
Abstract
Glioblastoma (GBM) is the most common primary malignant brain tumor in adults. Prior studies examining the mutational landscape of GBM revealed recurrent alterations in genes that regulate the same growth control pathways. To this regard, ~ 40% of GBM harbor EGFR alterations, whereas BRAF variants are rare. Existing data suggests that gain-of-function mutations in these genes are mutually exclusive. This study was designed to explore the clinical, pathological, and molecular differences between EGFR- and BRAF-mutated GBM. We reviewed retrospective clinical data from 89 GBM patients referred for molecular testing between November 2012 and December 2015. Differences in tumor mutational profile, location, histology, and survival outcomes were compared in patients with EGFR- versus BRAF-mutated tumors, and microarray data from The Cancer Genome Atlas was used to assess differential gene expression between the groups. Individuals with BRAF-mutant tumors were typically younger and survived longer relative to those with EGFR-mutant tumors, even in the absence of targeted treatments. BRAF-mutant tumors lacked distinct histomorphology but exhibited unique localization in the brain, typically arising adjacent to the lateral ventricles. Compared to EGFR- and IDH1-mutant tumors, BRAF-mutant tumors showed increased expression of genes related to a trophoblast-like phenotype, specifically HLA-G and pregnancy specific glycoproteins, that have been implicated in invasion and immune evasion. Taken together, these observations suggest a distinct clinical presentation, brain location, and gene expression profile for BRAF-mutant tumors. Pending further study, this may prove useful in the stratification and management of GBM.
Similar content being viewed by others


Introduction
Glioblastoma (WHO grade IV glioma, GBM) is a common malignancy of the central nervous system (CNS), constituting approximately 15% of all primarily brain tumors in adults1. The median survival rate of a GBM patient is approximately 14 months following the standard treatment of resection, irradiation, and chemotherapy2, a figure that has remained essentially unchanged despite significant advances in defining the molecular pathogenesis of these cancers3. With the dawn of the genomic era and the widespread implementation of clinical next-generation sequencing (NGS), there has been an intense search for sequence variants with diagnostic, prognostic, and/or therapeutic significance to inform optimized treatment protocols improve outcomes for GBM patients.
GBM harbor a wide array of molecular alterations, including combinations of point mutations, insertions and deletions (indels), copy number variants, and epigenetic modifications4. Some of these alterations correlate with differences in clinical outcome and are used to diagnose and classify tumors and/or inform treatment decisions. For example, mutations in IDH1 and IDH2 are the major stratifying features of diffuse gliomas4, and MGMT promoter methylation is associated with longer overall survival and response to temozolomide treatment2. The biological and clinical significance of other recurrent mutations has yet to be determined4; however, molecular data are being actively mined in search of features that could enhance the clinical stratification of GBM and reveal targets for existing and/or novel chemotherapeutic agents.
GBM frequently harbor mutations that activate mitogenic signaling pathways. Receptor tyrosine kinases (RTKs) regulate growth factor signaling to increase cell proliferation, metabolism, and survival in response to environmental cues5,6. Increased activation of the epidermal growth factor receptor (EGFR), a membrane bound RTK, is common in GBM. This may occur via increased protein expression or via mutations that lead to constitutive activity7. In a 2013 survey of 585 GBM, 45% had putative activating mutations (either copy number amplifications, exon or domain deletions, point mutations, or combinations thereof) in EGFR4. Some evidence (albeit inconsistent) suggests that gain-of-function alterations in EGFR could serve as an indicator of poor prognosis within the current treatment paradigm8,9,10. They are also being explored as a potential target for FDA-approved RTK inhibitors11.
The serine/threonine-protein kinase, B-Raf (encoded by the BRAF gene) is a member of the RAS/RAF/MEK/MAPK that functions downstream of EGFR. B-Raf inhibitors are approved for use in melanoma and non-small cell lung cancer12,13, and ongoing clinical trials are exploring their use in brain tumors (see ClinicalTrials.gov study IDs NCT03224767, NCT03973918, NCT02684058, etc.). The canonical BRAF p.V600E mutation occurs in roughly half of all epithelioid GBM14, but it is rare in classic GBM15. As such, the 585-patient cohort described above contained only 13 GBM specimens with putative gain-of-function BRAF amplifications and/or mutations4.
Activating alterations in EGFR and BRAF were reported in multiple GBM datasets, always appearing in a mutually exclusive pattern4,16. This observation led us to speculate that GBM with activating alterations in BRAF may constitute a biologically distinct entity. In this study, we sought to determine the clinical, pathologic, and molecular differences between GBM with each of these mutations to better characterize their impact on the diagnosis and management of GBM.
Materials and methods
Institutional case selection
Ninety-one consecutive formalin-fixed, paraffin-embedded (FFPE) GBM specimens from 89 unique patients were submitted to Genomics and Pathology Services at Washington University in St. Louis (http://gps.wustl.edu) for testing between November 2012 and December 2015 (Supplemental Table 1). All cases were initially reviewed by board-certified neuropathologists according to the WHO 2007 guidelines for classification of tumors of the CNS17 and appropriate ancillary diagnostic work-up was done wherever necessary. Cases were re-classified according to WHO 2016 classification guidelines for the purpose of this study18.
Panel-based next-generation sequencing assays
The cases described in this study were derived from three separate versions of the same comprehensive cancer assay. The assay version is listed for each case in Supplemental Table 1, and the reportable genes for each assay version are listed in Supplemental Table 2. Ten genes were present on all three assay versions: BRAF, CTNNB1, EGFR, IDH1, IDH2, KRAS, PDGFRA, PTEN, TP53, and WT1.
DNA preparation and next-generation sequencing
Next-generation sequencing was performed as previously described19,20. FFPE blocks were reviewed by board-certified neuropathologists to identify regions with sufficient neoplastic cellularity and tissue viability. Cores were punched from marked areas, and DNA was isolated using a Qiagen QIAamp DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). DNA was sonicated with a Covaris Ultrasonicator (Woburn, MA), and assessed by an Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA). NGS libraries were prepared using commercially available kits (Supplemental Table 3). Whole genome shotgun libraries were enriched by hybridization capture using custom-designed oligonucleotide baits (Supplemental Table 3) corresponding to the genes included on each version of the assay. Enriched libraries were multiplexed and sequenced on the Illumina HiSeq2500 platform (Illumina, San Diego, CA) to generate 2 × 100 bp reads.
Sample pre-processing and read alignment
Raw base call files (BCL files) were converted to fastq format using Cassava v1.8 (Illumina) and de-multiplexed using in-house scripts. Paired-end reads were aligned to the human reference assembly (UCSC hg19) using Novoalign (Novocraft Technologies, Selangor, Malaysia). Alignment files were converted to BAM format with samtools21, and duplicate reads were marked using Picard Tools MarkDuplicates (https://broadinstitute.github.io/picard/). Coverage metrics are provided in Supplemental Table 4.
Variant prediction, annotation, and interpretation
Variant calling methods are outlined in Supplemental Table 3. Aligned read depth at each chromosomal position was determined using SAMtools mpileup21,22. Insertion and deletion (indel) variants ≤ 21 bp were called using Genome Analysis Toolkit’s (GATK23) Unified Genotyper with the following cutoffs: depth ≥ 50 reads, Fisher strand bias ≤ 75, and not adjacent to a homopolymer run > 7 bp. Single nucleotide variants (SNVs) were called using either Genome Analysis Toolkit’s (GATK) Unified Genotyper or VarScan224 with the following cutoffs: depth ≥ 50 reads, base quality ≥ 20, mapping quality ≥ 30, Fisher strand bias ≤ 75, and variant allele fraction (VAF) > 3%. SNVs and indels present in reportable gene sets were subjected to annotation and clinical interpretation and were included in the patient’s medical record. Polymorphisms represented in public human genomic variation databases (gnomAD, ExAC, dbSNP, dbNSFP, NHLBI ESP) were discarded from this analysis. Remaining non-synonymous variants are listed in Supplemental Table 1. Variants were classified as pathogenic or likely pathogenic based on established criteria25.
Fluorescence in situ hybridization
Formalin fixed paraffin embedded blocks were sectioned at 5 µm and routinely processed using the Vysis EGFR/CEP7 FISH probe kit (Abbott Molecular Inc., Glean Oaks, IL) and counterstained with DAPI II. Stained slides were viewed using a BX61 fluorescent microscope (Olympus, Melville, NY, USA) and photographed with a Jai Progressive Scan camera and CytoVision Imaging System (Leica Biosystems, Wetzlar, Germany). Two-hundred interphase nuclei were examined in each specimen. Polysomies and monosomies were determined based on centromere enumerating probes. Focal gains and losses were determined based on signal ratios from locus-specific versus centromere enumerating probes.
Microarray-based gene expression studies
Primary, IDHwt, GBM specimens were selected from a TCGA dataset (Firehose Legacy, https://www.cbioportal.org/study?id=gbm_tcga) based on the presence of activating point mutations in EGFR and IDH1, and BRAF p.V600E mutations (Supplemental Table 5). CEL files from the selected cases were downloaded. Normalized signal values were generated using Expression Console v4.1.1 (Affymetrix, ThermoFisher Scientific, Waltham, MA) using the robust multichip average algorithm, and detection p-values were calculated using the MAS5 algorithm. Probe-sets were filtered based on the detection p-value; only those corresponding to coding genes were maintained in this analysis. Differentially expressed genes were determined using limma26 with fold change cutoff of ≥ 2 and a false-discovery rate (FDR) cutoff of ≤ 0.05.
Biological theme enrichment analysis
CompBio software (https://www.percayai.com/) was used to identify biological themes that were enriched among the differentially expressed genes. This is accomplished with an automated Biological Knowledge Generation Engine (BKGE) that extracts abstracts from PubMed that reference the genes contained in the input list (i.e., the differentially expressed genes identified in the microarray analysis) and uses contextual language processing and a biological language dictionary to compute the statistical enrichment of biological concepts that co-occur with the input genes more frequently than would be expected by random chance. Related concepts derived from the list of differentially expressed genes are further clustered into higher-level themes. Scoring of concept and theme enrichment is accomplished using a multi-component function referred to as the Normalized Enrichment Score (NES). The first component utilizes an empirical p-value derived from several thousand random entity lists of a size that is comparable to the users input entity list to define the rarity of a given entity-concept event. The second component, effectively representing the fold enrichment, is based on the ratio of the concept enrichment score to the mean of that concept’s enrichment score across the set of randomized entity data. As such, the NES reflects both the rarity of the co-occurrence of the input genes with the concept as well as the degree of overall enrichment. Based on these empirical criteria, observed entity-concept scores above 10.0, 100.0, and 1,000.0 are labeled as moderate, marked, or high in level of enrichment above random. Themes scoring above 500.0, 1,000.0, and 5,000.0 are labeled similarly. In this study, the top 25 most enriched themes with scores > 1000.0 for each condition were subjected to manual annotation and further analysis (Supplemental Fig. 1).

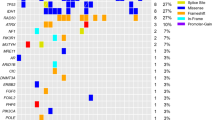
Mutations found in ninety-one specimens of glioblastoma. Ninety-one glioblastoma specimens from 89 unique patients were subjected to next-generation sequencing on a small gene panel used for routine clinical, diagnostic testing. Non-synonymous mutations are shown in nine genes that were included in all assay versions; common population polymorphism were excluded. Ancillary testing for EGFR amplification was performed via fluorescence in situ hybridization for 77 specimens as part of the diagnostic workup.
Ethics approval
The institutional review board of Washington University School of Medicine determined that this retrospective study of de-identified data did not fit the definition of human subjects research; thus, no IRB protocol was required.
Consent to participate
Not applicable.
Consent for publication
All authors of this manuscript have directly participated in the planning, analysis, and/or editing of this study, and approved the publication of this work.
Results
Specimens and clinical data
Ninety-one GBM (WHO grade IV) specimens from 89 unique patients were submitted for targeted NGS between November 2012 and December 2015 (Supplemental Table 1). These specimens included 87 primary GBM (IDH1 and IDH2 wild type, IDHwt) and four secondary GBM with mutations in IDH1. The male:female ratio among these patients was approximately 2:1 (56 M:33F). Patients ranged in age from 9 to 83 years at the age of biopsy (average 55.10 ± 14.97 years). Three pediatric/adolescent GBM (patients ≤ 18 years) were included in this cohort. One patient presented at age 12 with a GBM centered in the thalamic region and again at age 18 with a recurrence in the posterior fossa. The other two pediatric patients had been diagnosed with other cancers (desmoplastic infantile ganglioglioma and acute lymphoblastic leukemia) prior to the development of GBM.
EGFR mutations
Twenty-one of the specimens in our dataset (23.1%) harbored point mutations in EGFR (Fig. 1, Supplemental Table 1). Two specimens had EGFR p.G589V mutations; seven others had mutations in the extracellular domain (p.A289T, n = 1; p.A289D, n = 2; p.A289V, n = 4; Fig. 1, Supplemental Fig. 2). These mutations are common in GBM and are known to drive autophosphorylation and activation of EGFR in the absence of ligand7,27. The remaining 12 EGFR mutations were classified as variants of uncertain clinical significance (see methods for details); their impact on protein activity is unknown.

Magnetic resonance imaging and histological features of a representative BRAF mutated tumor. Patients with BRAF mutant GBMs were typically found adjacent to the ventricular system (A). BRAF mutated tumors did not show distinctive histomorphology compared to classical GBM (B).
Seventy-seven specimens were sent for analysis by FISH in addition to NGS. Thirty-three of these tested positive for EGFR gene amplification. Thirteen of the 33 had point mutations within the amplified gene (Fig. 1, Supplemental Table 1).
BRAF mutations
Five specimens from our cohort had point mutations in BRAF (Fig. 1, Supplemental Table 1). One of these was the canonical p.V600E mutation (VAF 50.2%), which is known to increase kinase activity and drive oncogenesis28. While the other four mutations classified as variants of uncertain functional/clinical significance according to standard criteria (see methods for details), they were all located within the tyrosine kinase domain (Supplemental Fig. 2). In absence of a germline control (typically unavailable in the clinical setting), we take the VAF of the mutations (ranging from 10.0–26.0%) and their absence or rarity in databases of human genomic variation (e.g., gnomAD, see methods) as soft indications that they are somatic rather than germline polymorphisms. A search of the COSMIC database29 showed that the p.K483E and p.H574Q mutations found in tumors GBM_01 and GBM_08, respectively, have both been reported in the context of other cancers, most frequently in melanoma. The BRAF p.G596R mutation found in GBM_10 is classified as a class III BRAF mutation and is known to activate ERK signaling in a RAS-dependent manner despite impaired kinase activity30. The fifth BRAF mutation, found in patient GBM_09, was a frameshift present at the beginning of the kinase domain, and would thus be predicted to result in loss of function.
Comparison of tumors with EGFR and BRAF mutations
As expected, BRAF and EGFR mutations were mutually exclusive in our cohort. EGFR gene amplifications and point mutations were only seen in primary GBMs, while one of the BRAF mutations was found alongside a gain-of-function mutation in IDH1. The three male and two female patients with SNVs in the BRAF gene were, on average, younger than the 21 patients with EGFR SNVs (mean, 41.82 ± 15.94 and 53.19 ± 11.33 years, respectively).
The location of the tumor within the brain was known for 87 of the 91 specimens analyzed. Tumors were found adjacent to the ventricular system in 3 of the 5 patients with BRAF mutations (Fig. 2A, Supplemental Table 1). Of the two remaining BRAF-mutant tumors, one was found in the frontal lobe involving the precentral gyrus and the location of the other was unknown. In comparison, tumors without BRAF mutations showed a range of locations, most commonly involving the frontal and temporal lobes and, less frequently, the parietal lobes, occipital lobes or midline structures (Supplemental Table 1).
The BRAF-mutant tumors in this cohort set did not exhibit distinctive histomorphology (Fig. 2B). None were characterized as epithelioid GBM.
Impact of BRAF and EGFR mutations on survival
Survival data was available for 85 of 91 patients. Kaplan–Meier curves were calculated to compare the outcomes of tumors with EGFR versus BRAF mutations (Fig. 3A). Excluding GBM_01 (a secondary GMB) and GBM_09 (harboring the likely-inactivating frameshift mutation), the average overall survival of three patients with BRAF-mutated tumors was longer than that of patients with EGFR-mutated tumors (average of 48.01 ± 24.42 months versus 16.13 ± 11.46 months, respectively; log-rank p-value = 0.02). These findings should be considered as preliminary given the small size of the BRAF-mutated cohort.

The impact of BRAF versus EGFR mutations on overall survival in glioblastoma. Kaplan–Meier plots indicate the difference in survival between BRAF and EGFR mutated tumors in our in-house cohort (A) and a dataset from TCGA (B). Considering only IDH1 wild-type tumors, the average overall survival of three patients with putative activating BRAF mutations was longer than that of 20 patients with EGFR mutations from the in-house cohort (average overall survival of 48.01 ± 24.42 months versus 16.13 ± 11.46 months, respectively; log-rank p = 0.02). One patient with an activating IDH1 mutation and another with a BRAF frameshift mutation (likely loss-of-function) were omitted from this analysis. Likewise, the average overall survival of 6 patients with gain-of-function BRAF mutations was longer compared to 77 patients with gain-of-function EGFR mutations in a cohort from TCGA (average overall survival of 27.68 ± 26.95 and 14.19 ± 11.28 months, respectively; log-rank p = 0.09).
We leveraged a TCGA dataset (Firehose Legacy, https://www.cbioportal.org/study?id=gbm_tcga) to further explore the implications of BRAF mutations on survival outcomes. Mutation data was provided for 290 of the 619 specimens included in this cohort. Six of the 290 profiled tumors harbored gain-of-function mutations in BRAF (5 p.V600E, 1 p.G596D) while 77 had gain-of-function mutations in EGFR. Consistent with our in-house dataset, primary GBM patients with BRAF-mutated tumors survived somewhat longer than patients with EGFR-mutated tumors (Fig. 3B; mean, 27.68 ± 26.95 and 14.19 ± 11.28 months, respectively; log-rank p-value = 0.09).
Differential gene expression in IDHwt-GBM with BRAF and EGFR mutations
Microarray data from the TCGA cohort described above was used to assess gene expression profiles in GBM with distinct mutation signatures (Table 1). mRNA expression data was only available for three IDHwt tumors with BRAF p.V600E mutations. These were compared to expression data from 12 randomly selected IDHwt tumors with gain-of-function EGFR mutations (individuals described in Supplemental Table 5). We included a third group of tumors with IDH1 mutations to help determine the relative directional change of the observed expression differences. Counts of genes considered differentially expressed across the three comparisons are listed in Table 1.
Biological theme enrichment analysis indicated that tumors with BRAF gain-of-function mutations show increased expression of genes implicated in MET pathway signaling, protein processing, immune function, and invasion, among other features that might be expected in a tissue exhibiting intense metabolic activity and cell growth/division (Fig. 4, Supplemental Fig. 3, Supplemental Video 1). Of note, we observed elevated expression of several genes typically thought to be selectively expressed in trophoblasts, including a complement of pregnancy-specific glycoproteins (PSGs) and HLA-G (Supplemental Fig. 4).

Biological concepts and themes enriched among genes differentially expressed between tumors gain-of-function mutation in BRAF, EGFR, or IDH1. A pseudo-heatmap was constructed from the comparison of biological themes associated with the differentially expressed genes identified from a microarray based study of gene expression profiles among glioblastoma with gain-of-function mutations in BRAF, EGFR and IDH1. Related biological themes that were associated with each list of differentially expressed genes (illustrated in Supplemental Fig. 1) were grouped into higher-level clusters to facilitate comparison between the three subgroups. Red indicates increased expression; blue indicates decreased expression.
Compared to the other two groups, tumors with IDH1 mutations showed a strong stem-like gene expression profile (Fig. 4). This included increased expression of genes related to cell cycle / mitosis, transformation to a mesenchymal phenotype, and Wnt signaling, as well genes involved in neurogenesis.
Little overlap was seen in the comparison of tumors with EGFR gain-of-function mutations to tumors with BRAF versus IDH1 mutations (Fig. 4).
Compared to tumors with BRAF mutations, EGFR-mutant tumors showed elevated expression of genes involved in cell cycle function, regulation of apoptosis, and diverse signaling cascades. They also showed increased expression of gene in the ERBB2 signaling cascade relative to tumors with IDH1 mutations.
Discussion
In this study, we combined data from an in-house cohort of 89 patients with data from TCGA to explore the clinical significance of gain-of-function mutations in BRAF versus EGFR. As previously shown, activating mutations in these genes were mutually exclusive, suggesting that the two subtypes may arise through a distinct mechanism that leads to different pathological and clinical features. Although BRAF-mutant GBM are rare, patients with BRAF-mutant tumors were, on average, younger and survived longer relative to those with EGFR-mutant tumors. Despite similar histomorphology, BRAF-mutant tumors often arose centrally in the brain, adjacent to the ventricular system. Microarray studies indicated that BRAF-mutant tumors have a unique mRNA expression profile compared to EGFR- and IDH1-mutant tumors, with upregulation of genes related to immune tolerance and invasion, suggesting a unique mechanism for tumorigenesis in this subset of GBM.
The aim of this study was not to describe the genomic landscape of GBM tumors; this has been reported elsewhere4,10. Relatively few genes were sequenced and analyzed in our in-house cohort, as the NGS panel was selected for clinical and diagnostic purposes rather than research, and most clinical NGS panels are limited in scope to minimize cost and analytical time. Furthermore, the presently described assay was not suitable for the detection of gene fusions, some of which are known to activate BRAF and the MAPK pathway31. The size and scope of the clinical NGS panel remains a limitation of this work though our results indicate that the in-house cohort was similar to those described in the literature, particularly with respect to the frequency of EGFR and BRAF mutations4,32,33. Further work will be needed to fully characterize the genomic profile of these two groups.
Patients with GBM harboring BRAF mutations presented earlier in life compared to those patients whose tumors had EGFR mutations. In other studies, BRAF p.V600E mutations have been associated with epithelioid features in GBM34. BRAF p.V600E mutations are common (~ 50% frequency) in epithelioid GBM, a sub-type that often occurs in young adults and children35. In contrast, only one of our BRAF mutated tumors was found in a pediatric patient (GBM_08, Supplemental Table 1) and none were characterized as the epithelioid subtype. The histological appearance was conventional in all BRAF-mutated specimens from our in-house cohort (Fig. 2).
While the histological appearance of the BRAF-mutant tumors included in this study was unremarkable, we did note that three of the four patients with available clinical data had BRAF-mutant tumors located adjacent to the ventricular system. Other studies have indicated that overall survival is decreased for GBM bordering the lateral ventricles36. Despite the small cohort size, our analysis suggested that patients with BRAF-mutant tumors may survive longer compared to tumors with EGFR mutations, which were most often found in the cerebral lobes (Supplemental Table 1). This difference in overall survival should be considered as a preliminary finding, as statistical significance was not achieved (likely due to the paucity of BRAF-mutant tumors). Furthermore, the rarity of BRAF-mutated tumors precluded multi-variate analyses that could help elucidate the role of other impactful features such as age or sex. Indeed, patient age may be a key factor in survival since patients with BRAF mutant tumors were, on average, younger than patients with EGFR mutated tumors. Nonetheless, the potential difference in outcomes is interesting, particularly in the absence of targeted treatments (e.g., FDA-approved BRAF inhibitors like dabrafenib and vemurafenib)37,38,39.
The most striking differences between BRAF mutant tumors compared to other groups of GBM were seen at the level of gene expression. BRAF-mutant tumors showed a unique mRNA expression profile, with increased expression of genes involved in immune modulation, including the HLA-G gene. HLA-G is a non-classical human leukocyte antigen class I molecule that functions in fetal-maternal immune tolerance40. It is possible that this profile serves a similar function in tumors, suppressing the anti-tumor response through interaction with inhibitory receptors on immune cells41. These findings could have implications for traditional checkpoint inhibition therapy (anti-CTLA-4 or anti-PD-1), in that the expression of HLA-G could serve as an alternate route for immune evasion in BRAF mutated GBM.
A host of PSGs were also upregulated in BRAF-mutated tumors, which resulted in a trophoblast-like gene expression profile. PSGs are a collection of secreted glycoproteins abundant in maternal blood during pregnancy. Like HLA-G, they play an important role in embryonic development42, and have been associated with upregulated expression in various cancers43,44,45,46. For example, studies have shown that PSG9 serves as a driver of angiogenesis in colorectal and hepatocellular cancers47,48. There are marked similarities between tumor invasion and trophoblast implantation49,50, so our results may suggest that BRAF-mutant GBM may co-opt trophoblast-like properties to facilitate their growth and spread.
While we have presented evidence that EGFR- and BRAF-mutated classical GBM are biologically distinct, we cannot state with certainty whether these differences are directly caused by the mutations identified in these two genes. It is possible that these mutations represent markers of particular tumor types, rather than the direct cause of their distinction. This is particularly true of EGFR mutations, which are known to correlate with various other impactful alterations (e.g., TERT promoter mutations, gain of chromosome 7, loss of chromosome 10, etc.)10. It is also possible that BRAF-mutated tumors are representative of a larger sub-type of non-EGFR-mutated GBM rather than a unique entity of their own. Regardless, the unique features of these two groups warrant further exploration.
Conclusions
BRAF-mutant GBM are very rare, especially in adult populations; however, differences in tumor location, survival rates, and global gene expression profiles set this set of tumors apart from other GBM. Striking differences were seen in comparison to tumors with EGFR gain-of-function mutations, another member of the same signaling cascade. Although this cohort was modest in size, BRAF-mutant GBM constituted a unique subgroup with potential implications for tumor biology and treatment.
Data availability
The clinical data generated from this study are available in the associated supplemental materials.
References
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro. Oncol. https://doi.org/10.1093/neuonc/nov189 (2015).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352(10), 987–996. https://doi.org/10.1056/NEJMoa043330 (2005).
Tanaka, S., Louis, D. N., Curry, W. T., Batchelor, T. T. & Dietrich, J. Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end?. Nat Rev Clin Oncol 10(1), 14–26. https://doi.org/10.1038/nrclinonc.2012.204 (2013).
Brennan, C. W. et al. The somatic genomic landscape of glioblastoma. Cell 155(2), 462–477. https://doi.org/10.1016/j.cell.2013.09.034 (2013).
Geschwind, A., Fischer, O. M. & Ullrich, A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer 4(5), 361–370. https://doi.org/10.1038/nrc1360 (2004).
Lemmon, M. A., Schlessinger, J. & Ferguson, K. M. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 6(4), a020768. https://doi.org/10.1101/cshperspect.a020768 (2014).
Xu, H. et al. Epidermal growth factor receptor in glioblastoma. Oncol. Lett 14(1), 512–516. https://doi.org/10.3892/ol.2017.6221 (2017).
Quan, A. L. et al. Epidermal growth factor receptor amplification does not have prognostic significance in patients with glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 63(3), 695–703. https://doi.org/10.1016/j.ijrobp.2005.03.051 (2005).
Tini, P. et al. Epidermal growth factor receptor expression may correlate with survival through clinical and radiological features of aggressiveness in glioblastoma treated with radiochemotherapy. Anticancer Res. 35(7), 4117–4124 (2015).
McNulty, S. N. et al. Beyond sequence variation: assessment of copy number variation in adult glioblastoma through targeted tumor somatic profiling. Hum. Pathol. https://doi.org/10.1016/j.humpath.2018.12.004 (2018).
Westphal, M., Maire, C. L. & Lamszus, K. EGFR as a target for Glioblastoma treatment: an unfulfilled promise. CNS Drugs 31(9), 723–735. https://doi.org/10.1007/s40263-017-0456-6 (2017).
Odogwu, L. et al. FDA approval summary: dabrafenib and trametinib for the treatment of metastatic non-small cell lung cancers harboring BRAF V600E mutations. Oncologist 23(6), 740–745. https://doi.org/10.1634/theoncologist.2017-0642 (2018).
Kim, G. et al. FDA approval summary: vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin. Cancer Res.. 20(19), 4994–5000. https://doi.org/10.1158/1078-0432.CCR-14-0776 (2014).
Kleinschmidt-DeMasters, B. K., Aisner, D. L., Birks, D. K. & Foreman, N. K. Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am. J. Surg. Pathol. 37(5), 685–698. https://doi.org/10.1097/PAS.0b013e31827f9c5e (2013).
Dahiya, S. et al. BRAF-V600E mutation in pediatric and adult glioblastoma. Neuro. Oncol. 16(2), 318–319. https://doi.org/10.1093/neuonc/not146 (2014).
Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, Chakravarty D, Daian F, Gao Q, Bailey MH, Liang WW, Foltz SM, Shmulevich I, Ding L, Heins Z, Ochoa A, Gross B, Gao J, Zhang H, Kundra R, Kandoth C, Bahceci I, Dervishi L, Dogrusoz U, Zhou W, Shen H, Laird PW, Way GP, Greene CS, Liang H, Xiao Y, Wang C, Iavarone A, Berger AH, Bivona TG, Lazar AJ, Hammer GD, Giordano T, Kwong LN, McArthur G, Huang C, Tward AD, Frederick MJ, McCormick F, Meyerson M, Cancer Genome Atlas Research N, Van Allen EM, Cherniack AD, Ciriello G, Sander C, Schultz N (2018) Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173 (2):321–337 e310. doi:https://doi.org/10.1016/j.cell.2018.03.035
Louis, D. N. et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 114(5), 547–547. https://doi.org/10.1007/s00401-007-0278-6 (2007).
Louis, D. N. et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131(6), 803–820. https://doi.org/10.1007/s00401-016-1545-1 (2016).
Cottrell, C. E. et al. Validation of a next-generation sequencing assay for clinical molecular oncology. J. Mol. Diagn. 16(1), 89–105. https://doi.org/10.1016/j.jmoldx.2013.10.002 (2014).
Cimino, P. J., Bredemeyer, A., Abel, H. J. & Duncavage, E. J. A wide spectrum of EGFR mutations in glioblastoma is detected by a single clinical oncology targeted next-generation sequencing panel. Exp. Mol. Pathol. 98(3), 568–573. https://doi.org/10.1016/j.yexmp.2015.04.006 (2015).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25(16), 2078–2079. https://doi.org/10.1093/bioinformatics/btp352 (2009).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27(21), 2987–2993. https://doi.org/10.1093/bioinformatics/btr509 (2011).
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA (2013) From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43:11 10 11–33. doi:https://doi.org/10.1002/0471250953.bi1110s43
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22(3), 568–576. https://doi.org/10.1101/gr.129684.111 (2012).
Li, M. M. et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the association for molecular pathology, american society of clinical oncology, and college of american pathologists. J. Mol. Diagn. 19(1), 4–23. https://doi.org/10.1016/j.jmoldx.2016.10.002 (2017).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47. https://doi.org/10.1093/nar/gkv007 (2015).
Lee, J. C. et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 3(12), e485. https://doi.org/10.1371/journal.pmed.0030485 (2006).
Pakneshan, S., Salajegheh, A., Smith, R. A. & Lam, A. K. Clinicopathological relevance of BRAF mutations in human cancer. Pathology 45(4), 346–356. https://doi.org/10.1097/PAT.0b013e328360b61d (2013).
Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, Campbell PJ (2015) COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43 (Database issue):D805–811. doi:https://doi.org/10.1093/nar/gku1075
Schreck KC, Grossman SA, Pratilas CA (2019) BRAF Mutations and the utility of RAF and MEK inhibitors in primary brain tumors. Cancers (Basel) 11 (9). doi:https://doi.org/10.3390/cancers11091262
Behling F, Schittenhelm J (2019) Oncogenic BRAF alterations and their role in brain tumors. Cancers (Basel) 11 (6). doi:https://doi.org/10.3390/cancers11060794
Eckel-Passow, J. E. et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N. Engl. J. Med. 372(26), 2499–2508. https://doi.org/10.1056/NEJMoa1407279 (2015).
Sturm, D. et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 14(2), 92–107. https://doi.org/10.1038/nrc3655 (2014).
Behling, F. et al. Frequency of BRAF V600E mutations in 969 central nervous system neoplasms. Diagn. Pathol. 11(1), 55. https://doi.org/10.1186/s13000-016-0506-2 (2016).
Korshunov, A. et al. Epithelioid glioblastomas stratify into established diagnostic subsets upon integrated molecular analysis. Brain Pathol. 28(5), 656–662. https://doi.org/10.1111/bpa.12566 (2018).
Chaichana, K. L. et al. Relationship of glioblastoma multiforme to the lateral ventricles predicts survival following tumor resection. J. Neurooncol. 89(2), 219–224. https://doi.org/10.1007/s11060-008-9609-2 (2008).
Woo, P. Y. M. et al. Regression of BRAF (V600E) mutant adult glioblastoma after primary combined BRAF-MEK inhibitor targeted therapy: a report of two cases. Oncotarget. 10(38), 3818–3826 (2019).
Johanns, T. M., Ansstas, G. & Dahiya, S. BRAF-targeted therapy in the treatment of BRAF-mutant high-grade gliomas in adults. J. Natl. Compr. Canc. Netw. 16(4), 451–454. https://doi.org/10.6004/jnccn.2018.7029 (2018).
Johanns, T. M., Ferguson, C. J., Grierson, P. M., Dahiya, S. & Ansstas, G. Rapid clinical and radiographic response with combined dabrafenib and trametinib in adults with BRAF-mutated high-grade glioma. J. Natl. Compr. Canc. Netw. 16(1), 4–10. https://doi.org/10.6004/jnccn.2017.7032 (2018).
Ferreira, L. M. R., Meissner, T. B., Tilburgs, T. & Strominger, J. L. HLA-G: at the interface of maternal-fetal tolerance. Trends Immunol. 38(4), 272–286. https://doi.org/10.1016/j.it.2017.01.009 (2017).
Carosella, E. D., Rouas-Freiss, N., Tronik-Le Roux, D., Moreau, P. & LeMaoult, J. HLA-G: an immune checkpoint molecule. Adv. Immunol. 127, 33–144. https://doi.org/10.1016/bs.ai.2015.04.001 (2015).
Moore, T. & Dveksler, G. S. Pregnancy-specific glycoproteins: complex gene families regulating maternal-fetal interactions. Int. J. Dev. Biol. 58(2–4), 273–280. https://doi.org/10.1387/ijdb.130329gd (2014).
Salahshor, S., Goncalves, J., Chetty, R., Gallinger, S. & Woodgett, J. R. Differential gene expression profile reveals deregulation of pregnancy specific beta1 glycoprotein 9 early during colorectal carcinogenesis. BMC Cancer 5, 66. https://doi.org/10.1186/1471-2407-5-66 (2005).
Shahinian, J. H. et al. Pregnancy specific beta-1 glycoprotein 1 is expressed in pancreatic ductal adenocarcinoma and its subcellular localization correlates with overall survival. J. Cancer 7(14), 2018–2027. https://doi.org/10.7150/jca.15864 (2016).
Houston, A. et al. Pregnancy-specific glycoprotein expression in normal gastrointestinal tract and in tumors detected with novel monoclonal antibodies. MAbs 8(3), 491–500. https://doi.org/10.1080/19420862.2015.1134410 (2016).
Marrero-Rodriguez, D. et al. Kruppel-like factor 10 participates in cervical cancer immunoediting through transcriptional regulation of Pregnancy-Specific Beta-1 Glycoproteins. Sci. Rep. 8(1), 9445. https://doi.org/10.1038/s41598-018-27711-8 (2018).
Yang, L. et al. Pregnancy-specific glycoprotein 9 (PSG9), a driver for colorectal cancer, enhances angiogenesis via activation of SMAD4. Oncotarget 7(38), 61562–61574. https://doi.org/10.18632/oncotarget.11146 (2016).
Rong, W. et al. PSG9 promotes angiogenesis by stimulating VEGFA production and is associated with poor prognosis in hepatocellular carcinoma. Sci. China Life Sci. 60(5), 528–535. https://doi.org/10.1007/s11427-016-0226-7 (2017).
Ferretti, C., Bruni, L., Dangles-Marie, V., Pecking, A. P. & Bellet, D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum. Reprod. Update 13(2), 121–141. https://doi.org/10.1093/humupd/dml048 (2007).
Costanzo, V., Bardelli, A., Siena, S. & Abrignani, S. Exploring the links between cancer and placenta development. Open Biol https://doi.org/10.1098/rsob.180081 (2018).
Acknowledgements
The authors gratefully acknowledge the contributions of the GPS@WUSTL team, who performed initial analyses of the clinical cases used in this study, the Washington University Cytogenetics and Molecular Pathology Laboratory where the FISH and IHC assays were performed, and the Washington University Division of Neuropathology where morphological assessments were performed. We also thank Jinsheng Yu for expert analysis of the microarray data and the Genome Technology Access Center at Washington University School of Medicine for use of CompBio software. We thank the Alvin J. Siteman Cancer Center at Washington University School of Medicine and Barnes-Jewish Hospital in St. Louis, MO for the use of its Biostatistics Core and Dr. Jingqin Luo for assistance with statistical analyses.
Funding
This study was supported by research funds from the Division of Neuropathology, Department of Pathology and Immunology, Washington University School of Medicine. The Siteman Cancer Center is supported in part by the NCI Cancer Center Support Grant #P30 CA091842. Sonika Dahiya, M.D received a one-year award from the Alvin J. Siteman Cancer Shared Resource Fund towards this work.
Author information
Authors and Affiliations
Contributions
SM and KS wrote the manuscript. SM, KS, CF, and CS collected data and performed experiments. SM and CS analyzed the data. SD performed the histopathologic evaluations. GA, AHK, DHG, and JBR provided the cases. SM, CS, RH, and SD interpreted data, designed and supervised the research, and supervised the writing of the manuscript. All authors have reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
McNulty, S.N., Schwetye, K.E., Ferguson, C. et al. BRAF mutations may identify a clinically distinct subset of glioblastoma. Sci Rep 11, 19999 (2021). https://doi.org/10.1038/s41598-021-99278-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-99278-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
