
Abstract
Genetics-based pest management processes, including the sterile insect technique, are an effective method for the control of some pest insects. However, current SIT methods are not directly transferable to many important pest insect species due to the lack of genetic sexing strains. Genome editing is revolutionizing the way we conduct genetics in insects, including in Tribolium castaneum, an important genetic model and agricultural pest. We identified orthologues of β2Tubulin, Rad50-ATPase and enolase in T. castaneum. Using RT-PCR, we confirmed that these genes are predominantly expressed in the testis. PiggyBac-based transformation of T. castaneum cis-regulatory regions derived from Tc-β2t, Tc-rad50 or Tc-eno resulted in EGFP expression specifically in the T. castaneum testis. Additionally, we determined that each of these regulatory regions regulates EGFP expression in different cell types of the male gonad. Cis-regulatory regions from Tc-β2t produced EGFP expression throughout spermatogenesis and also in mature sperms; Tc-rad50 resulted in expression only in the haploid spermatid, while Tc-eno expressed EGFP in late spermatogenesis. In summary, the regulatory cis-regions characterized in this study are not only suited to study male gonadal function but could be used for development of transgenic sexing strains that produce one sex in pest control strategies.
Similar content being viewed by others

Introduction
Infestation of plant crops by insect pests causes more than 45 billion US dollars in agricultural losses each year1, including damage caused by beetles. Insect pest management provides a number of different methods by which crop damage can be reduced. Conventional sterile insect technique (SIT) programs use radiation to induce male sterility in insects prior to releasing them in field2. In such genetics-based pest management programs, several approaches are used for the sex separation of insects so that only males are released3. For SIT programs for insects such as the mosquito Aedes aegypti, male and female sexes can be separated using differences in pupal body size. Some sex-specific phenotypes, for example body size or development rate are also influenced by environmental factors, and thus special care is required for such systems to be used effectively. However, many of these methods are not directly transferable to other important insect species. Irradiation may also cause reduced male mating competitiveness, potentially reducing the efficiency of traditional SIT approaches.
Several genetic approaches have been developed or are in development for the efficient sex separation of insects3. Genetically engineered systems for male sterilization through carrying a dominant lethal trait in males is a promising alternative to conventional SIT methods4,5. A precision-guided SIT strategy was recently demonstrated in Drosophila melanogaster in which complete male sterility was achieved by the directed mutagenesis of Dm-β2t using genome editing with CRISPR/Cas96. A transgenic sperm-marking strain was established by HDR-based genome editing in the pest Drosophila suzuki7. The development of such a “genetic-sexing” strain (GSS) is an alternative approach for improving the efficiency of SIT that could facilitate the mass scale separation of males and females for new pest species, including coleopterans8. However, the development of genetic sexing approaches in a new pest species may require knowledge about gonad differentiation, sex-biased gene expression and/or regulatory elements capable of efficient and conditional heterologous gene expression in the targeted organisms9,10,11,12. Tissue or stage-specific transgene expression is of particular value in the field of insect biotechnology, with enhancer/promoter elements used to drive the expression of fluorescent proteins or effector molecules in agricultural pests and disease vectors for sexing, monitoring, and reproductive biology studies7,8,11,12,13,14.
The testis-specific β2-tubulin gene in D. melanogaster has been studied in detail to determine its role in spermatogenesis15,16. Mutant β2-tubulin disrupts meiosis in the testis, generating impaired sperm and thus producing male sterility17. Study of the male gonad in insects is important for understanding the mechanism of how sexual identity impacts the processes of tissue organogenesis to create sexual dimorphism. Differential gene expression and regulation in the gonads is essential for producing either male sperm or female eggs required for sexual reproduction18, while variation in gene regulatory networks have been a major driving force in the production of the diverse morphology and phenotypes in different organisms19. The characterization of cis-regulatory regions is also critical for understanding conditional gene expression, assessing the impact of genetic variation on different phenotypes in evolutionary biology and for controlling transgene expression. Established cis-regulatory regions enable strategies to induce sterility by linking insect regulatory elements to lethal effector genes without compromising mating behavior. For example, Yamamoto et al.20 reported that the β2-tubulin promoter from Anopheles stephensi has been used to express a pro-apoptotic factor and thus impair male sterility, resulting in normal mating with control females. To breed lines in the lab with genetically-encoded male sterility, the sterility genes can be switched off/on via the tTA system, which has been established in several pests21,22. Transgenic male sterile strains can also be used to study reproductive biology and mating behavior, including sperm transfer, storage and sperm competition12,23,24.
Coleoptera (Beetles) are the most diverse animal group on earth and contain one fourth of all species described and includes many major pests of crop plants25. Coleoptera consists of approximately 380,000 known species, representing ca. 40% of insect diversity26, including T. castaneum, a model for functional genetics and developmental studies in agricultural pests. T. castaneum has a good quality annotated genome27,28, large scale RNA interference (RNAi)-based screens29,30, as well as efficient transgenesis and genome editing methods31,32,33. In many respects, T. castaneum is more representative of insects than D. melanogaster34. However, there have been very few reports about the stage-specific transgene expression in coleopteran insects, particularly in T. castaneum32. In this study we describe the identification and use of cis-regulatory regions derived from three genes (β2Tubulin, rad50 and enolase) expressed predominantly in the testis of T. castaneum. These DNA cis-regulatory regions enable testes-specific EGFP expression when introduced into the T. castaneum germline via piggyBac-mediated transformation. Stage, uni-sex and tissue specific gene expression is vital for the development of novel pest insect control approaches and also for experiments plan to enhance or improve our knowledge of insect molecular biology.
Materials and methods
RT-PCR analysis
Beetle pupae were separated based on sex prior to eclosion. Ovaries, testes and carcasses from male and female adult beetles were collected at 7–8 days post eclosion (30 beetles in each replicate processed in a single day per sample). Samples were snap frozen in liquid nitrogen and then transferred to − 80 °C prior to RNA extraction. RNA-extraction and cDNA preparation were performed simultaneously with all samples. Total RNA from these tissues was extracted with Trizol (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Single-strand cDNA was synthesized following the manufacturer’s instructions (Fermentas). Primers for RT-PCR were designed by using the software Primer-3 (http://frodo.wi.mit.edu/). Primers were designed by the rules of highest maximum efficiency, and sensitivity rules were followed to avoid formation of self and hetero-dimers, hairpins, self-complementarity and specific to two exons spanning an intron boundary. The primer sequences used in this study are given in Table S1. In brief, single-stranded cDNA was synthesized as follows: 500 ng of total RNA in 11 µl of sterile deionized water previously treated with DNase using the DNA-free kit (Ambion www.ambion.com) following the manufacturer’s instructions. The reverse-transcriptase reaction to generate the cDNA for use in RT-PCR was carried out using the First Strand cDNA Synthesis kit (Fermentas) as follows: 1 μl of oligo d (T) primer was added to the 11 µl of total RNA. The mixture was heated at 65 °C for 5 min, and then placed on ice, and the following were added: 4 μl of 5× first-strand buffer, 2 μl of dNTPs, 1 µl of RNase inhibitor, and 1 µl of reverse transcriptase. cDNA synthesis was performed at 42 °C for 30 min and 50 °C for 60 min. Reactions were stopped by heating samples at 95 °C for 2 min.
RT-PCR amplification conditions were 10 min at 95 °C to activate the polymerase, followed by 30 cycles at 95 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s. Ribosomal protein RpSL32 was used as reference gene, RpSL32 gene specific primer sequences used in this study are given in Table S1. RT-PCR was performed by using Q5 High fidelity DNA Polymerase (NEB).
Plasmid construction
Gene cassettes containing β2t-tTA-P2A-EGFP, Rad50-tTA-P2A-EGFP and Eno-tTA-P2A-EGFP, were synthesized (Epoch life sciences) and cloned into pBluescript-II (SK) (+). Gibson assembly35 (NEB), was used to clone each expression cassette into the donor plasmid pBac-3XP3-DsREDafm36. tTA was used in constructs to assess its expression effect in T. castaneum as part of future research. An overlap of 20 nt was used in primer sequences for the assembly of two fragments (Supplementary Table S1). PCR was performed with Q5 High-Fidelity DNA Polymerase. A total of 0.03–0.2 pmols of each DNA fragment was used in the assembly with 0.01 pmols of vector. The completed plasmids were verified by sequencing.
Development of transgenic lines
Prior to embryo collection, beetles were kept overnight on whole grain flour (29 °C) and switched to instant flour during the next day. Embryos were collected within two hours of oviposition and washed with luke-warm tap water at room temperature to remove any attached flour. Embryos were injected through the chorion with a mixture of phspBac helper37 ~ 300 ng/µl and donor ~ 500 ng/µl plasmid at the posterior end, injections completed within three hours of embryos collection. Injected embryos on each slide were transferred into a petri dish (without lid) and placed on a stand in a sealed plastic container with 100 ml of 2% salt solution (table salt in tap water; 99% relative high humidity) and incubated at 29 °C. At day 3 (injection day is day 0), later in the evening the petri dish was transferred into another plastic container with saturated salt solution (70% relative low humidity) and incubated at 29 °C until hatching. Larvae successfully hatched were counted, collected with a fine brush and transferred into a container with flour. G0 beetles surviving to adulthood were outcrossed to the white-eyed mutant strain, and G1 progeny assayed for DsRED expression using a Leica MZ165FC stereo fluorescence microscope. G1 DsRED + adult beetles were crossed with white-eyed beetles to establish each transgenic line.
Microscopy
Confocal microscopy was performed at the TAMU Microscopy and Imaging Center using a Leica SP8 laser scanning confocal microscope (Leica Microsystems, Wetzlar, Germany) equipped with white laser, AOBS beam splitter and HyD detectors. HC Plan Apo 10×/0.4 dry objective and HC PL APO 20×/0.75 IMM CORR CS2 multi-immersion objective, used in a water immersion mode, were employed for imaging, with pinhole set to 1 Airy unit, excitation 488 nm, fluorescence emission 493–533 nm. Transmitted light images were collected at the same time.
Genomic insertion loci of transgenes
Inverse PCR (iPCR)38 was performed for the isolation of inserted piggyBac elements in T. castaneum. For each transgenic strain, ten beetles were collected and placed into 1.5 ml Eppendorf tubes and frozen in liquid nitrogen. Total genomic DNA was extracted from transgenic lines using the Macherey–Nagel Nucleospin Tissue Kit and quantified with the Spectramax i3x. Genomic DNA of 1–3 µg from transgenic lines was digested with restriction enzymes Sau3AI, HaeII, HinP1I, HhaI, RsaI or HpyCH4III overnight at 37 °C; digested fragments were purified with the Nucleospin Tissue Kit and eluted in 35 µl of elution buffer. In ligation reaction 1 µg of purified digested DNA was self-ligated using T4 DNA Ligase (NEB) overnight at 16 °C, followed by a second purification step and collection in 30 µl of Elution buffer. First round PCR was performed using 2.5 µl of purified, circularized DNA, primers listed in Table S1, and Q5 polymerase (NEB). Cycle conditions were: 98 °C for 1 min, 54 °C for 45 s, and 72 °C for 1 min for 30 cycles. If no product was observed, a second, nested PCR was performed using 2.5 µl of 1st round PCR material (annealing temperature was shifted to 59 °C).
PCR products from different samples were purified from an agarose gel using the Macherey–Nagel Nucleospin Gel and PCR Clean up Kit; purified PCR products were quantified by Nanodrop and sequenced for insert and flanking genomic sequences. Insert sequences were aligned to the 5′ or 3′ piggyBac terminal sequences, with additional sequences as genomic flanking sequences. Genomic insertion sites were identified by comparison with the T. castaneum genome (Tcas5.2) using the blastn function as implemented by the i5K workspace (https://i5k.nal.usda.gov/). Putative insertions at specific sites in the T. castaneum chromosome were further confirmed by PCR amplification of genomic DNA using high fidelity Phusion polymerase (NEB) along with one primer landing within the 3’ UTR of the respective transgenic construct and with the other located in the genomic DNA of flour beetle (Supplementary Table S1).
T. castaneum strains and rearing
The white-eyed T. castaneum strain used in this study arose from an unknown mutation present in the wild-type beetle population which we used previously for detailed transcriptomic analysis39. This sub-strain was selected for transformation experiments, as it lacks black eye pigments that would interfere with our ability to detect eyespecific red fluorescence. White-eyed and wild-type black-eyed beetles were reared separately on flour medium (95% flour, 5% yeast by weight), and caged in glass jars with tight-fitting fine mesh closures. Beetles were housed in a growth chamber at 29 °C with 60–80% relative humidity and 12/12-h light/dark cycling. Populations of beetles were moved to fresh flour medium once per month with initial population densities of approximately 1–2 beetles/1 g flour medium.
Results
Identification and characterization of β2Tubulin, Rad50-ATPase and enolase orthologues in T. castaneum
We previously identified at least eighteen genes whose transcripts were substantially enriched in male testes compared to the rest of the body, female ovaries/body and early embryos39. We reasoned that these genes would be good candidates to donate cis-regulatory sequences that might be capable of driving transgene expression specifically in the testes. In order to select candidate genes, we calculated the distance from each testis-enriched gene to the next upstream and downstream gene in the T. castaneum genome (Supplementary Table S2). We reasoned that focusing on candidate genes with close neighbors would help ensure that the genomic fragments selected would contain the necessary cis-regulatory elements needed for testes-specific expression. We also considered intron length, focusing on genes with only short introns. Based on these criteria, we selected three testes-enriched genes for evaluation: TC009035 (β2-tubulin), TC006703 (rad50), and TC011729 (enolase).
Tubulin is the major constituent of microtubules, and testes-specific β-tubulin genes have been described in Drosophila, Bombyx, medfly and mosquitoes11,12,13,23. We identified four different β-tubulin orthologues TC009589, TC034766, TC010829, and TC009035 in the T. castaneum reference genome (Supplementary Fig. S1, Supplementary Table S3). However, only TC009035 (β2-tubulin) was found to have high expression in T. castaneum testes39, and we refer to this gene as Tc-β2t for simplicity. Tc-β2t is 95% identical at the amino acid level to the D. melanogaster orthologues β-Tub85D (CG9359; FBgn0003889) and CG9222 (FBgn0031784), and groups with the β-Tub85D gene highly expressed in the D. melanogaster testis (Supplementary Fig. S1). Rad50 forms a dimer with Mre11 nuclease and is required for dsDNA break repair, telomere maintenance, and ataxia telangiectasia mutated kinase checkpoint signaling40. TC015093, referred to here as Tc-rad50, is the only rad50 gene in the T. castaneum genome, and encodes a 1:1 orthologue of the vertebrate Rad50 protein (Supplementary Fig. S2, Supplementary Table S4). Enolase metallo-enzyme is responsible for the conversion of 2-phosphoglycerate into phosphoenolpyruvate, the second to last step in glycolysis process41. Unlike Drosophila, which encodes a single enolase gene, three enolase orthologues are present in T. castaneum (Supplementary Fig. S3, Supplementary Table S5), though only TC011729, which we refer to as Tc-eno, was strongly expressed in the testes39.
To confirm the expression pattern of Tc-β2t, Tc-rad50 and Tc-eno as testis-enriched, we performed reverse transcriptase PCR (RT-PCR) on total mRNA extracted from dissected adult tissues of T. castaneum (Fig. 1). PCR based analysis showed that Tc-β2t, Tc-rad50 and Tc-eno transcripts could be found only in male testes and were not detectable in any other tested tissues such as female ovaries and both male and female carcasses (Fig. 1). Based on the tissue-restricted expression and amenable gene structure, potential cis-regulatory regions from the Tc-β2t, Tc-rad50 and Tc-eno loci were selected for generating transposon-based transformation vectors.

Tc-β2t, Tc-rad50 and Tc-eno are expressed specifically in the testes of T. castaneum. RT-PCR-based transcript analysis of cDNA prepared from white-eye mutant T. castaneum adults. T. castaneum RpSL32 housekeeping gene was used to confirm the quality of each cDNA. Lane (M) indicates 50 bp DNA Ladder Catalog No. (N3236S) New England Biolabs.
PiggyBac-based transformation of candidate regulatory sequences into the T. castaneum genome
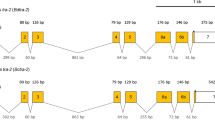
To determine if genomic sequences derived from Tc-β2t, Tc-rad50 and Tc-eno could drive the expression of an EGFP reporter gene specifically in the testis we constructed three independent transformation plasmids based on the piggyBac transposon (Fig. 2). In all cases, genomic fragments corresponding to the entire genomic region upstream and downstream of the respective ORF were cloned upstream/downstream of the selected reporter gene (tTA-P2A-EGFP, Supplementary Table S6), bounded only by the sequence coding for the ORF of each neighboring gene (Fig. 2).

Schematic diagram of Tc-β2t, Tc-rad50 and Tc-eno loci and corresponding transformation constructs. Schematic diagrams of the Tc-β2t genomic locus and piggyBac-β2t donor plasmid (A), the Tc-rad50 locus and piggyBac-Rad50 donor plasmid (B), and the Tc-eno locus and piggyBac-Eno donor plasmid (C). All three constructs contained the 3XP3-DsRED cassette as a visual marker and piggyBac inverted repeats (RH and L) for transposase mediated integration in T. castaneum genome. tTA adv and EGFP were separated by the P2A site in all tested plasmids. P, putative promoter region; T, putative terminator region. Dotted lines indicate the upstream and downstream genomic regions used as promoter and terminator respectively to drive the tTA-P2A-EGFP expression in vector construction.
In addition, all constructs contained the 3xP3-DsRED cassette to serve as a visual marker for transformation and piggyBac inverted repeats for transposase mediated integration into the T. castaneum genome. As eye-specific DsRED expression was anticipated to be difficult to detect in wild-type beetles, we performed germline transformation in a white-eyed T. castaneum strain (Supplementary Fig. S4). Embryos from the white-eyed T. castaneum strain were injected using an hsp70-driven transposase helper plasmid42 in conjunction with each of the three piggyBac donor plasmids. In each case, transgenic founder events were recovered (Table 1).
Spatial and temporal expression of EGFP in Tc-β2t-EGFP, Tc-Rad50-EGFP and Tc-Eno-EGFP lines
Three independent transgenic T. castaneum lines, which we refer to as Tc-β2t-EGFP#1, Tc-β2t-EGFP#2 and Tc-β2t-EGFP#3 were produced following injection with piggyBac-β2t. Male and female beetles were separated based on black spots on the first pair of legs of male adults which are absent in females43. While some autofluorescence was visible in the beetle eyes in the green channel (Supplementary Fig. S5A), EGFP was visible only in the abdomen of male adults (Fig. 3). DsRED fluorescence was detected in the insect compound eyes, regardless of the beetle sex, while EGFP fluorescence was detectable exclusively in the male gonad (Fig. 4). This tissue specific expression pattern of EGFP in dissected testes from male adults from the Tc-β2t-EGFP line was similar to that reported for β2-tubulin in other insects12,23,44. As was expected, we did not detect EGFP in the ovaries of transgenic beetles in any Tc-β2t-EGFP lines (Supplementary Fig. S6). While reliable identification of EGFP expression in adult male beetles was feasible, we were not able to observe EGFP in pupae or late stage larvae due to auto-fluorescence (Supplementary Fig. S5B) for any Tc-β2t-EGFP lines. This is different from reports in mosquitoes, where detection of the reporter protein through the body wall was obvious in all developmental stages12,44.

Fluorescent microphotographs showing sex-specific β2t drive EGFP expression in T. castaneum. (A) Transgenic adult male and female beetles viewed under bright field (B) DsRED filter and (C) GFP filter. Arrows indicate the abdomen where EGFP expression respectively was expected for transgenic male beetles in Tc-β2t-EGFP lines in gfp field.

Tc-β2t cis-regulatory regions drive EGFP expression in testes. (A) Testes were dissected from Tc-β2t-EGFP#2 (top right) and white eye mutant (lower left) beetles and viewed under bright field, EGFP or DsRED filters.
Three independent transgenic T. castaneum lines were generated by using the piggyBac-Rad50 construct, Tc-Rad50-EGFP#1, Tc-Rad50-EGFP#2 and Tc-Rad50-EGFP#3. Once again, EGFP was detectable only in the male gonads in beetles from Tc-Rad50-EGFP lines (Fig. 5), and we did not detect EGFP fluorescence in ovaries from transgenic females (Supplementary Fig. S7). Unlike the Tc-β2t-EGFP lines, EGFP was not detectable in whole adults from Tc-Rad50-EGFP lines, while the marker gene DsRED was visible in the eyes at all developmental stages, irrespective of sex. Interestingly, the pattern of EGFP fluorescence was unique in Tc-Rad50-EGFP testes as compared to Tc-β2t-EGFP lines. The EGFP in these beetles appeared less intense as compared to the testes from Tc-β2t-EGFP beetles, and was not as widely distributed along the spherical shape of the testes (Fig. 5). For both Tc-β2t-EGFP and Tc-Rad50-EGFP constructs, the pattern and specificity of EGFP expression was similar for separate transgenic events, with EGFP fluorescence detectable only in the gonads of males (Supplementary Fig. S8).

Tc-rad50 regulatory regions drive EGFP expression in testes. (A) Testes from transgenic beetles (upper right) and non-transgenic beetles (lower left) viewed under bright field, EGFP or DsRED filters. (B) Adult transgenic (left) and non-transgenic (right) viewed under bright field, EGFP or DsRED filters. Arrows indicate the eyes and testes where DsRED and EGFP expression, respectively, were expected for transgenic Tc-Rad50-EGFP#1 line beetles.
Finally, a single transgenic T. castaneum line was generated using the piggyBac-Enolase construct; Tc-Eno-EGFP#1. In all tested beetles, the EGFP was detectable from dissected transgenic male adults, corresponding to the region where spermatogenesis45 is completed. As in adult beetles from the Tc-Rad50-EGFP lines, EGFP was detectable only in the male dissected gonad (Fig. 6). The intensity of EGFP from dissected testes appeared to be less than that observed for Tc-β2t-EGFP and Tc-Rad50-EGFP lines, and no fluorescence was detected in testes and ovaries collected from wild-type beetles. However, we interpret these data with caution as only a single line was developed.

Tc-eno regulatory regions drive EGFP expression in testes. (A) Testes from transgenic beetles (upper right) and non-transgenic beetles (lower left) viewed under bright field, EGFP or DsRED filters. (B) Adult transgenic (left) and non-transgenic (right) viewed under bright field, EGFP or DsRED filters. Arrows indicate the eyes and testes where DsRED and EGFP expression respectively were expected for transgenic Tc-Eno-EGFP#1 line beetles.
In addition to measuring EGFP fluorescence, the expression of EGFP transcripts was examined by RT-PCR in transgenic beetles. In all cases, EGFP transcripts were detectable in the gonads of males, but not in ovaries, or male and female carcasses (Fig. 7), mimicking the expression pattern of the endogenous Tc-β2t, Tc-rad50 and Tc-eno loci in T. castaneum male testes (Fig. 1).

Transcription of the EGFP reporter in transgenic strains is testes-specific. cDNA from Tc-β2t-EGFP#2, Tc-Rad50-EGFP#1 and Tc-Eno-EGFP#1 transgenic lines was used to perform the RT-PCR. T. castaneum RpSL32 served as a control for cDNA quality (same samples as shown in Fig. 1). Lane#M is 50 bp DNA Ladder Catalog No. (N3236S) New England Biolabs.
To further analyze the expression pattern of the fluorescent reporter within the male beetle gonad, confocal microscopic analyses were performed on dissected testes from transgenic beetles from Tc-β2t-EGFP, Tc-Rad50-EGFP and Tc-Eno-EGFP lines. Dissected testes from Tc-β2t-EGFP individuals confirmed a very strong and widespread distribution of EGFP fluorescence along the longitudinal axis, ranging from the gonial (primitive germ cells) amplification stages, developing spermatocytes, spermatids, and spermatozoa45 (Fig. 8), up to individual mature sperm cells (Fig. 9). However, EGFP was not detectable in the apical tip of the testes (Fig. 8), indicating that the cloned cis-regulatory regions from the Tc-β2t locus in piggyBac-β2t plasmid did not direct EGFP expression in hub cells, male germline stem cells and somatic stem cells.

Confocal analysis of EGFP expression in transgenic male gonad. Expression of EGFP from transgenic males in whole testes and close up of single spherical testis dissected from Tc-β2t-EGFP#2, Tc-Rad50-EGFP#1, and Tc-Eno-EGFP#1 beetles. Black circle around the area in the whole testis corresponds to the close up image. Scale bar indicates 50 µm.

Tc-β2t drives EGFP expression in mature sperm. Sperm from testis of Tc-β2t-EGFP#2 viewed under EGFP, DsRED and Bright field filters. Green fluorescence can be visualized among single sperm as indicated by arrows. Scale bar indicates 50 µm.
In Tc-Rad50-EGFP beetles, EGFP fluorescence was detected only in elongated spermatid cells (Fig. 8). EGFP was not detectable in all stages of spermatogenesis including the germ stem cells in the apical tip, thus indicating that the cis-regulatory region from the Tc-rad50 locus was active specifically in spermatid cells during spermatogenesis (Figs. 8, 10). Below the elongated spermatid cells, round spermatogonia, spermatocytes and germ stem cells, presumably the somatic stem cells surrounding the germ stem cells did not reveal any EGFP fluorescence in dissected testes from Tc-Rad50-EGFP male testes. No EGFP was detected in mature sperm from the beetles in Tc-Rad50-EGFP beetles again supporting the idea that the cis-regulatory regions derived from the Tc-rad50 gene regulated EGFP expression in the T. castaneum testes differently than cis-regulatory regions from the Tc-β2t locus (Figs. 8, 9, 10). Similarly, testis in Tc-Eno-EGFP beetles were also examined in confocal microscopic analysis where we observed that EGFP fluorescence was entirely localized to cells transformed from spermatids into spermatozoa by the process of spermiogenesis (Figs. 8, 10). As in Tc-Rad50-EGFP beetles, Tc-Eno-EGFP was not detectable in mature sperm. No EGFP fluorescence was detected in the testes and ovaries from the wild-type black eye or untransformed white-eyed beetles (Supplementary Fig. S9).

Diagram of the organization of a T. castaneum testis. The stem cell niche which is maintained by Stromal hub cells adheres to the apical tip of the testis. Hub cells are surrounded by germ line stem cells and somatic stem cells. Germ cells differentiate into spermatogonia and begin the development of spermatogenesis process to become mature sperm. Relative expression patterns of Tc-β2t, Tc-eno and Tc-rad50 are noted.
Insertion site detection
Insertion junctions for each line were subsequently determined using inverse PCR (iPCR)38 (Fig. 11). Sequences analysis confirmed that all insertions terminate correctly with expected piggyBac inverted repeats and that all are flanked by the normal piggyBac (TTAA) target sequence. Transgenes insertion were further confirmed by direct amplification of genomic DNA using Phusion polymerase (NEB) along with one primer located within the 3′ UTR of the respective transgenic construct, with the other located in the genomic DNA of host T. castaneum. We conclude that each element did indeed integrate into unique locations in the T. castaneum genome.

Inverse PCR strategies to isolate and sequence the piggyBac vector and insertion sites in different transgenic lines. (A) Schematic diagram (not to scale) of the piggBac vector insertion in the T. castaneum genome. The vector contained the marker cassette as a visual marker and piggyBac inverted repeats (RH and L) for transposase mediated integration in T. castaneum genome. P putative promoter region, T putative terminator region. Duplicated TTAA flanked the piggyBac elements in the host genome. (B) Below are shown the TTAA position in the T. castaneum genome where the transgenes were inserted and the flanked piggyBac transgenes insertion site sequences from the transgenic lines.
Discussion
Here, we report on the development of transgenic strains expressing a fluorescent marker specifically in the male gonads of T. castaneum, a model for coleopterans and an important pest of stored grain. While several promoters have been characterized from or for T. castaneum to control transgene expression, these have been restricted to activity only in embryos/muscles such as twist46, caudal47, hunchback47, nubbin32, hairy48, and tailless49, hsp6850 or lack tissue-specificity as with the constitutive promoter Polyubiquitin51,52. In this report we show that cis-regulatory regions derived from three different T. castaneum genes were capable of controlling transgene expression specific to the testes.
The β2-tubulin promoter has been successfully used in other insects for transgenic male sexing. In Drosophila melanogaster and mosquitos, β2-tubulin transcripts are detectable in the male gonads from late larval developmental stages throughout later stages of sperm development44,53. In Drosophila, this gene is transcribed in late third larval instar before the onset of meiosis in the developing testis and remains active throughout adulthood54. Low level expression of β-Tub85D was also reported in other tissues in the fly such as in adult carcass and larval fat body55. In A. aegypti fourth instar larvae and pupae were easily scored as positive or negative for DsRED driven by the β2-tubulin promoter, these were confirmed as males upon adult emergence12. Similarly, in An. stephensi, EGFP driven by β2-tubulin promoter was used in automated sex sorting. Mosquitoes were separated during the larval stages and all larvae identified with green fluorescence phenotypes developed into males, while all larvae lacking EGFP were confirmed as female44. Like mosquitoes12,44, in the beetle we also found that EGFP expressed by the Tc-β2t regulatory regions could be detected through the body wall in male adults. The male gonad specific expression of EGFP under the tight control of the Tc-β2t cis regulatory region in T. castaneum provides an efficient, male-specific marker that can be used for sex sorting. In dissected testes, Tc-β2t controlled EGFP signals were not observed until spermatogonia reached the primary spermatocyte stage, with EGFP remaining present upon completion of spermatogenesis and in mature sperm.
Rad50 plays a key role in double stranded DNA break repair40,56, and to our knowledge our work represents the first use of a rad50 promoter to drive transgene expression in insects. Interestingly, low Rad50 expression was linked with spermatogenic failure in humans57, suggesting a potential conserved role in this process. In spermatogenesis, meiotic cell division is a vital step during which diploid spermatocytes generate haploid spermatids. This process is initiated by the formation of DNA double-strand breaks at specific sites58, which may need to be repaired using a complex containing of certain proteins such as MRE11, Rad50, ATM, NBS1 and Rad5159. Though meiotic cell division also takes place in eggs, Rad50 is not expressed in the ovary, suggesting other repair complexes may be dominant in the female germline. While much additional work is required to evaluate the role of DNA repair in beetle gonadal development, in our study we were unable to detect Tc-rad50 driven EGFP in dissected ovaries. Like in B. mori where the Bmβ4-promoter was found to drive EGFP expression only in the microtubule of testes13, similarly the Tc-rad50 cis-regulatory region expressed EGFP in dissected testes but we did not observe EGFP signals in mature sperm.
The structure of Drosophila enolase has been characterized and the mature protein forms a homodimer with conserved residues at the dimer interface60. Fly enolase has an open conformation in its structure and has conserved residue elements for catalytic activity60. Enolase contains conserved key amino acid residues for metal binding (magnesium ion binding) and substrate binding (phosphopyruvate hydratase activity). The fly genome encodes one enolase and the B. mori genome has two enolase orthologues, in which one was shown to have high testis expression61. T. castaneum encodes three enolase genes, in which only the Tc-eno used in this study has high expression in testes39. The pattern of EGFP expression from the Tc-eno regulatory regions in beetle testes was distinct from the other two tested regulatory elements. While Tc-eno EGFP was not observed in mature sperm, the endogenous gene product may assist with providing energy to fuel sperm mobility.
Our investigation evaluating the Tc-β2t, Tc-rad50 and Tc-eno loci cis-regulatory regions for male gonad expression and function in the T. castaneum model system and could be extended to other related insects. While the β2t promoter12,13,15,62 is well studied in other insects, here we report the rad50 and eno based reporter gene expression in insect testis for the first time. EGFP fluorescence was readily detected in Tc-β2t adult beetles and could potentially be used for non-lethal approaches in sex separation in the adult stage. In our experiments, EGFP expression alone could not be used as a marker to predict sex in beetles at the larval and pupal stages. This complicates the use of simple reporter constructs as presented here from being used for non-lethal approaches in sex separation in early developmental stages as seen for the β2-tubulin promoter in Drosophila and different mosquitos’ species12,44,53. It is possible that this could be overcome with the use of alternative reporters and/or using filters sets that minimize autofluorescence. β4-Tubulin in transgenic silkworm also drives EGFP expression in testis from late stage larvae to adult stage13, however, these authors did not report if the EGFP signal from transgenic animals could be detected through non-lethal approaches, as was reported in mosquitos12,44. The EGFP pattern driven by Bmβ4p was different from the Tc-β2t, where EGFP was detectable in the microtubules of testis from the dissected late larval to adult stages13. All of our transgenic strains were designed to also express the tetracycline transactivator (tTA), in addition to EGFP, through the use of the P2A viral sequence. While have not yet analyzed these strains for their ability to drive the expression of a gene under the control of tetO, this is a priority for future research, where tTA could activate the expression of a lethal gene8 or impair mating ability specifically in males. We note that male beetles from each strain remain fertile, so the level of tTA expression in the testes is considered below the threshold for potential strong toxicity. In conclusion, we have successfully established three male-specific reporter transgene systems in T. castaneum. The regulatory elements we characterized could be used for functional analysis of processes occurring in the testes in this and other related insects as well as for the development of transgenic strategies for adult male sexing in important agricultural pest species such as T. castaneum.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
Change history
28 October 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41598-021-01236-z
References
Pretty, J. & Bharucha, Z. P. Integrated pest management for sustainable intensification of agriculture in Asia and Africa. Insects 6, 152–182. https://doi.org/10.3390/insects6010152 (2015).
Klassen, W. & Curtis, C. F. History of the sterile insect technique. In Sterile Insect Technique: Principles and Practice in Area-Wide Integrated Pest Management (eds Dyck, V. A. et al.) 3–36 (Springer, 2005).
Papathanos, P. A. et al. Sex separation strategies: Past experience and new approaches. Malar. J. https://doi.org/10.1186/1475-2875-8-s2-s5 (2009).
Benedict, M. Q. & Robinson, A. S. The first releases of transgenic mosquitoes: An argument for the sterile insect technique. Trends Parasitol. 19, 349–355. https://doi.org/10.1016/s1471-4922(03)00144-2 (2003).
Alphey, L. et al. Sterile-insect methods for control of mosquito-borne diseases: An analysis. Vector-Borne Zoonotic Dis. 10, 295–311. https://doi.org/10.1089/vbz.2009.0014 (2010).
Kandul, N. P. et al. Transforming insect population control with precision guided sterile males with demonstration in flies. Nat. Commun. https://doi.org/10.1038/s41467-018-07964-7 (2019).
Ahmed, H. M. M., Hildebrand, L. & Wimmer, E. A. Improvement and use of CRISPR/Cas9 to engineer a sperm-marking strain for the invasive fruit pest Drosophila suzukii. BMC Biotechnol. https://doi.org/10.1186/s12896-019-0588-5 (2019).
Yan, Y. & Scott, M. J. A transgenic embryonic sexing system for the Australian sheep blow fly Lucilia cuprina. Sci. Rep. https://doi.org/10.1038/srep16090 (2015).
Papathanos, P. A., Windbichler, N., Menichelli, M., Burt, A. & Crisanti, A. The vasa regulatory region mediates germline expression and maternal transmission of proteins in the malaria mosquito Anopheles gambiae: A versatile tool for genetic control strategies. Bmc Mol. Biol. https://doi.org/10.1186/1471-2199-10-65 (2009).
Schroder, R. vasa mRNA accumulates at the posterior pole during blastoderm formation in the flour beetle Tribolium castaneum. Dev. Genes Evol. 216, 277–283. https://doi.org/10.1007/s00427-005-0054-3 (2006).
Michiels, F., Gasch, A., Kaltschmidt, B. & Renkawitzpohl, R. A 14-bp promoter element directs the testis specificity of the Drosophila-beta-2 tubulin gene. Embo J. 8, 1559–1565. https://doi.org/10.1002/j.1460-2075.1989.tb03540.x (1989).
Smith, R. C., Walter, M. F., Hice, R. H., O’Brochta, D. A. & Atkinson, P. W. Testis-specific expression of the beta 2 tubulin promoter of Aedes aegypti and its application as a genetic sex-separation marker. Insect Mol. Biol. 16, 61–71. https://doi.org/10.1111/j.1365-2583.2006.00701.x (2007).
Xu, J. et al. Transgenic characterization of two testis-specific promoters in the silkworm, Bombyx mori. Insect Mol. Biol. 24, 183–190. https://doi.org/10.1111/imb.12144 (2015).
Schetelig, M. F. & Handler, A. M. A transgenic embryonic sexing system for Anastrepha suspensa (Diptera: Tephritidae). Insect Biochem. Mol. 42, 790–795. https://doi.org/10.1016/j.ibmb.2012.07.007 (2012).
Kemphues, K. J., Kaufman, T. C., Raff, R. A. & Raff, E. C. The testis specific beta tubulin subunit I Drosophila melanogaster has multiple functions in spermatogenesis. Cell 31, 655–670. https://doi.org/10.1016/0092-8674(82)90321-x (1982).
Fackenthal, J. D., Turner, F. R. & Raff, E. C. Tissue-specific microtubule functions in Drosophila spermatogenesis require the β2-tubulin isotype-specific carboxy terminus. Dev. Biol. 158, 213–227 (1983).
Kemphues, K. J., Raff, E. C., Raff, R. A. & Kaufman, T. C. Mutation in a testis-specific β-tubulin in Drosophila: Analysis of its effects on meiosis and map location of the gene. Cell 21, 445–451. https://doi.org/10.1016/0092-8674(80)90481-x (1980).
Whitworth, C., Jimenez, E. & Van Doren, M. Development of sexual dimorphism in the Drosophila testis. Spermatogenesis 2, 129–136 (2012).
Carroll, S. B. Evo-devo and an expanding evolutionary synthesis: A genetic theory of morphological evolution. Cell 134, 25–36. https://doi.org/10.1016/j.cell.2008.06.030 (2008).
Yamamoto, D. S. et al. A synthetic male-specific sterilization system using the mammalian pro-apoptotic factor in a malaria vector mosquito. Sci. Rep. https://doi.org/10.1038/s41598-019-44480-0 (2019).
Harvey-Samuel, T., Ant, T. & Alphey, L. Towards the genetic control of invasive species. Biol. Invas. 19, 1683–1703. https://doi.org/10.1007/s10530-017-1384-6 (2017).
Scott, M. J., Concha, C., Welch, J. B., Phillips, P. L. & Skoda, S. R. Review of research advances in the screwworm eradication program over the past 25 years. Entomol. Exp. Appl. 164, 226–236. https://doi.org/10.1111/eea.12607 (2017).
Scolari, F. et al. Fluorescent sperm marking to improve the fight against the pest insect Ceratitis capitata (Wiedemann; Diptera: Tephritidae). New Biotechnol. 25, 76–84. https://doi.org/10.1016/j.nbt.2008.02.001 (2008).
Scolari, F. et al. Polyandry in the medfly—Shifts in paternity mediated by sperm stratification and mixing. BMC Genet. 15, S10. https://doi.org/10.1186/1471-2156-15-s2-s10 (2014).
Hunt, T. et al. A comprehensive phylogeny of beetles reveals the evolutionary origins of a superradiation. Science 318, 1913–1916. https://doi.org/10.1126/science.1146954 (2007).
Slipinski, S. A., Leschen, R. A. B. & Lawrence, J. F. Animal biodiversity: An outline of higher-level classification and survey of taxonomic richness. Zootaxa 3148, 1–237 (2011).
Herndon, N. et al. Enhanced genome assembly and a new official gene set for Tribolium castaneum. BMC Genomics. https://doi.org/10.1186/s12864-019-6394-6 (2020).
Richards, S. et al. The genome of the model beetle and pest Tribolium castaneum. Nature 452, 949–955. https://doi.org/10.1038/nature06784 (2008).
Doenitz, J., Gerischer, L., Hahnke, S., Pfeiffer, S. & Bucher, G. Expanded and updated data and a query pipeline for iBeetle-Base. Nucleic Acids Res. 46, D831–D835. https://doi.org/10.1093/nar/gkx984 (2018).
Schmitt-Engel, C. et al. The iBeetle large-scale RNAi screen reveals gene functions for insect development and physiology. Nat. Commun. https://doi.org/10.1038/ncomms8822 (2015).
Johnathan, C. et al. Expanding the genetic toolkit of Tribolium castaneum. PLoS ONE 13, e0195977. https://doi.org/10.1371/journal.pone.0195977 (2018).
Lai, Y. T. et al. Enhancer identification and activity evaluation in the red flour beetle, Tribolium castaneum. Development. https://doi.org/10.1242/dev.160663 (2018).
Gilles, A. F., Schinko, J. B. & Averof, M. Efficient CRISPR-mediated gene targeting and transgene replacement in the beetle Tribolium castaneum. Development 142, 2832. https://doi.org/10.1242/dev.125054 (2015).
Brown, S. J. et al. The red flour beetle, Tribolium castaneum (Coleoptera): a model for studies of development and pest biology. Cold Spring Harb. Protoc. 2009(8), pdb.emo126. https://doi.org/10.1101/pdb.emo126 (2009).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343. https://doi.org/10.1038/nmeth.1318 (2009).
Horn, C. & Wimmer, E. A. A versatile vector set for animal transgenesis. Dev. Genes. Evol. 210, 630–637. https://doi.org/10.1007/s004270000110 (2000).
Handler, A. M. & Harrell, R. A. Germline transformation of Drosophila melanogaster with the piggyBac transposon vector. Insect Mol. Biol. 8, 449–457. https://doi.org/10.1046/j.1365-2583.1999.00139.x (1999).
Ochman, H., Gerber, A. S. & Hartl, D. L. Genetic applications of an inverse polymerase chain reaction. Genetics 120, 621–623 (1988).
Khan, S. A., Eggleston, H., Myles, K. M. & Adelman, Z. N. Differentially and co-expressed genes in embryo, germ-line and somatic tissues of Tribolium castaneum. G3: Genes Genomes Genet. 9, 2363. https://doi.org/10.1534/g3.119.400340 (2019).
Williams, G. J. et al. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat. Struct. Mol. Biol. 18, 423–431 (2011).
Gerlt, J. A., Babbitt, P. C. & Rayment, I. Divergent evolution in the enolase superfamily: The interplay of mechanism and specificity. Arch. Biochem. Biophys. 433, 59–70. https://doi.org/10.1016/j.abb.2004.07.034 (2005).
Lorenzen, M. D. et al. piggyBac-mediated germline transformation in the beetle Tribolium castaneum. Insect Mol. Biol. 12, 433–440. https://doi.org/10.1046/j.1365-2583.2003.00427.x (2003).
Beeman, R. W., Haas, S. & Friesen, K. Stored Product Insect and Engineering Research (2019). http://www.ars.usda.gov/Research/docs.htm?docid=12892. Accessed 15 May 2021.
Catteruccia, F., Benton, J. P. & Crisanti, A. An Anopheles transgenic sexing strain for vector control. Nat. Biotechnol. 23, 1414–1417. https://doi.org/10.1038/nbt1152 (2005).
Salazar, K., Dias, G., Boucher, S., Lino-Neto, J. & Serrao, J. E. Morpho-anatomy of the male reproductive tract and spermatogenesis of the South American Spasalus silvarum Kuwert (Coleoptera: Passalidae). Zoomorphology 135, 487–497. https://doi.org/10.1007/s00435-016-0321-z (2016).
Handel, K., Basal, A., Fan, X. & Roth, S. Tribolium castaneum twist: Gastrulation and mesoderm formation in a short-germ beetle. Dev. Genes Evol. 215, 13–31. https://doi.org/10.1007/s00427-004-0446-9 (2005).
Wolff, C., Schroder, R., Schulz, C., Tautz, D. & Klingler, M. Regulation of the Tribolium homologues of caudal and hunchback in Drosophila: Evidence for maternal gradient systems in a short germ embryo. Development 125, 3645–3654 (1998).
Eckert, C., Aranda, M., Wolff, C. & Tautz, D. Separable stripe enhancer elements for the pair-rule gene hairy in the beetle Tribolium. Embo Rep. 5, 638–642. https://doi.org/10.1038/sj.embor.7400148 (2004).
Schroder, R., Eckert, C., Wolff, C. & Tautz, D. Conserved and divergent aspects of terminal patterning in the beetle Tribolium castaneum. Proc. Natl. Acad. Sci. U.S.A. 97, 6591–6596. https://doi.org/10.1073/pnas.100005497 (2000).
Schinko, J. B., Hillebrand, K. & Bucher, G. Heat shock-mediated misexpression of genes in the beetle Tribolium castaneum. Dev. Genes Evol. 222, 287–298. https://doi.org/10.1007/s00427-012-0412-x (2012).
Siebert, K. S., Lorenzen, M. D., Brown, S. J., Park, Y. & Beeman, R. W. Tubulin superfamily genes in Tribolium castaneum and the use of a Tubulin promoter to drive transgene expression. Insect Biochem. Mol. 38, 749–755. https://doi.org/10.1016/j.ibmb.2008.04.007 (2008).
Lorenzen, M. D., Brown, S. J., Denell, R. E. & Beeman, R. W. Transgene expression from the Tribolium castaneum Polyubiquitin promoter. Insect Mol. Biol. 11, 399–407. https://doi.org/10.1046/j.1365-2583.2002.00349.x (2002).
Kemphues, K. J., Raff, E. C., Raff, R. A. & Kaufman, T. C. Mutation in a Testis-specific beta tubulin in Drosophila analysis of its effect of meiosis and map location of gene. Cell 21, 445–451. https://doi.org/10.1016/0092-8674(80)90481-x (1980).
Kemphues, K. J., Kaufman, T. C., Raff, R. A. & Raff, E. C. The testis specific beta tubulin subunit in Drosophila melanogaster has multiple functions in spermatogenesis. Cell 31, 655–670. https://doi.org/10.1016/0092-8674(82)90321-x (1982).
Gelbart, W. M. & Emmert, D. B. FlyBase High Throughput Expression Pattern Data (2013).
Hopfner, K. P. et al. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell 101, 789–800. https://doi.org/10.1016/s0092-8674(00)80890-9 (2000).
Hu, M. H. et al. Decreased expression of MRE11 and RAD50 in testes from humans with spermatogenic failure. J. Assist. Reprod. Genet. 37, 331–340. https://doi.org/10.1007/s10815-019-01686-5 (2020).
Lange, J. et al. The landscape of mouse meiotic double-strand break formation, processing, and repair. Cell 167, 695. https://doi.org/10.1016/j.cell.2016.09.035 (2016).
Gunes, S., Al-Sadaan, M. & Agarwal, A. Spermatogenesis, DNA damage and DNA repair mechanisms in male infertility. Reprod. Biomed. Online 31, 309–319. https://doi.org/10.1016/j.rbmo.2015.06.010 (2015).
Sun, C., Xu, B., Liu, X., Zhang, Z. & Su, Z. Crystal structure of enolase from Drosophila melanogaster. Acta Crystallogr. Sect. F Struct. Biol. Commun. 73, 228–234. https://doi.org/10.1107/s2053230x17004022 (2017).
Kikuchi, A. et al. Identification of functional enolase genes of the silkworm Bombyx mori from public databases with a combination of dry and wet bench processes. BMC Genomics. https://doi.org/10.1186/s12864-016-3455-y (2017).
Yan, Y., Schwirz, J. & Schetelig, M. F. Characterization of the Drosophila suzukii β2-tubulin gene and the utilization of its promoter to monitor sex separation and insemination. Gene 771, 145366. https://doi.org/10.1016/j.gene.2020.145366 (2021).
Acknowledgements
This work was supported by the Defense Advanced Research Projects Agency Project Number HR0011-16-2-0036 and the BRAG program at NIFA under Award Number 2019-33522-30063, as well as by Agrilife Research and the Department of Entomology at Texas A&M University. We thank Collin Valentin for assistance in insect rearing. The use of the Microscopy and imaging Center facility at Texas A&M University is acknowledged. The Olympus FV1000 confocal microscope acquisition was supported by the Office of the Vice President for Research at Texas A&M University.
Author information
Authors and Affiliations
Contributions
S.K. and Z.A. conceived and designed the analysis. S.K. and E.J. performed experiments and collected the data. S.K., E.J., Z.A. and K.M. analyzed the data. S.K. drafted the manuscript. E.J., Z.A. and K.M. edited the manuscript. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained errors in the Figure legends of Figure 3 and Figure 4. The legends of these Figures were inadvertently switched.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khan, S.A., Jakes, E., Myles, K.M. et al. The β2Tubulin, Rad50-ATPase and enolase cis-regulatory regions mediate male germline expression in Tribolium castaneum. Sci Rep 11, 18131 (2021). https://doi.org/10.1038/s41598-021-97443-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-97443-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
