennox medium (Difco) at 37 °C. E. coli strain MM290 carrying the helper plasmid pRK2013 was selected in kanamycin 40 µg/mL (Fisher Scientific). Donor strains of E. coli DH5α and B. cenocepacia K56-2 carrying the sgRNA plasmids were selected in trimethoprim 50 µg/mL and 100 µg/mL (Sigma), respectively.
Identification of EDC genes from Tn-Seq dataset
Candidate EDC genes were identified with a custom python script using the Tn-seq dataset17. The script analyzed every gene previously classified as “non-essential” by splitting it into two equal halves and counting the number of reads mapped to each half-gene. The script then used the “min ratio” and “min reads” as filtering criteria to call EDC genes. “Min ratio” was defined as the desired ratio of reads between the halves of the gene. “Min reads” was defined as the minimum number of reads in the non-empty end that is equal to a 14% of that half's length. Min reads was set to 0.14, while min ratio was set as 0. For each gene, 10% from each end of the gene was discarded from the analysis. The parameters can be changed to yield either more stringent or more general results. The script is available at https://github.com/cardonalab/EssentialDomains
Bioinformatic analysis
Orthologous essential genes were identified using BLASTx against DEG 1520. Multidomain information was fetched from the UniProt database based on Pfam8 and InterPro9 domain features. DUF containing genes were characterized using the Pfam tool available on the Pfam website (https://pfam.xfam.org/). Domain sequences were retrieved in FASTA format from the Pfam database8 and aligned by Clustal Ω39. Maximum-likelihood phylogenetic trees were generated with MEGA-X40 using a Jones-Taylor-Thornton (JTT)-based model41 applying 100 bootstrap values. Phylogenetic trees were visualized, edited and taxonomic labels were assigned using Interactive Tree Of Life (i-TOL)42. Bootstrap values are represented on a scale of 0 to 1. Taxonomic annotations were labelled based on the NCBI taxonomy database using UniProt identifiers.
Creating knockdown mutants of the candidate EDC genes with CRISPRi
CRISPRi mutants of the EDC genes were created as previously described19. Briefly, pSCB2-sgRNAv2, a modified plasmid from pSCB2-sgRNA19, was used as the template for inverse PCR to insert 20 bp target-specific sgRNA sequence. Inverse PCR was performed using Q5 high-fidelity polymerase (NEB), forward primers with individual sgRNAs as 5′ tail, and 1092 as the reverse primer. The resultant fragments were ligated to create circular plasmids by incubating 0.5µL of the respective PCR products with quick ligation buffer (NEB), 0.25 μL DpnI, 0.25 μL T4 polynucleotide kinase (NEB), and 0.25 μL T4 ligase (NEB) for 30 min at 37 °C. Resultant plasmids were transformed into E. coli DH5α, recovered for 2 h and selected in LB supplemented with trimethoprim 50 µg/mL (Sigma). The transformants were further confirmed by colony PCR using primers 1409 and 848. E. coli strains carrying the sgRNA plasmids were used as donors, and E. coli MM290/pRK2013 as the helper for triparental mating to introduce the sgRNA plasmids into B. cenocepacia K56-2 containing the chromosomally integrated dCas9 under the control of a rhamnose inducible promoter, as described previously43. Trimethoprim resistant colonies (100 µg/mL) were selected and screened by colony PCR using the primers 1409 and 848. The list of all the primers used in this study is provided in Supplementary Table 4.
Conditional growth phenotype analysis of the CRISPRi mutants
To determine the conditional growth phenotype of the candidate genes, overnight cultures of the CRISPRi mutants were back diluted to OD600nm 0.01. The cultures were grown at 37 °C for 20–24 h with continuous shaking in a 384-well plate containing LB broth supplemented with trimethoprim 100 µg/mL and with/without 1% rhamnose. OD600nm readings were taken at 1-h intervals using BioTek Synergy 2 microplate reader.
References
van Opijnen, T., Bodi, K. L. & Camilli, A. Tn-seq: High-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6, 767–772 (2009).
Akerley, B. J. et al. Systematic identification of essential genes by in vitro mariner mutagenesis. Proc. Natl. Acad. Sci. USA 95, 8927–8932 (1998).
Chao, M. C., Abel, S., Davis, B. M. & Waldor, M. K. The design and analysis of transposon insertion sequencing experiments. Nat. Rev. Microbiol. 14, 119–128 (2016).
Langridge, G. C. et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res. 19, 2308–2316 (2009).
Shields, R. C., Zeng, L., Culp, D. J. & Burne, R. A. Genomewide identification of essential genes and fitness determinants of Streptococcus mutans UA159. mSphere 3, e00031-18 (2018).
Forslund, S. K., Kaduk, M. & Sonnhammer, E. L. L. Evolution of protein domain architectures. in Evolutionary Genomics (ed. Anisimova, M.) vol. 1910 469–504 (Springer, 2019).
Schnoes, A. M., Brown, S. D., Dodevski, I. & Babbitt, P. C. Annotation error in public databases: Misannotation of molecular function in enzyme superfamilies. PLoS Comput. Biol. 5, e1000605 (2009).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Mitchell, A. L. et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360 (2019).
Bateman, A., Coggill, P. & Finn, R. D. DUFs: Families in search of function. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 66, 1148–1152 (2010).
Goodacre, N. F., Gerloff, D. L. & Uetz, P. Protein domains of unknown function are essential in bacteria. mBio 5, e00744-13 (2014).
DeJesus, M. A. et al. Bayesian analysis of gene essentiality based on sequencing of transposon insertion libraries. Bioinformatics 29, 695–703 (2013).
Zhang, Y. J. et al. Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog. 8, e1002946 (2012).
Michel, A. H. et al. Functional mapping of yeast genomes by saturated transposition. eLife 6, e23570 (2017).
Lluch-Senar, M. et al. Defining a minimal cell: Essentiality of small ORFs and ncRNAs in a genome-reduced bacterium. Mol. Syst. Biol. 11, 780 (2015).
Lu, Y. et al. A novel essential domain perspective for exploring gene essentiality. Bioinformatics 31, 2921–2929 (2015).
Gislason, A. S., Turner, K., Domaratzki, M. & Cardona, S. T. Comparative analysis of the Burkholderia cenocepacia K56-2 essential genome reveals cell envelope functions that are uniquely required for survival in species of the genus Burkholderia. Microb. Genomics 3, e000140 (2017).
Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Hogan, A. M., Rahman, A. S. M. Z., Lightly, T. J. & Cardona, S. T. A broad-host-range CRISPRi Toolkit for silencing gene expression in Burkholderia. ACS Synth. Biol. 8, 2372–2384 (2019).
Luo, H. et al. DEG 15, an update of the database of essential genes that includes built-in analysis tools. Nucleic Acids Res. 49, D677–D686 (2021).
The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 (2019).
Gao, M., Glenn, A. E., Blacutt, A. A. & Gold, S. E. Fungal Lactamases: Their occurrence and function. Front. Microbiol. 8, 1775 (2017).
Kanaan, S. P., Huang, C., Wuchty, S., Chen, D. Z. & Izaguirre, J. A. Inferring protein–protein interactions from multiple protein domain combinations. In Computational Systems Biology (eds. Ireton, R., Montgomery, K., Bumgarner, R., Samudrala, R. & McDermott, J.) vol. 541 43–59 (Humana Press, 2009).
Miravet-Verde, S., Burgos, R., Delgado, J., Lluch-Senar, M. & Serrano, L. FASTQINS and ANUBIS: Two bioinformatic tools to explore facts and artifacts in transposon sequencing and essentiality studies. Nucleic Acids Res. 48, e102 (2020).
Goodall, E. C. A. et al. The essential genome of Escherichia coli K-12. mBio 9, e02096-17 (2018).
Qayyum, M. Z., Dey, D. & Sen, R. Transcription elongation factor NusA is a general antagonist of rho-dependent termination in Escherichia coli. J. Biol. Chem. 291, 8090–8108 (2016).
Ha, K. S., Toulokhonov, I., Vassylyev, D. G. & Landick, R. The NusA N-terminal domain is necessary and sufficient for enhancement of transcriptional pausing via interaction with the RNA exit channel of RNA polymerase. J. Mol. Biol. 401, 708–725 (2010).
Wu, C.-C., Naveen, V., Chien, C.-H., Chang, Y.-W. & Hsiao, C.-D. Crystal structure of DnaK protein complexed with nucleotide exchange factor GrpE in DnaK chaperone system: Insight into intermolecular communication. J. Biol. Chem. 287, 21461–21470 (2012).
Barthel, T. K., Zhang, J. & Walker, G. C. ATPase-defective derivatives of Escherichia coli DnaK that behave differently with respect to ATP-induced conformational change and peptide release. J. Bacteriol. 183, 5482–5490 (2001).
Vogel, M., Mayer, M. P. & Bukau, B. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J. Biol. Chem. 281, 38705–38711 (2006).
Britton, R. A., Lin, D.C.-H. & Grossman, A. D. Characterization of a prokaryotic SMC protein involved in chromosome partitioning. Genes Dev. 12, 1254–1259 (1998).
Minnen, A. et al. Control of Smc coiled coil architecture by the ATPase heads facilitates targeting to chromosomal ParB/parS and release onto flanking DNA. Cell Rep. 14, 2003–2016 (2016).
Bikard, D. et al. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41, 7429–7437 (2013).
Hawkins, J. S., Wong, S., Peters, J. M., Almeida, R. & Qi, L. S. Targeted transcriptional repression in bacteria using CRISPR interference (CRISPRi). Methods Mol. Biol. 1311, 349–362 (2015).
Vigouroux, A., Oldewurtel, E., Cui, L., Bikard, D. & van Teeffelen, S. Tuning dCas9’s ability to block transcription enables robust, noiseless knockdown of bacterial genes. Mol. Syst. Biol. 14, e7899 (2018).
Bastard, K. et al. Revealing the hidden functional diversity of an enzyme family. Nat. Chem. Biol. 10, 42–49 (2014).
Dessailly, B. H. et al. PSI-2: Structural genomics to cover protein domain family space. Structure 17, 869–881 (2009).
Zhang, X. et al. Assignment of function to a domain of unknown function: DUF1537 is a new kinase family in catabolic pathways for acid sugars. Proc. Natl. Acad. Sci. 113, E4161–E4169 (2016).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Jones, D. T., Taylor, W. R. & Thornton, J. M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 8, 275–282 (1992).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Hogan, A. M. et al. Competitive fitness of essential gene knockdowns reveals a broad-spectrum antibacterial inhibitor of the cell division protein FtsZ. Antimicrob. Agents Chemother. 62, e01231-18 (2018).
Acknowledgements
This work was supported by grants from the Canadian Institutes of Health Research (CIHR), Cystic Fibrosis Foundation, Cystic Fibrosis Canada to STC; ASMZR was supported by a University of Manitoba Graduate Fellowship (UMGF). The authors thank Dr. Georg Hausner, Andrew Hogan, Dustin Maydaniuk and rest of the Cardona lab members for critically reading the manuscript.
Funding
Funding is provided by Canadian Institutes of Health Research (Grant no. 5211, project Grant), Cystic Fibrosis Canada (Grant no. 50501).
Author information
Authors and Affiliations
Contributions
A.S.M.Z.R.—performed the majority of the experiments and wrote the manuscript; L.T.—created the python script and contributed to manuscript editing; F.G.—created CRISPRi mutants and contributed to manuscript editing; S.T.C.—conceived the idea, supervised the work, provided financial support, and edited the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rahman, A.S.M.Z., Timmerman, L., Gallardo, F. et al. Identification of putative essential protein domains from high-density transposon insertion sequencing. Sci Rep 12, 962 (2022). https://doi.org/10.1038/s41598-022-05028-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-05028-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
