
Abstract
Congenital heart defects, one of the most common birth defects, affect approximately 1% of live birth globally and remain the leading cause of infant mortality in developed countries. Utilizing the pathogenicity score and inheritance mode from whole exome sequencing results, a heterozygous mutation (NM_001278939.1: c.1939G>T, p.Gly647Ter) in elastin (ELN) was identified among 6,440 variants in a female proband born with an atrial septal defect accompanied by pulmonary artery stenosis. Results of RT-PCR showed that the mutation (NM_001278939.1: c.1939G>T, p.Gly647Ter) did not affect the expression levels of ELN mRNA but increased protein level. The content of ELN truncate (functional component) was significantly lower in both the intracellular and extracellular compartments after mutation. These results indicate that the ELN mutation (NM_001278939.1: c.1939G>T, p.Gly647Ter) affected the protein truncate, which may be a functional component of ELN and play crucial roles for this pedigree. Here we report of an ELN heterozygous variant associated with congenital heart disease accompanied with pulmonary artery stenosis, which is less common. Based on our results, we speculate that this may be the main molecular mechanism underlying the mutation-led functional changes, and propose that the decrease of ELN protein level may cause this pedigree vascular abnormality, especially pulmonary artery stenosis, and reinforce the view that ELN insufficiency is the primary cause of these vascular lesions. This may be the main molecular mechanism underlying the mutation-led functional changes. Thus, systematic analysis not only enables us to better understand the etiology of this disease but also contributes to clinical and prenatal diagnosis.
Similar content being viewed by others


Introduction
Congenital heart defects (CHDs), one of the most common birth defects, affect approximately 1% of live birth globally and remain the leading cause of infant mortality in developed countries1. It has been reported that a strong genetic component, environmental causes, and familial forms play crucial roles in CHDs2. It has been estimated that de novo mutations in more than 400 genes likely contribute to ~ 10% of CHDs3, while chromosomal abnormalities are implicated in over 20% cases4. Heart failure is the most frequent complication of CHDs. With improved medical management and surgical outcomes, survival in cases of complex CHDs has improved significantly. However, many CHDs or complications with unknown etiology require urgent and targeted treatments.
Pulmonary artery stenosis is a narrowing (stenosis) that occurs in the pulmonary artery, which sends oxygen-poor blood into the lungs to be enriched with oxygen. There are four types of pulmonary artery stenosis: type I, type II, type III, and type IV5. CHDs, such as atrial septal defects, are sometimes accompanied with pulmonary artery stenosis. CHDs are mainly caused by a combination of genetic and environmental factors. Williams syndrome, is a rare hereditary disorder that affects many parts of the body, and it is most often due to deletions on chromosome 7q11.23. According to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar) and the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk), more than 100 pathogenic or presumed pathogenic variants of the Elastin (ELN) gene have been described6. The ELN gene sequence and its protein structure were first reported in 19857 and amended in 19918. Human ELN gene encodes for 786 amino acids and is composed of 34 coding exomes. It is well recognized that mutations in a functional domain or a protein translation modification site can alter protein function or protein–protein interaction9. ELN is the major structural protein of tissues such as the aorta and nuchal ligament, which must expand rapidly and recover completely. The ELN includes one conserved domain, Sporozoite_P67. The structure and function of ELN are not fully understood. The molecular defects in ELN have mainly been described in three conditions: supravalvular aortic stenosis10, autosomal-dominant cutis laxa11, and Williams syndrome6; it has rarely been reported ELN mutation in CHDs accompanied with pulmonary artery stenosis.
In this study, we report a female proband diagnosed of atrial septal defect accompanied with pulmonary artery stenosis. Whole exome sequencing (WES) was performed to identify possible disease-causing genes or variants. Paired-end reading was aligned with the GRCh37/hg19 human reference sequence. Through comprehensive Clinvar software and GATK analysis, BAM and VCF files were produced. An interesting heterozygous mutation of ELN (NM_001278939.1: c.1939G>T, p.Gly647Ter), identified in this female infant, is discussed along with the possible mechanism of gene mutation. Our results indicate that ELN mutation may be involved in CHDs accompanied with pulmonary artery stenosis and should be screened in prospective clinical practice.
Materials and methods
Clinical information and Ethics
A female infant was born with an atrial septal defect (ASD). There was no fever, cyanosis, cough, vomiting, or diarrhea, and no special treatment was given at that time. Subsequently, regular follow-up and reexamination were conducted in the outpatient department of our hospital. No signs or symptoms were identified during physical examinations. Ethics documentation of this case was waived with the approval of the Shanghai Children’s Hospital Institutional Review Board. The parents of the patient provided written informed consent for publication. All of the following studies were performed in accordance with the guidelines and regulations of the Ethics Committee of Experimental Research of Shanghai Children’s Hospital, Shanghai Jiaotong University.
WES
DNA library construction and WES assays were carried out in accordance with the manufacturer's instructions. Briefly, the genomic data of the patient and her parents were collected using a whole-blood genomic DNA extraction kit (Tiangen, China), and 1 µg DNA was used for the WES assay. The precise experimental procedures and experimental instruments are described in detail in our previous study12.
Sanger sequencing and mutation analysis
ELN mutation was confirmed via Sanger sequencing. Primers were designed using Primer 5 software to cover the known mutation sequence. The sequence of the forward primer was 5′-CCACTAGGAACTCCAGTTCTTC-3′, and that of the reverse primer was 5′-GGTCAGGCTGGTCTGGAACC-3′. PCR products were resolved and purified using the QIAquick kit (Qiagen, Germantown, MD USA). Sanger sequencing was carried out at Suzhou Hong Xun Biotechnology Co., Ltd.
The filtered and analyzed data are described in detail in our previous study 12. Briefly, data were filtered by self-developed software, and compared with the human genome database (GRCh 37/hg 19) using BWA-0.710 software. The single base mutation and insertion deletion mutation were identified, and compared with the 1,000 Genomes Project, Exome Aggregation Consortium databases, Exome Variant Server, gnomAD, Clinvar (http://www.ncbi.nlm.nih.gov/clinvar), OMIM, HGMD, and 370 samples of whole exome sequencing in our hospital. Variant filtration was performed after all variants were obtained, annotated, and assessed from the exome sequencing process. The mutation sites were evaluated with the COBALT homology alignment of amino acid sequences online tool among different species. The predicted effect of variants on protein function and conservation across species was assessed using SIFT, Polyphen-2, GERP (genomic evolutionary rate profiling), Mutation Taste, and combined annotation-dependent depletion. After identifying candidate genes, the frequency of the variant in the Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org) was reviewed. Rare variants for validation were polymerase chain reaction amplified. Illumina Variant Studio was used to filter the variants as per their frequency and presence or absence in the affected family members versus the healthy individuals.
Plasmid construction and transfection
A DNA fragment containing full-length ELN cDNA was obtained by PCR amplification. The primer sequences are listed in Table 1 (ELN-F, R). Plasmid pEGFP-C1 was purchased from Bioeagle Biotech Company, Ltd, Wuhan, China. Restriction sites and full-length ELN (besides of introne) were inserted into the pEGFP-C1 plasmid to construct the recombinant vector pEGFP-ELN-wt. Using site-directed mutation kit (Fast site-directed mutagenesis kit, Tiangen, Beijing), the ELN mutation site (NM_001278939.1: c.1939G>T, p.Gly647Ter) was introduced in the above recombination carrier to construct the recombination carrier pEGFP-ELN-mut. The primer sequences used in these experiments are listed in Table 1 (ELN-F1, R1). The above two recombination plasmids were confirmed by sequencing.
Human embryonic kidney 293T cells were purchased from the Cell Bank of the Chinese Academy of Sciences and maintained in a humidified incubator maintained at 37 °C and 5% CO2 atmosphere. 293T cells were transiently transformed with pEGFP-ELN-wt/mut plasmid using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Transfected 293T cells were cultured on a confocal dish, and the expression of pEGFP-ELN-wt/mut was examined using an inverted fluorescence microscope (Zeiss, Germany) with excitation at 488 nm and emission at 507 nm. Total RNA and proteins were extracted and verified by real-time-PCR (qPCR) and western blotting.
Real-time PCR (RT-PCR)
RT-PCR was used to detect the expression of ELN mRNA. Briefly, 293T cells were transiently transformed with pEGFP-ELN-wt/mut plasmids, and the RNA of the cells was extracted according to the manufacturer’s protocol (Takara, Japan). The primer sequences are listed in Table 1(ELN-qPCR-F, R).
Translation inhibition
A translation inhibitor cycloheximide (CHX) was used to detect the ELN translation13. Briefly, the constructed wild-type and mutant-type eukaryotic recombinant expression vectors were transiently transfected into human embryonic kidney 293 T cells then treated with cycloheximide (500 μM) for 0, 4, 8, and 24 h. Cell proteins were extracted and analyzed by western blotting.
Western blotting
Western blotting method was described in detail in our previous work14. Proteins were collected and quantified using the BCA reagent (Thermo Fisher Scientific, Waltham, MA, USA). The proteins were resolved on a sodium dodecyl sulfate 10% polyacrylamide gel, transferred onto a polyvinylidene fluoride membrane (Millipore, Bedford, MA, USA), and incubated with primary antibodies (1:1000 dilution) against ELN (ABclonal, Wuhan, China) and GFP (Cell Signaling Technology, Danvers, MA, USA) at 4 °C overnight. Blots were incubated with secondary antibodies for another hour at room temperature. After washing, the blots were visualized using a chemiluminescent substrate, and then analyzed by image J software.
Statistical analysis
Results are expressed as the mean ± SEM. Statistical analysis was performed using SPSS software, version 21.0 (SPSS, Inc., Chicago, IL, USA). Comparisons among groups were performed using one-way ANOVA. Paired data were evaluated by two-tailed Student’s t-test. Statistical significance was considered when P < 0.05.
Results
General clinical information and mutation characteristics
A 12-lead electrocardiogram (ECG) during the resting time showed sinus arrhythmia and minor hypertrophy in the right ventricle (Fig. 1A). Biochemical metabolism, myocardial enzyme, and cardiac computed tomography angiography were also performed and showed normal results. Further ultrasound cardiogram showed that the atrial septum was continuously interrupted, with a size of approximately 1.20 cm × 1.56 cm (Fig. 1B–E). The internal diameter of the right pulmonary artery opening was 1.14 cm, and the blood flow was 2.2 m/s. The internal diameter of the left pulmonary artery opening was 0.8 cm, the flow rate was 4.21 m/s, and the pressure difference was 71 mmHg (Fig. 1F–I). The infant showed symptoms of atrial septal defect and pulmonary artery stenosis.

12-lead ECG and ultrasonic cardiogram in the patient. (A) 12-lead ECG in resting time showing sinus arrhythmia; slight hypertrophy (RV5/SV1 = 1.51/0.00 mV) evident in the right ventricular. (B–I) Ultrasonic cardiogram of the patient. The red arrow indicates the site of an atrial septal defect in the left and right pulmonary arteries.
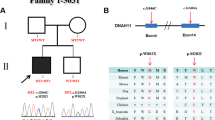
As shown in Fig. 2A,B, the outermost layer, 81,567 mutation sites, the total number of mutations detected from 20,999 genes, which was annotated and filtered by Ingenuity Variant Analysis (MAF < 0.05) according to the standards of the Exome Aggregation Consortium, 1,000 Genomes Project, Exome Sequencing Project, or gnomAD. In the second layer, 6,440 mutations were found in 4,497 genes screened for population frequency. In the third layer, 1,378 mutations were found in 1,315 genes at sites predicted to be pathogenic. In the fourth layer, 13 mutations were found in 5 genes, which were analyzed based on co-segregation of families. Fifth layer, 2 mutations occur in 2 genes (ELN and CHRNG), phenotypic related genetic variations. Lastly, we find that ELN was the most consistent with the phenotype, the ELN mutation was identified and selected after rigorous analysis linked to the infant phenotype (Fig. 2B). These rare phenotype-related variants were classified following the guidelines of the American College of Medical Genetics and Genomics/Association for Molecular Pathology.

Family pedigree and the filtering process for WES data. (A) Family pedigree consists of one proband. I-1 represents the proband’s father; I-2 represents the proband’s mother; II-1 represents the proband. (B) The filtering process for WES data, containing 81,567 total coding variants. Filtering results: 1315 deleterious variations, two variants selected from genetic analysis, final gene associated with this phenotype variation.
A heterozygous mutation of the ELN gene (NM_001278939.1: c.1939G>T, p.Gly647Ter) was associated with a CHD with pulmonary artery stenosis pedigree through WES. The mutation (NM_001278939.1: c.1939G>T, p.Gly647Ter) is located in exon 27 of ELN. The infant and her father were confirmed to be heterozygous carriers of 1939G>T (NM_001278939.1: c.1939G>T, p.Gly647Ter), and her mother was homozygous negative for the mutation as shown through Sanger sequencing (Fig. 3A,B).

The ELN mutation site and its conservation. (A) Human ELN gene maps to chromosome 7q11.23 and contains 34 exomes. The base pair mutation site is c.1939G>T, which is located on the exome 27 of ELN. (B) The proband and her father were confirmed to be heterozygous carriers of 1939G>T hybridization, and her mother was homozygous negative for this mutation, as shown by Sanger sequencing.
ELN mutation analysis
RT-PCR was utilized to detect expression of ELN mRNA. 200 µL of whole-blood sample was used to extract RNA according to protocol (PrimeScript™ RT Master Mix, takara). Primers were designed before and after the mutation to explore whether the mutation altered its expression. Before the mutation forward primer: 5′-AGCTAAAGCAGCAGCAAAGT-3′ and reverse primer: 5′-CTGCAGCAGCTCCATACTTG-3′. After the mutation forward primer: 5′-CTTGGAGTTCCAGGTGTTGG-3′ and reverse primer: 5′-TGGGAAAATGGGAGACAATC-3′. We find that ELN mRNA levels remained unchanged before and after mutation. The pEGFP-ELN-wt/mut sites are shown in Fig. 4A. Results of RT-PCR showed that ELN mRNA expression levels did not change after mutation, which suggests that the mutation did not affect ELN expression at the mRNA level (Fig. 4B). The results of the relative fluorescence intensity of GFP and the western blotting suggested that the mutation (NM_001278939.1: c.1939G>T, p.Gly647Ter) increased ELN protein levels. The pEGFP-ELN-wt group ELN staining indicated a molecular weight of approximately 113 kDa, whereas the pEGFP-ELN-mut group ELN staining showed a molecular weight of approximately 98 kDa (Fig. 4C,D). The RT-PCR and western blotting indicated that the mutation (NM_001278939.1: c.1939G>T, p.Gly647Ter) can lead to increasing intracellular ELN protein expression.

The mutation site (NM_001278939.1: c.1939G>T, p.Gly647Ter) affects ELN expression. (A) The pEGFP-ELN-wt/mut sites as showed through Sanger sequencing. (B) ELN mRNA levels before and after mutation. (C) Micrographs of fluorescence microscope images of ELN in 293 T. Scale bar, 200 µm. ELN was stained with green, the nucleus with blue (N = 3). (D) Immunoblot of GFP protein levels and quantification of ELN relative protein levels in 293 T. GAPDH served as loading control. Values are means ± SEM. *P < 0.05, **P < 0.01 was considered significant (N = 6).
After 4 h of treatment with CHX, the ELN expression level of the pEGFP-ELN-wt group was significantly reduced and was difficult to detect after 24 h. However, the degradation of the pEGFP-ELN-mut group was considerably slower, and the protein bands were visible even after 24 h (Fig. 5A,B). Different exposure times showed the relative quantity of ELN and ELN truncate. The band showing larger molecular weight indicates full-length expressed proteins, such as pEGFP-ELN-wt at 113 kDa and pEGFP-ELN-mut at 98 kDa. With the increase in exposure time, protein truncates (68 kDa and 53 kDa bands) were noted in the two groups, and these two truncated body proteins were expected to be the ELN truncates. The relative amount of ELN truncates (68 kDa and 53 kDa bands) decreased after introduction of the mutation (Fig. 5C). These results suggest that the mutation may affect the processing of ELN protein, which occurs before its exocytosis. Millipore Amicon ultra-4 10K was used to concentrate approximately 6 mL cell culture super plasma to 200 μL, after which the relative expression of ELN protein was again detected by western blotting. In these extracellular samples, no full-length ELN bands, namely 113 kDa and 98 kDa bands, were detected; only the ELN truncate, namely 68 kDa and 53 kDa bands, were found (Fig. 5D). The exocytosis of ELN in the pEGFP-ELN-wt group was significantly higher than that in the pEGFP-ELN-mut group (Fig. 5E).

Effects of CHX on ELN expression and its relative secreted expression. (A) Effects of CHX on ELN expression in transfected cells. Immunoblot of GFP protein levels between the pEGFP-ELN-wt and pEGFP-ELN-mut group after incubation with CHX (500 μM) for 0, 4, 8, and 24 h. (B) Quantification of relative ELN protein levels in transfected cells. (C) The relative quantity of ELN and its truncated body under different exposure times. (D) Immunoblot of secreted ELN protein levels. (E) Quantification of ELN shear body relative protein levels. Values are means ± SEM. *P < 0.05, **P < 0.01 was considered significant (N = 5).
Taken together, our results shown that the mutation (NM_001278939.1: c.1939G>T, p.Gly647Ter) did not affect the mRNA expression and that the expression level of ELN was significantly higher in mutated variants than that of the wild-type group. However, the content of ELN truncates (functional component) was significantly lower in both the intracellular and extracellular compartments than in the wild-type group.
Discussion
In this study, clinical phenotype and genotype of a mutated CHD with pulmonary artery stenosis pedigree were collected and analyzed to investigate a potential disease-causing variant. Three interesting findings are as follows: (1) The heterozygous mutation of ELN (NM_001278939.1: c.1939G>T, p.Gly647Ter) is found in this pedigree; (2) The mutation did not alter ELN gene mRNA levels, while increase its protein levels; (3) The content of ELN truncate (functional component) was significantly lower both in intracellular and extracellular than in the wild-type group. The ELN (NM_001278939.1: c.1939G>T, p.Gly647Ter) mutation could affect its functional active component expression, and may play a crucial role in this case.
It is well established that extensive heterogeneity of CHDs is due to a combination of genetic and environmental factors that serve as phenotype modulators. TBX5, NKX2.5, GATA4, ZIC, MYH6, NOTCH1, CRELD1, and FOG2 have been reported to be associated with CHDs15,16. Within the known non-syndromic CHD cases, the incidence of causative gene mutations is less than 7%, which hinders the understanding of the disease mechanism and the development of treatment strategies.
To explore the possibility of the role of functional effects caused by the ELN mutation (NM_001278939.1: c.1939G>T, p.Gly647Ter) on disease occurrence, we constructed both wild type and mutant plasmids of ELN and analyzed the ELN mRNA and protein levels. We found that the mutation site had no significant impact on its transcriptional level, while it significantly increased the protein levels (Fig. 4). The ELN heterozygous variant (NM_001278939.1: c.1939G>T, p.Gly647Ter), is located in the exon 27 and plays a vital role in modulating elastin. Humans are extremely sensitive to reduced ELN expression, and develop profound arterial thickening, which in turn markedly increases the risk of obstructive vascular disease17,18. Moreover, the aorta, septum, and pulmonary artery contains high levels of elastin, indicating that the ELN heterozygous variant may be one of the probable reasons for this pedigree.
A paradoxical increase in arterial elasticity along with abnormalities in elastic fibers was observed in patients with Williams–Beuren syndrome19. To investigate why a deletion mutation in the ELN gene causes an inherited obstructive arterial disease, such as supravalvular aortic stenosis, Li et al. generated a transgenic mouse (Eln+/−). They found that, during the arterial development, ELN hemizygosity in mice and humans induces a compensatory increase in the number of rings of elastic lamellae and smooth muscle. Humans are sensitive to reduced ELN expression, development to deep arterial thickening and significantly increased risk of obstructive vascular disease20. Micale Lucia et al. then used minigene and cycloheximide experiments to show that some selected frameshift mutant alleles (c.1161delC, c.838_839insG, c.1195delG) are the substrates of nonsense-mediated mRNA decay (NMD), which confirmed that the functional haploinsufficiency of the ELN gene is the main pathological mechanism of supravalvular aortic stenosis. Their results indicate the importance of screening for the ELN gene in patients with vascular abnormalities, especially SVAS and pulmonary artery stenosis, and reinforce the view that haploinsufficiency at ELN is the primary cause of these vascular lesions21.
In the present study, the mutation site increased ELN protein level (Fig. 4D), which is not normally seen in this pedigree (pulmonary artery stenosis). This proband is diagnosed with ASD accompanied with left and right pulmonary artery stenosis. It is well recognized that ELN mutation may act via impacting protein dosage and function, and may cause diseases such as supravalvular aortic stenosis10, autosomal-dominant cutis laxa11, and Williams syndrome6. In non-syndromic supravalvular aortic stenosis with a nonsense mutation, mRNA degradation of mutant alleles leads to a large numbers of premature termination codon mutations in ELN, resulting in insufficient ELN levels22. While reviewing the literature, we found that increased protease activity in the elastin-rich tissues leads to elastin degradation23. The peptides produced due to the degradation (elastokines, elastin-derived peptides, and elastin-related peptides) have been shown to be biologically active17. Their activities can be either physiologically beneficial or part of a pathological process. Accordingly, we increased the exposure time and found truncated body proteins (68 kDa and 53 kDa) in both pEGFP-ELN-wt and pEGFP-ELN-mut groups, and the relative amount of truncated body proteins (68 kDa and 53 kDa) decreased after the mutation effect (Fig. 5C). These results indicate that the mutation may affect the processing of ELN protein, which occurs before exocytosis of ELN protein.
Next, we detected the protein content secreted in the cell culture medium. The concentration of the cell culture medium indicated that there was no full-length ELN protein in the two groups, and the expression of truncated ELN decreased in the mutant group (Fig. 5D,E). In the arteries of patients with Williams-Beuren syndrome, increased proliferation of arterial smooth muscle cells in a quiescent contractile state is due to decreased deposition of elastin22. Therefore, beyond elasticity, elastin can also act as an autocrine factor in smooth muscle cells. A reduction in ELN expression was also observed in cutaneous fibroblasts and aortic smooth muscle cells in affected non-syndromic supravalvular aortic stenosis patients, thus, supporting the role of ELN haploidy as a pathogenesis of vascular lesions24. Overall, the content of truncated ELN (functional component) processed after translation was significantly lower both in intracellular and extracellular compartments after mutation. Our data are consistent with previous findings21. We propose that the decrease of ELN protein level may cause this pedigree vascular abnormality, especially pulmonary artery stenosis, and reinforce the view that ELN insufficiency is the primary cause of these vascular lesions. This may be the main molecular mechanism underlying the mutation-led functional changes.
In conclusion, genetic diagnosis of CHDs before the onset of symptoms is essential. The ELN heterozygous mutation (NM_001278939.1: c.1939G>T, p.Gly647Ter) was associated with the CHD accompanied with a pulmonary artery stenosis family. ELN truncate (functional component) was significantly lower both in intracellular and extracellular regions, and this may be the main molecular mechanism responsible for the mutation leading to the disease phenotype. Systematic analysis not only allows us to gain a better understanding of this disease etiology, but also contributes to clinical and prenatal diagnosis.
References
Liu, Y. et al. Global birth prevalence of congenital heart defects 1970–2017: Updated systematic review and meta-analysis of 260 studies. Int. J. Epidemiol. 48, 455 (2019).
Zaidi, S. & Brueckner, M. Genetics and genomics of congenital heart disease. Circ. Res. 120, 923 (2017).
Jin, S. C. et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 49, 1593 (2017).
Soemedi, R. et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am. J. Hum. Genet. 91, 489 (2012).
Ngo, M. L., Aggarwal, A. & Knudson, J. D. Peripheral pulmonary artery stenosis: an unusual case and discussion of genetic associations. Congenit. Heart Dis. 9, 448 (2014).
Duque, L. M. & Kozel, B. A. Elastin-driven genetic diseases. Matrix Biol. 71–72, 144 (2018).
Emanuel, B. S. et al. Chromosomal localization of the human elastin gene. Am. J. Hum. Genet. 37, 873 (1985).
Fazio, M. J. et al. Human elastin gene: new evidence for localization to the long arm of chromosome 7. Am. J. Hum. Genet. 48, 696 (1991).
Wu, W. et al. Whole Exome sequencing identifies a novel pathogenic RET variant in hirschsprung disease. Front. Genet. 9, 752 (2018).
Curran, M. E. et al. The elastin gene is disrupted by a translocation associated with supravalvular aortic stenosis. Cell 73, 159 (1993).
Tassabehji, M. et al. Williams syndrome: Use of chromosomal microdeletions as a tool to dissect cognitive and physical phenotypes. Am. J. Hum. Genet. 64, 118 (1999).
Xie, L. et al. A compound heterozygosity of Tecrl gene confirmed in a catecholaminergic polymorphic ventricular tachycardia family. Eur. J. Med. Genet. 62, 103631 (2019).
Qiu, C. et al. A novel TSC1 frameshift mutation c.1550_1551del causes tuberous sclerosis complex by aberrant splicing and nonsense-mediated mRNA degradation (NMD) simultaneously in a Chinese family. Mol. Genet. Genomic Med. 8, e1410 (2020).
Hou, C. L. et al. Protective effects of hydrogen sulfide in the ageing kidney. Oxid. Med. Cell Longev. 2016, 7570489 (2016).
Garg, V. et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424, 443 (2003).
Wessels, M. W. & Willems, P. J. Genetic factors in non-syndromic congenital heart malformations. Clin. Genet. 78, 103 (2010).
Fulop, T., Khalil, A. & Larbi, A. The role of elastin peptides in modulating the immune response in aging and age-related diseases. Pathol. Biol. (Paris) 60, 28 (2012).
Duca, L. et al. Matrix ageing and vascular impacts: Focus on elastin fragmentation. Cardiovasc. Res. 110, 298 (2016).
Aggoun, Y. et al. Mechanical properties of the common carotid artery in Williams syndrome. Heart 84, 290 (2000).
Li, D. Y. et al. Elastin is an essential determinant of arterial morphogenesis. Nature 393, 276 (1998).
Micale, L. et al. Identification and characterization of seven novel mutations of elastin gene in a cohort of patients affected by supravalvular aortic stenosis. Eur. J. Hum. Genet. 18, 317 (2010).
Urban, Z. et al. Connection between elastin haploinsufficiency and increased cell proliferation in patients with supravalvular aortic stenosis and Williams-Beuren syndrome. Am. J. Hum. Genet. 71, 30 (2002).
Pierre, A. et al. Impact of aging on inflammatory and immune responses during elastin peptide-induced murine emphysema. Am. J. Physiol. Lung Cell. Mol. Physiol. 316, L608 (2019).
Urban, Z. et al. Isolated supravalvular aortic stenosis: functional haploinsufficiency of the elastin gene as a result of nonsense-mediated decay. Hum. Genet. 106, 577 (2000).
Acknowledgements
This work is supported by Shanghai Jiaotong University Medical Technology Crossing Project (YG2017ZD26, ZH2018ZDA26), Shanghai Science and Technology Committee (18411965800, 19411963600). No benefits in any form have been or will be received from a commercial organization directly or indirectly.
Author information
Authors and Affiliations
Contributions
C.H. and T.X. designed and operated the project. C.H. designed the project and wrote the manuscript with the input from L.X., X.S., W.L., and Y.Z.. M.X., Y.L., and J.Z. provided the clinical analyses.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hou, C., Zheng, J., liu, W. et al. Identification and characterization of a novel ELN mutation in congenital heart disease with pulmonary artery stenosis. Sci Rep 11, 14154 (2021). https://doi.org/10.1038/s41598-021-93736-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-93736-1
This article is cited by
-
Three-dimensional molecular architecture of mouse organogenesis
Nature Communications (2023)
-
Sudden Cardiac Arrest During a Sedated Cardiac Magnetic Resonance Study in a Nonsyndromic Child with Evolving Supravalvar Aortic Stenosis Due to Familial ELN Mutation
Pediatric Cardiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
