
Abstract
Neritidae is one of the most diverse families of Neritimorpha and possesses euryhaline properties. Members of this family usually live on tropical and subtropical coasts and are mainly gregarious. The phylogenetic relationships between several subclasses of Gastropoda have been controversial for many years. With an increase in the number of described species of Neritidae, the knowledge of the evolutionary relationships in this family has improved. In the present study, we sequenced four complete mitochondrial genomes from two genera (Clithon and Nerita) and compared them with available complete mitochondrial genomes of Neritidae. Gene order exhibited a highly conserved pattern among three genera in the Neritidae family. Our results improved the phylogenetic resolution within Neritidae, and more comprehensive taxonomic sampling of subclass Neritimorpha was proposed. Furthermore, we reconstructed the divergence among the main lineages of 19 Neritimorpha taxa under an uncorrelated relaxed molecular clock.
Similar content being viewed by others


Introduction
The mitochondrial genome (mitogenome) is typically circular in invertebrates and generally approximately 15–20 kb in size1. It usually contains 37 genes, divided into one control region, 13 protein-coding genes, two rRNA genes, and 22 tRNA genes, in which the number of tRNA genes is highly variable2. Due to rapid evolution, cellular abundance, and an absence of introns, mitochondrial sequences can be easily amplified. In addition, they have a compact size, maternal inheritance, conserved features in their gene organization, a lack of extensive recombination, and a higher mutation rate than nuclear sequences3,4,5. These sequences been extensively used in comparative and evolutionary genomics6, species identification, population genetics7, molecular evolutionary and phylogenetic analyses and taxonomic diagnosis in marine biological studies8,9,10. In particular, phylogenetic analysis based on complete mitogenomes proved that the resolution of inferred phylogenetic trees was improved compared with that of trees based on partial gene fragments11. With the rapid development of sequencing and amplification technology for complete mitogenomes, they have been widely used to reconstruct phylogenetic relationships in different gastropod groups12,13,14.
Gastropods from family Neritidae (Rafinesque, 1815) are the most diverse species of Neritimorpha. They are euryhaline, meaning that they occur in marine, brackish, and freshwater systems15,16. Members of this family live on tropical and subtropical coasts and usually inhabit the middle to upper intertidal rocky zones17. Neritidae graze on algae on rock surfaces18,19. This family is ecologically important in freshwater and marine ecosystems because it manages the growth of certain algae and acts as a food source for other organisms. Family Neritidae includes marine genera such as Nerita, whereas species from Clithon and Neritina prefer to inhabit estuaries, mangrove streams and intertidal muddy sand banks15,16,20. Species in the genus Clithon are common in brackish estuarine areas with little tidal influence15 and are often used to study morphological patterns due to their shell color and pattern variations21,22,23. The genus Nerita is the most prominent intertidal group along tropical shores. It is relatively abundant in the fossil record, first appearing in the Cretaceous. In addition, Nerita species display extensive dispersal potential, producing veliger larvae that stay in the plankton stage for weeks to months24.
This family and the whole subclass of Neritimorpha are unique branches of marine gastropods in terms of morphology, structure and phylogeny, and international research on these species is ongoing. There is a long history of taxonomic studies on family Neritidae. In 1815, Rafinesque formally established Neritidae at the rank of family25. In addition, synonyms caused by the classification of shell shape often occur, such as Neritina zebra, which was initially defined as a species of Nerita by Bruguière but was classified as a species of Neritina in later studies26,27. Subsequently, Haynes identified the genus by studying the differences between male and female reproductive systems in Clithon and Neritina28. Currently, with the development of molecular biology technology, mitogenome sequencing analysis is being increasingly applied in the phylogenetic analysis of family Neritidae. Moises et al. reconstructed the phylogeny of three species of snails by comparing their mitogenome sequences with those of other gastropods29. Feng et al. carried out sequence analysis, phylogenetic reanalysis and divergence time estimation of Nerita undata and Nerita balteata and eight other species of neritids30. To date, more than 14 entire Neritidae mitogenomes have been sequenced (https://www.ncbi.nlm.nih.gov). However, two-thirds of them belong to the genus Nerita; currently, only one complete mitogenome dataset is available for the genus Clithon.
In the present study, two new sequences of Nerita were obtained, and two sequences of Clithon were also provided, which will further clarify the phylogenetic relationships among different genera and even within the whole Neritimorpha subclass. We determined the complete mitogenomes of four Neritidae species, namely, Clithon oualaniense (Lesson 1831), Clithon sowerbianum (Récluz, 1843), Nerita chamaeleon (Linnaeus, 1758) and Nerita japonica (Dunker, 1860), which are widely distributed in the southeastern China Sea. The characteristics of the species were compared, and we evaluated the variation in and conservation of mitogenomes among Neritidae species. To better understand the functions of related genes, we analyzed the relative synonymous codon usage (RSCU) and AT skew values of protein coding genes (PCGs). Furthermore, the phylogeny of subclass Neritimorpha and related species was reconstructed, and the relationships between these taxa were discussed. The divergence time of four species in subclass Neritimorpha was evaluated, and selective pressure analysis was performed.
Results and discussion
Genome structure, organization, and composition
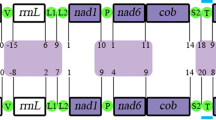
The entire mitogenome sequences of the four Neritimorpha species have lengths of 15,706 bp for C. oualaniense, 15,919 bp for C. sowerbianum, 15,716 bp for N. chamaeleon and 15,875 bp for N. japonica (GenBank accessions MT568501, MT230542, MT161611 and MN747116, respectively) (Table 1). The four circular molecules encode seven PCGs, eight tRNA genes on the forward strand, and 22 other mitochondrial genes on the reverse strand in the same orientation (Table 2). The control region is located between the cox3 and trnE genes, similar to the pattern in other previous reports on Neritidae species29,30,31,32,33,34,35 (Fig. 1). The genome structures of the four species were identical to those of other Neritimorpha taxa, without gene rearrangement, which may be related to their life history and habitat.

Gene map of the complete mitogenomes of Clithon oualaniense (GenBank accession No. MT568501), Clithon sowerbianum (MT230542), Nerita chamaeleon (MT161611) and Nerita japonica (MN747116). The ring indicates the gene arrangement and distribution. The largest ring is for C. oualaniense, and the smallest ring is for N. japonica. ND1-6 NADH dehydrogenase subunits 1–6, COX1-3 cytochrome c oxidase subunits 1–3, ATP6 and ATP8 ATPase subunits 6 and 8, CYTB cytochrome b, rRNA ribosomal RNA gene, tRNA transfer RNA gene.
The nucleotide compositions of the four whole mitogenomes were A: 29.81% to 33.79%, T: 30.67 to 35.36, G: 15.24 to 21.18, and C: 13.66 to 20.30 (Table 3). The contents of A and T exhibited high values, indicating codon usage bias towards A and T. The G and C contents of the four species were low, indicating an obvious bias against G and C. Moreover, the base compositions of 14 species in family Neritidae of the Neritimorpha were compared (Table 4). The AT contents of the 14 entire mitogenomes ranged from 61.67% to 66.28%, while the AT skew of most species was negative (− 0.1117 to − 0.0438), indicating the occurrence of fewer A than T nucleotides, except in C. sowerbianum (0.0484).
PCGs, tRNA genes, rRNA genes and codon usage
The AT contents of PCGs (− 0.2014 to − 0.0577) and tRNAs (− 0.0365 to − 0.0044) in the 14 Neritidae species had the same base bias as the entire genome (Table 4); however, the AT skew of the rRNAs (0.0614 to 0.0970) was slightly positive. All AT skew values were negative, while most GC skew values were positive. The AT content values of PCGs ranged from 60.43% to 65.64% in the 14 Neritidae species, indicating strong AT bias. All PCGs in the four mitogenomes started with the conventional initiation codon ATG or ATT and stopped with TAA or TAG.
The most frequently utilized amino acids in the four species were Leu2, Lys, Phe, Ser1 and Val (with frequencies ranging from 6.17% and 7.60%) (Fig. 2). The least common amino acid was Arg (all frequencies less than 2%), which is similar to the pattern previously reported in two Neritidae species (N. undata and N. balteata)30. Relative synonymous codon usage (RSCU) values for the 13 PCGs showed that UUA (Leu2) and CCU (Pro) were the two most frequent codons in the Clithon species (Fig. 3), and the most frequent codons in the Nerita species were CCU (Pro) and GCU (Ala). The 13 PCGs ranged in size from 165 bp (atp8 of all Neritidae) to 1717 bp (nad5 of C. sowerbianum). It is noteworthy that the atp8 gene is the smallest PCG in all currently described neritids. These comparative analyses showed that codon usage patterns are conserved among Neritidae species.

Percentage of each amino acid for proteins coded by PCGs in the four newly obtained mitochondrial genomes of C. oualaniense, C. sowerbianum, N. chamaeleon, and N. japonica.

The relative synonymous codon usage (RSCU) in the mitochondrial genomes of four Neritidae species. Co indicates the RSCU of C. oualaniense, Cs indicates the RSCU of C. sowerbianum, Nc indicates the RSCU of N. chamaeleon, and Nj indicates the RSCU of N. japonica.
The lengths of the tRNA genes were almost identical among the four Neritidae species, ranging from 57 (trnL1 of N. chamaeleon) to 74 bp (trnN of two Nerita species). The AT contents of tRNA genes ranged from 62.06% to 63.93% in the 14 Neritidae species (Table 4). The rrnL genes of the four Neritidae species were 1318 to 1334 bp in length, while the rrnS genes were 863 to 870 bp. In general, the A and T contents were greater than the G and C contents in the two rRNA genes (Table 3).
Selective pressure analysis
To investigate the evolutionary relationships among and selective pressure on 16 Neritimorpha species, we used the nonsynonymous to synonymous substitution (Ka/ Ks) ratio. The result showed that the average Ka/Ks ratio ranged from 0.060 for cox1 to 0.766 for nad4. This result indicated that the 13 PCGs of all Neritimorpha mitogenomes evolved under purifying selection (Fig. 4). The Ka/Ks ratio for all PCGs was below one, indicating that the mutations yielded synonymous substitutions. The cox1 gene has the lowest Ka/Ks ratio among studied genes and little change in amino acids; hence, it is widely used as a molecular marker for species identification and phylogenetic analysis36,37. The substitution saturation index value for the combined dataset of the 13 PCGs in all species (Iss = 0.685) was significantly lower than the critical values (Iss. cSym = 0.859 or Iss.cAsym = 0.847, p = 0.000) (Fig. 5). Thus, the combined sequence substitution was unsaturated, making the sequences suitable for phylogenetic analysis.

The purple line indicates the mean pairwise divergence of the Ka/Ks ratio for 13 PCGs among 16 Neritimorpha mitochondrial genomes. The 16 species of Neritimorpha are listed in Table 1. The pink and blue boxes indicate the number of nonsynonymous substitutions per nonsynonymous site (Ka) and the number of synonymous substitutions per synonymous site (Ks), respectively.

Saturation plots for 13 PCGs. The plots show the uncorrected pairwise divergence in transitions (s) and transversions (v) against the divergence calculated using the GTR model.
Phylogenetic relationships
Phylogenetic analyses were conducted on the concatenated alignment of 13 PCGs covering 88 gastropod species from thirty families of six subclasses (Vetigastropoda, Neomphaliones, Caenogastropoda, Neritimorpha, Patellogastropoda and Heterobranchia). We selected two Veneridae species (Bivalvia) as the outgroup. Maximum likelihood (ML) and Bayesian inference (BI) analyses produced almost identical topologies, with strong bootstrap and posterior probability values. However, family Lottidae of Patellogastropoda exhibited potential long-branch attraction (LBA) when we construct a Bayesian tree. Due to the large difference in branch length between members of this family and other related species, systematic errors occurred, and the true placements of these Lottidae taxa were not revealed38,39. This is the same as the result previously reported for the mitogenome of two limpets40. Finally, we combined the two methods to obtain a consistent evolutionary tree (Fig. 6).

Phylogenetic tree inferred using Bayesian inference (BI) and maximum likelihood (ML) methods based on concatenated sequences of 13 PCGs from 88 gastropod mitogenomes. The sequences of two Veneridae species were chosen as the outgroups. The blue dots indicate the four Neritidae species sequenced in this study. The number at each node is the bootstrap probability.
Our phylogenetic analysis indicated that all species representing subclass Neritimorpha clustered on the same branch; meanwhile, all posterior probability values were 1, and the bootstraps values were greater than 80. Within the Gastropoda class, the six subclasses exhibited the following phylogenetic relationships: ((((Vetigastropoda + Neomphaliones) + Caenogastropoda) + Neritimorpha) + Patellogastropoda) + Heterobranchia. Neritimorpha is closely related to Caenogastropoda and Patellogastropoda. Strikingly, we found that the branching orders of Neritimorpha and Caenogastroopoda were slightly different due to the increasing abundance of Neritimorpha species.
In Neritimorpha, whole mitogenomes are available for only two families, and Helicinidae forms an independent branch. The main evolutionary pattern in the Neritimorpha was the division of Neritidae into three genera, namely, Clithon, Neritina and Nerita. The Clithon and Neritina species clustered together and then with the genus Nerita. This indicated that the genus Clithon has a closer genetic relationship with the genus Neritina. The newly sequenced species C. sowerbianum was the closest relative of Clithon retropictus and then clustered with the new experimental species Clithon oualaniense, followed by Neritina usnea and Neritina violacea. In the genus Nerita, Nerita melanotragus was located on a separate branch and then clustered with Nerita albicilla. Furthermore, two new species of the genus Nerita, i.e., Nerita chamaeleon and Nerita japonica, were close to Nerita balteata and Nerita yoldii, respectively.
Divergence times
The time-calibrated phylogeny indicated that Neritimorpha originated approximately 232.16 million years ago (Mya) (95% highest posterior density [HPD] interval = 268.41–231.69 Mya) (Fig. 7), in agreement with the finding of a previous study suggesting that Neritimorpha appeared in the Triassic period30. The Triassic was the first period of the Mesozoic, which was the transitional period of the formation of the modern biota after the disappearance of the Paleozoic biota. Great changes have taken place in marine invertebrate groups41. In Neritidae, the differentiation time between Nerita and the other three genera was the earliest (97.65 Mya). However, the estimate provided by this analysis was slightly older than the origin of the Neritidae estimated in our previous analyses (76.17–83.25 Mya)30. This is probably due to misidentification in the fossil record, which is determined by various taxonomic methods and influenced by different levels of experience and expertise42. According to our findings, especially the attribution of fossils to different genera, the fossil record of Neritidae requires a complete revision. In the genus Nerita, the divergence time between N. melanotragus and other Nerita species was the earliest (68.18 Mya). For years, studies on the divergence time of neritids have shown that N. melanotragus was the first species differentiated from Nerita43. There were 7.64 million gaps between N. melanotragus and N. albicilla and 4.31 between N. albicilla and other Nerita species.

Divergence time estimation for Neritimorpha inferred via Bayesian relaxed dating methods (BEAST) based on the nucleotide sequences of 12 PCGs (excluding the cox3 gene). Fossil samples used to calibrate internal nodes are indicated by an asterisk. The 95% HPD is reported as blue bars, and Bayesian posterior probabilities are reported for each node. The accession numbers of the sequences used in the time-calibrated tree analysis are listed in Supplementary Table S1.
In our study, the addition of N. chamaeleon and N. japonica changed the divergence time of Nerita. N. balteata and N. chamaeleon split approximately 41.46 Mya, and N. japonica and N. yoldii were differentiated approximately 16.89 Mya. Moreover, the observations for other Nerita species were consistent with our previous estimates of divergence time30. Most Neritidae species differentiation was concentrated in the Cenozoic Paleogene (approximately 2.4–65 Mya). This is the period when continental transgression was rapidly reduced and marine sediments appeared in the marginal areas of China. On the other branch, the differentiation time of Theodoxus species was the earliest (82.44 Mya), followed by those of Neritina and Clithon species. N. usnea and N. violacea differentiated approximately 51.74 Mya. There were 16.17 million gaps between C. oualaniense and the other two Clithon species, and C. retropictus and C. sowerbianum differentiated approximately 34.79 Mya. This geographical isolation resulting from geological movement provided environmental conditions suitable for the divergence of Neritidae, and marine sediments provided a food source for Neritidae growth.
Conclusion
We obtained the mitogenome sequences of C. oualaniense, C. sowerbianum, N. chamaeleon and N. japonica by high-throughput sequencing, and their lengths were 15,706 bp, 15,919 bp, 15,716 bp and 15,875 bp, respectively. Each mitogenome is composed of a control region, 2 rRNAs, 13 PCGs and 22 tRNAs. The genome size, gene order and nucleotide composition of these four mitogenomes are similar to those of other neritids reported previously. Most PCGs were initiated with the ATG codon and terminated with the TAA codon. The Ka/Ks ratio indicated that these Neritimorpha species were subjected to purifying selection. Phylogenetic trees contributed to the scientific classification of Neritimorpha species. This study provides information on the genetic characteristics, phylogenetic relationships and evolution of neritids as well as a basis for resource management and selective breeding in aquaculture. These four species differentiated in the late Paleogene and early Neogene, and their evolution may be related to the geological events that changed their living environments.
Materials and methods
Samples and DNA extraction
Wild specimens of C. oualaniense (March 2020, E114°65, N22°73) were collected in the Pearl River Estuary, Guangdong Province; C. sowerbianum (October 2019, E110°34′, N20°08′) and N. chamaeleon (October 2019, E110°34′, N20°08′) were collected in Haikou, Hainan Province; and N. japonica (November 2018, E119°64′, N26°19′) were collected in Lianjiang, Fujian Province. All specimens were collected in the southeastern China Sea and were then preserved in absolute ethyl alcohol. The samples were identified via a published taxonomic book44, and we consulted taxonomists from the marine biology museum of Zhejiang Ocean University. Genomic DNA was extracted from small pieces of foot tissue taken below the operculum using the salting-out method and was stored at − 20 °C before sequencing. Only one specimen of each species was used for sequencing. All animal experiments were conducted in accordance with the guidelines and approval of the Animal Research and Ethics Committees of Zhejiang Ocean University.
DNA sequencing and genome assembly
The mitogenomes of four Neritidae species were submitted to Origingene Bio-pharm Technology Co., Ltd. (Shanghai, China), for Illumina PE library construction and high-throughput sequencing by the Illumina HiSeq X Ten platform. Sequencing libraries with average insert sizes of approximately 400 bp were prepared. Each library generated approximately 5 Gb of raw data. Removing the low-quality and contaminated reads resulted in higher ‘N’ ratio sequences and adapters. The clean reads of the four species were de novo assembled separately using NOVOPlasty software (https://github.com/ndierckx/NOVOPlasty)45.
Gene annotation and sequence analysis
Four newly assembled mitogenomes were annotated with the MITOS web server (http://mitos2.bioinf.uni-leipzig.de/index.py) based on the invertebrate genetic code46. Start and stop codons were confirmed using previously published Neritidae mitogenomes as references29,30. The circular genomes of the four Neritidae species were visualized with the CGView Server (http://stothard.afns.ualberta.ca/cgview_server/index.html)47. The nucleotide composition of the mitogenome for each species in family Neritidae; PCGs, tRNA genes, and rRNA genes; A and T content values; and relative synonymous codon usage (RSCU) and codon usage of PCGs were determined using MEGA 7.048. The base skew values were calculated with the formulas AT skew = (A − T)/(A + T) and GC skew = (G − C)/(G + C)49. To test for evolutionary adaptation, rates of nonsynonymous (Ka) and synonymous (Ks) substitutions in the mitogenomes of all species of Neritidae were estimated with DnaSP 6.050.
Phylogenetic inference and divergence time estimation
Evolutionary relationships were reconstructed with the PCGs from 88 gastropod mitogenomes, the four species (C. oualaniense, C. sowerbianum, N. chamaeleon and N. japonica) newly sequenced here and two representatives of the bivalves (Dosinia troscheli and Paphia undulata) as outgroups (Table 1). Phylogenetic trees were reconstructed using BI and ML methods. The nucleotide sequences for each PCG were adjusted by DAMBE 5.3.1951, and substitution saturation was tested for using the GTR substitution model. Sequences for each PCG were aligned using ClustalW of MEGA 7.048. Phylogenetic analyses incorporated both the maximum likelihood (ML) method using IQ-TREE52 and Bayesian inference (BI) using MrBayes v3.253. The best-fitting model (GTR + F + R7) selected by the BIC criteria implemented in ModelFinder54 was used for the ML analyses. In ultrafast likelihood bootstrapping, 1000 bootstrap replicates were applied to reconstruct a consensus tree. The MrBayes settings for the best substitution model (GTR + I + G) were determined by MrModeltest 2.355 under the AIC. The BI analyses involved two Markov chain Monte Carlo (MCMC) runs with 2,000,000 generations, sampling every 1000 generations and a discarded burn-in of 25%.
The estimates of divergence times among subclass Neritimorpha species were based only on nucleotide level (12 PCGs, with cox3 excluded due to this gene being incomplete in some species) and obtained using a Bayesian framework with an uncorrelated relaxed clock and lognormal relaxed molecular clock model in BEAST v1.8.456. The Yule process of speciation was used for the tree prior. For divergence time calibration, two calibration points were used as the prior for the corresponding split divergence time. Priors for fossil ages were drawn from normal distributions, and the root Pleuropoma jana was constrained between 235 and 223 million years ago (MYA)57. The 80 Ma point calibration was set as the root rate of Nerita based on the fossil of Nerita melanotragus (95–80 MYA)58. The final Markov chain was run twice for 100 million generations, with sampling every 1000 generations and 10% of samples discarded as a burn-in by TreeAnnotator v1.8.4 software (in the BEAST package). Then, using Tracer v. 1.659, chain convergence was confirmed, and the majority of the values exceeded an effective sample size (ESSs) of 200. The phylogenetic tree and divergence times were visualized using FigTree v1.4.3 software60.
Data availability
The mitochondrial genome data has been submitted to NCBI GenBank under the following accession numbers: Clithon oualaniense (MT568501), Clithon sowerbianum (MT230542), Nerita chamaeleon (MT161611), Nerita japonica (MN747116).
References
Tyagi, K. et al. The gene arrangement and phylogeny using mitochondrial genomes in spiders (Arachnida: Araneae). Int. J. Biol. Macromol. 146, 488–496 (2020).
Yang, M., Song, L., Zhou, L., Shi, Y. & Zhang, Y. Mitochondrial genomes of four satyrine butterflies and phylogenetic relationships of the family Nymphalidae (Lepidoptera: Papilionoidea). Int. J. Biol. Macromol. 145, 272–281 (2019).
Ballard, J. W. O. & Whitlock, M. C. The incomplete natural history of mitochondria. Mol. Ecol. 13, 729–744 (2004).
Gissi, C., Iannelli, F. & Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 101, 301–320 (2008).
Kurabayashi, A. et al. Phylogeny, recombination, and mechanisms of stepwise mitochondrial genome reorganization in mantellid frogs from Madagascar. Mol. Biol. Evol. 25, 874–891 (2008).
Saccone, C., De, G. C., Gissi, C., Pesole, G. & Reyes, A. Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system. Gene 238, 195–209 (1999).
Ye, Y. Y., Wu, C. W. & Li, J. J. Genetic population structure of Macridiscus multifarius (Mollusca: Bivalvia) on the basis of mitochondrial markers: Strong population structure in a species with a short planktonic larval stage. PLoS ONE 10, e0146260 (2015).
Zhang, Y., Gong, L., Lu, X., Jiang, L. & Zhang, X. Gene rearrangements in the mitochondrial genome of Chiromantes eulimene (Brachyura: Sesarmidae) and phylogenetic implications for Brachyura. Int. J. Biol. Macromol. 162, 704–714 (2020).
Kumar, V., Tyagi, K., Chakraborty, R., Prasad, P. & Chandra, K. The Complete mitochondrial genome of endemic giant tarantula, Lyrognathus crotalus (Araneae: Theraphosidae) and comparative analysis. Sci. Rep. 10, 74 (2020).
Curole, J. P. & Kocher, T. D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 14, 394–398 (1999).
Ruan, H., Li, M., Li, Z., Huang, J. & Zou, K. Comparative analysis of complete mitochondrial genomes of three Gerres fishes (Perciformes: Gerreidae) and primary exploration of their evolution history. Int. J. Mol. Sci. 21, 1874 (2020).
Yang, H., Zhang, J. E., Xia, J., Yang, J. & Luo, M. Comparative characterization of the complete mitochondrial genomes of the three apple snails (Gastropoda: Ampullariidae) and the phylogenetic analyses. Int. J. Mol. Sci. 19, 3646 (2018).
Lee, S. Y., Lee, H. J. & Kim, Y. K. Comparative analysis of complete mitochondrial genomes with Cerithioidea and molecular phylogeny of the freshwater snail, Semisulcospira gottschei (Caenogastropoda, Cerithioidea). Int. J. Biol. Macromol. 135, 1193–1201 (2019).
Yang, Y., Liu, H., Qi, L., Kong, L. & Li, Q. Complete mitochondrial genomes of two toxin-accumulated Nassariids (Neogastropoda: Nassariidae: Nassarius) and their implication for phylogeny. Int. J. Mol. Sci. 21, 3545 (2020).
Tan, S. K. & Clements, R. Taxonomy and distribution of the Neritidae (Mollusca: Gastropoda) in singapore. Zool. Stud. 47, 481–494 (2008).
Chen, Z. Y., Lian, X. P., Tan, Y. H. & Zhang, S. P. Progress in the systematics of Neritidae (Mollusca, Gastropoda, Cycloneritimorpha). Mar. Sci. 40, 168–173 (2016).
Takenori, S. & Hiroshi, I. The first occurrence of a Neritopsine gastropod from a, phreatic community. J. Molluscan. Stud. 68, 286–288 (2002).
Park, S. Y. et al. Transcriptomic analysis of the endangered neritid species Clithon retropictus: De novo assembly, functional annotation, and marker discovery. Genes 7, 1–19 (2016).
Hamish, G. S., Jonathan, M. W. & Thomas, E. E. Taxonomy and nomenclature of black nerites (Gastropoda: Neritimorpha: Nerita) from the South Pacific. Invertebr. Syst. 21, 229–237 (2007).
Chen, Z. Y., Lian, X. P. & Tan, Y. H. Comparative study on several confused species of Nerita (Gastropoda, Cycloneritimorpha, Neritidae). J. Trop. Oceanogr. 37, 1009–5470 (2018).
Sasaki, T. Macro- and microstructure of shell and operculum in two Recent gastropod species, Nerita (Theliostyla) albicilla and Cinnalepeta pulchella (Neritopsina: Neritoidea). Paleontol. Res. 5, 21–31 (2001).
Grüneberg, H. Micro-evolution in a polymorphic prosobranch snail (Clithon oualaniensis). Proc. R. Soc. Lond. B 200, 419–440 (1978).
Grüneberg, H. Pseudo-polymorphism in Clithon oualaniensis. Proc. R. Soc. Lond. B 216, 147–157 (1982).
Frey, M. A. & Vermeij, G. J. Molecular phylogenies and historical biogeography of a circumtropical group of gastropods (Genus: Nerita): Implications for regional diversity patterns in the marine tropics. Mol. Phylogenet. Evol. 48, 1067–1086 (2008).
Rafinesque, C. S. Analyse de la nature: Ou Tableau de l'univers et des corps organisés. Palerme 144 (1815).
Barroso, C. X. & Matthews-Cascon, H. Spawning and intracapsular development of Neritina zebra (Bruguiere, 1792) (Mollusca: Gastropoda: Neritidae). Invertebr. Reprod. Dev. 53, 137–143 (2009).
Barroso, C. X., Matthews-Cascon, H. & Simone, L. R. L. Anatomy of Neritina zebra from Guyana and Brazil (Mollusca: Gastropoda: Neritidae). J. Conchol. 41, 49–64 (2012).
Haynes, A. An evaluation of members of the genera Clithon Montfort, 1810 and Neritina Lamarck 1816 (Gastropoda: Neritidae). Molluscan Res. 25, 75–84 (2005).
Arques, M., Colgan, D. & Castro, L. R. Sequence and comparison of mitochondrial genomes in the genus Nerita (Gastropoda: Neritimorpha: Neritidae) and phylogenetic considerations among gastropods. Mar. Genome 5, 45–54 (2014).
Feng, J. T. et al. Sequence comparison of the mitochondrial genomes in two species of the genus Nerita (Gastropoda: Neritimorpha: Neritidae): Phylogenetic implications and divergence time estimation for Neritimorpha. Mol. Biol. Rep. 47, 7903–7916 (2020).
Castro, L. R. & Colgan, D. J. The phylogenetic position of Neritimorpha based on the mitochondrial genome of Nerita melanotragus (Mollusca: Gastropoda). Mol. Phylogenet. Evol. 57, 918–923 (2010).
Fukumori, H., Itoh, H. & Kano, Y. The complete mitochondrial genome of the stream snail Clithon retropictus (Neritimorpha: Neritidae). Mitochondrial DNA B 1, 820–821 (2016).
Xie, J. Y., Feng, J. T., Guo, Y. H., Ye, Y. Y. & Guo, B. Y. The complete mitochondrial genome and phylogenetic analysis of Nerita yoldii (Gastropoda: Neritidae). Mitochondrial DNA B 4, 1099–1100 (2019).
Feng, J. T. et al. The complete mitochondrial genome of Nerita albicilla (Neritimorpha: Neritidae). Mitochondrial DNA B 4, 1597–1598 (2019).
Wang, P., Zhu, P., Wu, H. P., Xu, Y. H. & Zhang, H. The complete mitochondrial genome of Neritina violacea. Mitochondrial DNA B 4, 2942–2943 (2019).
Astrin, J. J., Hfer, H., Spelda, J., Holstein, J. & Muster, C. Towards a DNA barcode reference database for spiders and harvestmen of Germany. PLoS ONE 11, e0162624 (2016).
Barrett, R. D. H. & Hebert, P. D. N. Identifying spiders through DNA barcodes. Can. J. Zool. 83, 481–491 (2005).
Uribe, J. E., Irisarri, I., Templado, J. & Zardoya, R. New patellogastropod mitogenomes help counteracting long-branch attraction in the deep phylogeny of gastropod mollusks. Mol. Phylogenet. Evol. 133, 12–23 (2019).
Williams, S. T., Foster, P. G. & Littlewood, D. T. J. The complete mitochondrial genome of a turbinid Vetigastropod from MiSeq Illumina sequencing of genomic DNA and steps towards a resolved gastropod phylogeny. Gene 533, 38–47 (2014).
Feng, J. T. et al. Comparative analysis of the complete mitochondrial genomes in two limpets from Lottiidae (Gastropoda: Patellogastropoda): Rare irregular gene rearrangement within Gastropoda. Sci. Rep. 10, 19277 (2020).
Alan, L. Triassic Period, Encyclopædia Britannica. https://www.britannica.com/science/Triassic-Period (2020).
Sepkoski, J. J. Rates of speciation in the fossil record. Philos. Trans. R. Soc. Lond. Ser. 353, 315–326 (1988).
Frey, M. A. Molecular phylogenies and historical biogeography of a circumtropical group of gastropods (Genus: Nerita): Implications for regional diversity patterns in the marine tropics. Mol. Phylogenet. Evol. 48, 1067–1086 (2008).
Zhang, S. P. Seashells of China 46–50 (China Ocean Press, 2008).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18 (2017).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Grant, J. R. & Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 36, 181–184 (2008).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Hassanin, A., Léger, N. & Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst. Biol. 54, 277–298 (2005).
Rozas, J. & Rozas, R. DnaSP, DNA sequence polymorphism: An interactive program for estimating population genetics parameters from DNA sequence data. Comput. Appl. Biosci. 11, 621–625 (1995).
Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 30, 1720–1728 (2013).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587 (2017).
Nylander, J. A., Ronquist, F., Huelsenbeck, J. P. & Nieves-Aldrey, J. Bayesian phylogenetic analysis of combined data. Syst. Biol. 53, 47–67 (2004).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Uribe, J. E., Colgan, D. & Castro, L. Phylogenetic relationships among superfamilies of Neritimorpha (Mollusca: Gastropoda). Mol. Phylogenet. Evol. 104, 21–31 (2016).
Postaire, B., Bruggemann, J. H., Magalon, H. & Faure, B. Evolutionary dynamics in the southwest Indian Ocean marine biodiversity hotspot: A perspective from the rocky shore gastropod genus Nerita. PLoS ONE 9, e95040 (2014).
Rambaut, A., Suchard, M. A., Xie, D. & Drummond, A. J. Tracer v1.6. http://tree.bio.ed.ac.uk/software/tracer/ (2014).
Rambaut, A. FigTree, Version 1.4.3. http://tree.bio.ed.ac.uk/software/figtree/ (accessed 1 July 2016).
Acknowledgements
This work was financially supported by the Project of Bureau of Science and Technology of Zhoushan (2020C21026), NSFC Projects of International Cooperation and Exchanges (42020104009) and National Natural Science Foundation of China (41976111, 42076119).
Author information
Authors and Affiliations
Contributions
F.J.T. analyzed the data, wrote the paper, and prepared the figures and tables. X.L.P., Y.C.R. and M.J. collected field material and processed the samples. Y.Y.Y. conceived and designed the experiments, reviewed drafts of the paper. L.J.J. contributed analysis tools, reviewed drafts of the paper. G.B.Y and L.Z.M supervised and directed the work, all authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Feng, Jt., Xia, Lp., Yan, Cr. et al. Characterization of four mitochondrial genomes of family Neritidae (Gastropoda: Neritimorpha) and insight into its phylogenetic relationships. Sci Rep 11, 11748 (2021). https://doi.org/10.1038/s41598-021-91313-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-91313-0
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
