
Abstract
The alfalfa weevil Hypera postica, native to the Western Palearctic, is an invasive legume pest with two divergent mitochondrial clades in its invading regions, the Western clade and the Eastern/Egyptian clade. However, knowledge regarding the native populations is limited. The Western clade is infected with the endosymbiotic bacteria Wolbachia that cause cytoplasmic incompatibility in host weevils. Our aim was to elucidate the spatial genetic structure of this insect and the effect of Wolbachia on its population diversity. We analyzed two mitochondrial and two nuclear genes of the weevil from its native ranges. The Western clade was distributed in western/central Europe, whereas the Eastern/Egyptian clade was distributed from the Mediterranean basin to central Asia. Intermediate mitotypes were found from the Balkans to central Asia. Most Western clade individuals in western Europe were infected with an identical Wolbachia strain. Mitochondrial genetic diversity of the infected individuals was minimal. The infected clades demonstrated a higher nonsynonymous/synonymous substitution rate ratio than the uninfected clades, suggesting a higher fixation of nonsynonymous mutations due to a selective sweep by Wolbachia. Trans-Mediterranean and within-European dispersal routes were supported. We suggest that the ancestral populations diversified by geographic isolation due to glaciations and that the diversity was reduced in the west by a recent Wolbachia-driven sweep(s). The intermediate clade exhibited a body size and host plant that differed from the other clades. Pros and cons of the possible use of infected-clade males to control uninfected populations are discussed.
Similar content being viewed by others
Introduction
Recent invasion events and routes of alien agricultural pests are of particular importance for the management and control of pests1,2,3,4,5,6. The knowledge of historical diversification and dispersal of agricultural pests in their native range provides insights to understand their natural and biological selective environments, including the role played by endosymbionts in pest emergence7,8,9.
The alfalfa weevil Hypera postica (Gyllenhal) (Coleoptera: Curculionidae: Hyperini), native to the Western Palearctic region, is a serious pest of alfalfa and other beneficial legumes in its invading territories, such as Medicago, Vicia, Trifolium, and Astragalus10,11,12 (Nearctic, Japan, Southeast Asia, and Oceania13,14,15). Invading populations in the USA comprise the Western (North American) type that invaded Utah in 190416, the Egyptian type that invaded Arizona in 193917, and the Eastern (North American) type that invaded Maryland in 195118. These types are different in their ecological, behavioral, and defensive traits (pupation site and aggregation during aestivation13, defensive behavior19, and encapsulation of immature endoparasitoid20,21,22) but are morphologically indistinguishable. Allozyme and mitochondrial DNA markers distinguish the Western type from the Egyptian and Eastern types22,23,24, whereas the Egyptian and Eastern types are distinguishable only by a slight difference (1–2 SNPs in tRNASer) in a mitochondrial gene sequence22,24,25. The nuclear DNA polymorphism indicates that the three types share a gene pool, namely, a single species25,26. They also mate with each other to reproduce (but see later for incompatibility27,28).
Wolbachia (Alphaproteobacteria: Rickettsiales: Rickettsiaceae) are maternally (vertically) transmitted intracellular bacteria that infect approximately 40% of insects and other arthropods alongside nematodes29. These endosymbiotic bacteria can manipulate host reproduction via reproductive cells and the genetic mechanism of this manipulation has recently been uncovered30. Cytoplasmic incompatibility (CI), or postzygotic incompatibility between infected and uninfected gametes, is the most commonly observed phenotype of Wolbachia. Theoretically, bidirectional CI most strongly accelerates host speciation. Unidirectional CI, or postzygotic isolation of gametes between infected males and uninfected females, can also promote host speciation. While infection by Wolbachia is favored in females in populations with high Wolbachia prevalence, loss of Wolbachia can also occur through incomplete inheritance from mothers with low Wolbachia density29. The loss (or incomplete transmission) rate of Wolbachia in insect hosts is slightly higher than the gain rate31. Maternal transmission and unidirectional CI eventually reduce host mitochondrial diversity over generations. The selective sweep of mitochondria leads to a close association between the mitochondrial clade and Wolbachia infection32,33,34,35. Several studies have discovered that Wolbachia may also accelerate the fixation of nonsynonymous mutations in hosts29,36,37,38. Various positive fitness effects of endosymbionts on their hosts have been revealed, such as viral suppression and metabolic provisioning39,40. The effect of endosymbionts on maternal mitochondria may also influence coevolution between mitochondria and nuclear genomes41,42.
In H. postica, the Western clade is found to be infected by Wolbachia that induces unidirectional CI27,43,44. Several invading populations of the Western clade are free of Wolbachia, and a cross between uninfected Western males and Egyptian/Eastern females within these populations does produce viable offspring28. This reconfirms that these clades, while genetically distant, remain conspecific. The CI effect between infected Western males and uninfected Eastern females is almost perfect (only 0.1% of hybrid eggs hatch), while 29.5% of hybrid eggs between uninfected Eastern males and infected Western females hatch27.
The presence of diverged clades and Wolbachia infection history in Palearctic H. postica in its primary range is not known to date. Here, this study aims to explore the process of selection and diversification in H. postica in its native range by revealing and testing mitochondrial and nuclear genetic variation geographically and phylogenetically. We also aim to test if the endosymbiont Wolbachia affected evolution in host weevils. The benefits and risks of the Incompatible Insect Technique45 using infected clade males to control the uninfected clade populations are discussed.
Results
Haplotype networks and diversity
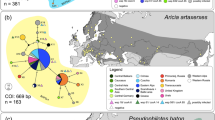
Sequenced segments were 2001 bp; 527 bp for COI-tRNALeu-COII, 281 bp for Cyt b-tRNASer-ND1 (n = 149), 801 bp for 28S (n = 122) and 392 bp for EF-1α (n = 62) (Table 1). Despite our sizable effort, PCR failed for nuclear gene segments (especially EF-1α) for a part of the specimens. The mitotype network revealed two main clades alongside intermediate variants (Fig. 1). The first group corresponded to the Eastern/Egyptian clade (Fig. 1) and consisted of diverse mitotypes with multiple connections, which contributed to a high mitochondrial genetic diversity (Table 2). This clade was widely distributed from central Asia to the Mediterranean region (Fig. 2). Within this clade, populations from the Balkan peninsula displayed high mitochondrial and nuclear genetic diversity (Table 3). The second clade corresponded to the Western clade and exhibited substantially fewer mitotypes, one dominant mitotype and rarer, closely related mitotypes in a star-shape topology, which corresponds to low genetic diversity in both gene fragments (Fig. 1, Table 2). This clade was distributed in western and central Europe, north of the Alps and Pyrenees (Fig. 2). Within this clade, 50.0% of individuals and 50.0% of populations were infected with Wolbachia (Fig. 2). The Wolbachia-infected populations demonstrated lower mitochondrial and nuclear genetic diversity than the uninfected populations (Table 2). Compared with the uninfected Eastern/Egyptian clade, the infected Western clade displayed 22 (Cyt b-tRNASer-ND1) to 82 (COI-tRNALeu-COII) times lower mitochondrial genetic diversity (Table 2). The intermediate clade was distributed from the Balkans to central Asia (Fig. 2). The network for the nuclear fragments, 28S and EF-1α, appeared incongruent with the mitochondrial network (Figs. 1, 3), but as in mitotype variation, there was a significant difference in nuclear haplotype variation between individuals belonging to the different mitochondrial clades (EF-1α, Table 2).

Statistical parsimony network of mitochondrial COI-tRNALeu-COII and Cyt b-tRNASer-ND1 of Hypera postica in its native range. Generated using TCS 1.2180.

Geographic distribution of mitochondrial clades of Hypera postica in its native range. Pie chart sizes for clades are proportional to sample sizes. Wolbachia infection (dark gray ring: infected; white (partial) ring: uninfected) is only indicated for the Western clade individuals because none of the Egyptian/Eastern clade or the intermediate clade were infected. The background map was obtained from Fotolla, https://stock.adobe.com/jp/photos/.

Wolbachia infection
The Western clade individuals in northern France, the Netherlands, the Czech Republic, and Poland were infected with Wolbachia (Fig. 2), and all of them had identical ftsZ (699 bp), coxA (432 bp), and hcpA (463 bp) sequences as reported previously46, which corresponds to the prevailing strain wHypera147. In contrast, Western clade individuals found in coastal southern France, Spain, Latvia, Hungary, and Croatia were uninfected (Fig. 2). Additionally, we confirmed that Wolbachia was absent from all native populations studied in the Eastern/Egyptian clade. A reconstructed phylogenetic relationship confirms that wHypera1 belongs to Supergroup B (Fig. 4). The Wolbachia strain closest to wHypera1 to date is the one that infects the mite Bryobia praetiosa (Acari: Tetranychidae)48.

Bayesian consensus tree of Wolbachia strain based on ftsZ, coxA, and hcpA. Strain codes, if available, followed by host species and Wolbachia supergroups are shown. The Wolbachia strain, wHypera1, infecting Hypera postica in its native range, is shown in bold. Bayesian support values (posterior probabilities > 0.7) are shown near nodes. The outgroup is Anaplasma marginale (Alphaproteobacteria: Rickettsiales: Anaplasmataceae). Generated using MrBayes 3.2.673. Host strains, Wolbachia isolates and GenBank accession numbers are listed in Supplementary Table S3.
Body size
Elytral length was significantly different among different clades (χ22 = 10.96, p = 0.004) and between sexes (χ21 = 7.52, p = 0.006) and marginally different between infected and uninfected individuals (χ21 = 3.77, p = 0.052). The elytron was longer in the intermediate variants (3.83 ± 0.110 mm, mean ± SE, n = 21) than in the Egyptian/Eastern (3.48 ± 0.025 mm, n = 64, p = 0.003) and Western (3.50 ± 0.057 mm, n = 29, p = 0.041) clades (no difference between the Egyptian/Eastern and Western clades, p = 0.934), longer in females (3.62 ± 0.042 mm, n = 60) than in males (3.47 ± 0.044 mm, n = 52), and marginally longer in uninfected individuals (3.57 ± 0.032 mm, n = 102) than in infected individuals (3.36 ± 0.111 mm, n = 11).
Selective neutrality test and positive selection test
For the Western clade, selective neutrality for mitochondrial segments was rejected by all indexes with minus values (D, D*, and F*), while for nuclear genes, selective neutrality was rejected by none of the indices (Table 4), suggesting recent sudden population growth after bottleneck event(s) in the mitochondrial lineage. For the Eastern/Egyptian clade, selective neutrality for both mitochondrial and nuclear segments was rejected by all indexes (Table 4), suggesting recent sudden population growth after bottleneck event(s) in the mitochondrial lineage and nuclear variants. For the intermediate clade, selective neutrality for mitochondrial segments was rejected by none of the indexes (Table 4). Selective neutrality for nuclear segments was untested because of the insufficient sample size.
A model with different ω (dN/dS) assigned for infected and uninfected clades improved the model fit most, compared to models with different ω for two or three different clades, although the improvement was nonsignificant (Table 5). The ω for infected clade was three times higher than the ω for uninfected clade even though both were < 1 (Table 5).
Geographic history
Isolation by distance
Isolation by distance (IBD) was supported for all populations (p = 0.003, number of pairwise comparisons n = 528) and for the Eastern/Egyptian clade excluding populations with intermediate mitotypes (p = 0.050, n = 153), but not for the Western clade (p = 0.102, n = 55). When the populations with intermediate mitotypes were included, the IBD within the Eastern/Egyptian clade was not supported (p = 0.215, n = 231).
Phylogeography
Based on available samples, the Balkan/Italian peninsulas and the Middle East are the most likely area of the origin of H. postica, from which the Western clade diversified via France (Fig. 5a). France is the likely area where the ancestral population was first infected with Wolbachia (Fig. 5a, right). We found two connections between regions that were highly supported with BF > 3: France and western Europe (BF = 7.98) and Balkan/Italy and North Africa (BF = 7.90) (BSSVS analysis, Fig. 5b).

(a) The maximum clade credibility (MCC) tree of Hypera postica resulting from a discrete Bayesian phylogeographic analysis, based on one sample per clade per country. The outgroup is H. miles. The nodes and branches are colored according to the most probable region (left panel; see Fig. 1 for colors indicating geographic regions and Table 1 for the country codes) and Wolbachia infection state (right panel; red line: infected). The numbers on the nodes indicate posterior probabilities of the most likely ancestral states [geographic region (left panel) or Wolbachia infection state (right panel)]. Generated using BEAST 1.10.490 and visualized using FigTree 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/). (b) Map of colonization routes of H. postica. Thick lines: supported by BF (Bayes factor) > 7.0. Generated using SpreaD3 v0.9.7.1rc92 and visualized using Google Earth 7.3.0 (https://www.google.com/earth/).
Discussion
This study revealed the large-scale geographic distribution and genetic diversity of H. postica in its native range. Intermediate mitotypes with larger body sizes were found from the Balkans to central Asia. The observed reduced diversity within the Western clade is likely due to a high percentage of Wolbachia infection within this clade, which is known in other species. We also identified a higher substitution rate of nonsynonymous mutations, suggesting promoted (fixation of) nonsynonymous mutations in the infected Western clade. In the Western clade, recent sudden population growth after a bottleneck was suggested only for mitochondrial genes and not for nuclear genes, supporting a recent selective sweep on mitochondria by Wolbachia infection.
Our results demonstrated a clear pattern of geographic distribution of the two divergent mitochondrial clades across the area of study, the Eastern/Egyptian and Western clades. The low genetic variation and star-like haplotype network within the Western clade is a signature of a recent demographic expansion from a few founders49. If the populations experienced ancient demographic bottlenecks, mitochondrial and nuclear genes are expected to have a concordant population structure. There was a weak concordance (Table 2), suggesting that these genes may have shared similar evolutionary trajectories. Two bottleneck events are likely; postglacial recolonization (see next section) and a recent mitochondrial sweep by Wolbachia. The former may also serve as a major driver of IBD that was supported for overall geographic populations (two clades distributed separately in the north and south with intermediate mitotypes in between). The latter likely accelerates the fixation of nonsynonymous mutations in the Western clade33,35,50,51,52,53.
The asymmetric inheritance of maternal mitochondria of an infected host caused by unidirectional CI-inducing Wolbachia can eventually lead to a sweep, which likely explains the low mitochondrial genetic variation among infected individuals. The infected clade demonstrated accelerating nonsynonymous mutations or fixation. This result is consistent with a general trend of Wolbachia infected insect groups38, suggesting fixation of nonsynonymous mutations in mitochondria promoted by its small effective population size under the CI-inducing Wolbachia infection. Furthermore, Wolbachia infection is advantageous for H. postica by enhancing resistance against its adult parasitoid Microctonus aethiopoides22.
In the southernmost populations of the Western clade or the geographic contact zone between the two clades, most individuals were uninfected or had lost Wolbachia. The imperfect maternal transmission was observed in the interclade crosses of H. postica27; fitness costs incurred by cytoplasmic incompatibility and stochasticity during the invasion process45,54 may lower the Wolbachia infection rate. Environmental causes (e.g., extreme temperatures) may also accelerate the endosymbiont loss55,56. Resulting uninfected H. postica populations (or with lowered Wolbachia density) must have regained reproductive compatibility between clades and enabled crosses between the diverged clades.
The intermediate variants exhibited a large body size. Larger genitalia of the males with these mitotypes may inhibit mating with the females of other clades and promote reproductive isolation. These mitotypes also were associated with an ecological niche that differed from the niche of other clades. Bulgaria populations used Vicia cracca as a host plant, whereas other populations used Medicago and Trifolium. Vicia cracca has high contents of cyanamide57 and canavanine58 that are toxic to insect herbivores59,60.
The genetic structure of most European biota has been strongly influenced by glacial oscillations of the Holocene61,62, and most temperate species exhibit northward post-glacial recolonization from glacial refugia located in southern Europe through central Asia during the last glacial maxima (southern genetic richness/northern purity63; in beetles64). In H. postica, we observed mtDNA differentiation for all clades and within the Eastern/Egyptian clade. Based on the estimated ancestral states in mitochondrial phylogeography and mitochondrial/nuclear genetic diversity, the Balkan and Italian peninsulas are a possible candidate for the origin of the Eastern/Egyptian clade and western Europe (France) for that of the Western clade. The primary center of genetic and species diversity of the main host Medicago is the Caucasus (north-western Iran and north-eastern Turkey)65, which may also consist of the area of origin of H. postica.
The recent dispersal routes that include the north Mediterranean were highly supported. Anthropogenic factors may allow occasional dispersal of H. postica to Europe and North Africa with alfalfa traded for livestock feed (by 2,600 years ago66). More recent international trade of alfalfa meal and pellets may continue to aid the weevil’s opportunistic long-distance dispersal; France, Spain, and Italy are the major alfalfa exporters among H. postica’s native ranges67.
Conclusion
While geographic isolation assisted continental diversification of the weevil H. postica, recent Wolbachia infection reduced diversity in a mitochondrial clade in the host weevil in western Europe. Wolbachia-infected males could be used as a control agent for the Incompatible Insect Technique on uninfected populations, however, the risk of heterosis in interclade crosses following accidental cure of Wolbachia must be assessed before application.
Methods
Sampling
Hypera postica (n = 149) were obtained from 33 localities covering most of its native distribution range (Table 1). Adults were collected from cultivated and wild legume vegetation of Medicago47, Trifolium in Egypt, and Vicia cracca in Bulgaria, mostly during the latest decade. The samples were then stored in ethanol at 4 °C until DNA extraction.
PCR and sequencing
DNA was extracted from all specimens using a DNeasy Blood & Tissue kit (Qiagen, Tokyo, Japan). We amplified and sequenced two mitochondrial fragments, COI-tRNALeu-COII and Cyt b-tRNASer-ND1 and two nuclear fragments, 28S and EF-1α. The primers used were C1-J-279768 and C2-N-338024 for the COI-tRNALeu-COII fragment, CB-J-11545 and N1-N-1184168 for Cyt b-tRNASer-ND1, 28S-01 and 28SR-01 for 28S69, and ef415F (5′-AACCAGAGAACATGCTCTTCTCG-3′) and ef862R (5′-CTCAATTTTTTAAGTTTGTTCAATTTG-3′) (this study) for EF-1α. PCRs were performed using GoTaq Green Master Mix (Promega, Tokyo, Japan). Cycling conditions for COI-tRNALeu-COII amplification consisted of preheating at 95 °C for 2 min, followed by 38 cycles of denaturation at 94 °C for 50 s, annealing at 50 °C for 1 min, and an extension at 60 °C for 1 min. Amplification conditions for Cyt b-tRNASer-ND1 were identical, except that annealing was performed at 55 °C for 1 min. Cycling conditions for 28S amplification were as follows; preheating as above, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 52 °C for 40 s, and an extension at 70 °C for 1 min. Those for EF-1α consisted of preheating as above, followed by 38 cycles of denaturation at 94 °C for 30 s, annealing at 51 °C for 40 s, and an extension at 68 °C for 1 min. Sequencing was carried out using a BigDye Terminator v3.1 Cycle Sequencing kit (Life Technologies/Applied Biosystems, Foster City, CA, USA) on a 3730 DNA Analyzer (Applied Biosystems).
Wolbachia infection and phylogeny
We used PCR to screen for possible Wolbachia infections. The Wolbachia ftsZ coding fragment was amplified using the primers fts-Z-f and fts-Z-r70. PCRs were performed using preheating as above, followed by 32 cycles at 94 °C for 40 s, 55 °C for 45 s, and 70 °C for 1 min. As a positive control, we used Wolbachia-infected Callosobruchus chinensis71. Blurred and extremely weak signals compared with the positive control were considered uninfected, which differs from a previous study47. Wolbachia-positive H. postica were further subjected to PCR and sequencing of the genes, coxA and hcpA, in addition to ftsZ for multilocus sequence typing of Wolbachia72.
For the phylogenetic reconstruction of Wolbachia, we used sequences from representative supergroups of Wolbachia (nr/nt database, Supplementary Table S1) with sequences of Anaplasma marginale as an outgroup. We used the GTR model, which was selected as the best fit model of nucleotide substitution by MrBayes3.2.673, based on the AICc, using MrAIC.pl 1.3.174. The three gene segments were partitioned. Markov chain Monte Carlo (MCMC) simulations were performed for one million generations, with sampling conducted every 1,000 generations. The convergence of independent parallel runs was checked using Tracer 1.675, and the first 25% of trees were discarded as burn-in.
Body size
After collecting specimens, the right elytron lengths of the samples were measured to the precision of 0.01 mm with a microscope (VH-5500, Keyence, Osaka, Japan). The sex of the samples was determined by both external and genital morphology. The effect of sex, clades (Egyptian/Eastern, Western, and intermediate), and Wolbachia infection on elytral lengths were tested by nonparametric Wilcoxon/Kruskal-Wallis signed rank tests. Posthoc multiple comparison was performed on the significant factor using the Steel-Dwass test. JMP 14.2.0 was used for statistical analyses.
Selective neutrality test and positive selection test
Selective neutrality was tested in each clade with Tajima76′’s D, Fu and Li77′’s D* and F*, using DNASP 6.12.0378. P values were derived by coalescent simulations with 2,000 replications. For the coalescent simulations for nuclear segments, an intermediate recombination rate was assumed. We used all mitochondrial (808 bp) or nuclear sequences (1,193 bp) of two individuals sampled per clade from each population to avoid sample size bias between populations.
The equal nonsynonymous/synonymous substitution rate ratio (dN/dS ratio, ω) between infected and uninfected clades and between the two major clades was tested with a phylogenetic analysis using the maximum likelihood method (likelihood ratio test) employed by the codeml program in PAML 4.9i79. The models with three or more different ω for each branch were compared with a reference (basal) model with two different ω (one for the root and the other for both the Western and Egyptian/Eastern clades). We concatenated all the open reading frames (protein-coding fragments) and removed potential stop codons (leading to 215 codons) of the mitochondrial sequences of two individuals randomly sampled per clade from each population to avoid sample size bias between populations. Codons for invertebrate mitochondria were used.
Haplotype networks and diversity
All mitochondrial sequences were assembled using Sequencher 5.0 (Gene Codes Corp, Ann Arbor, MI, USA), and we checked for the presence of pseudogenes using commonly employed methods80,81. Statistical parsimony networks were reconstructed based on mitochondrial and nuclear fragments using TCS 1.2182, in which we allowed connections between haplotypes of 20 steps for mitochondrial genes and 95% for the nuclear genes, to elucidate the maximal divergence observed among haplotypes. Nucleotide diversity (average over loci) and п (mean number of pairwise differences)83 were estimated for each geographic area, using Arlequin 3.5.2.284.
Geographic history
Isolation by distance
We assessed spatial mitochondrial differentiation by testing for isolation by distance (IBD)85. With a sweep (e.g., by Wolbachia) followed by rapid spread or frequent anthropogenic long-distance dispersal events, the IBD correlation is predicted to be weak at most. We tested if, as predicted by IBD, pairwise geographic distances and pairwise genetic differences were positively correlated using a one-tailed Mantel test86 based on 2,000 permutations with the ISOLDE program implemented in Genepop 4.287. For pairwise genetic differences, we employed corrected average pairwise differences between populations X and Y, [πXY − (πX + πY)/2]88 and their p values were derived using Arlequin.
Phylogeography
We estimated the historical dispersal patterns of H. postica, using a Bayesian discrete phylogeographic approach89 with a Bayesian skyride framework implemented in the software package BEAST 1.10.490. We used two mitochondrial segments (808 bp). To avoid sample size bias, we selected only one individual per clade from a given locality but excluded intermediate mitotypes, which reduced the data set to 34 individuals, with H. miles as an outgroup. We used default settings, applied the same molecular evolution model as presented above, and used an uncorrelated relaxed clock model assuming lognormal rate distribution91. We assigned each sequence to one of the seven geographic regions, and the symmetric exchanges between the geographic regions throughout the entire phylogeny were modeled with the Bayesian stochastic search variable selection (BSSVS). MCMC runs were performed for 50 million generations, sampling one tree every 25,000 generations. After confirming the stationarity of parameter estimates using Tracer, the first 40% of trees were discarded as burn-in, and maximum clade credibility (MCC) tree was built using TreeAnnotatorv1.10.4. As each node in each MCMC sample is annotated with a geographic region and Wolbachia infection status, we assessed the certainty of the geographic reconstruction by looking at the distribution of node states across the MCMC using FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/). Bayes factor (BF) values for exchange rates between each pairwise regions were retrieved from the log file from the BSSVS analysis using SpreaD3 v0.9.7.1rc92.
Data availability
GenBank accessions KX372573–372592, 372596–372619 and MW 393902–393920 for COI-tRNALeu-COII, KX372620–372639, 372643–372666 and MW393921–393939 for Cyt b-tRNASer-ND1, KX372667 and MW383443–383462 for 28S, MW389094–389112 and 392102 for EF-1α, and MW389113–389118 for ftsZ, coxA and hcpA.
References
Bonizzoni, M. et al. On the origins of medfly invasion and expansion in Australia. Mol. Ecol. 13, 3845–3855 (2004).
Tuda, M., Kagoshima, K., Toquenaga, Y. & Arnqvist, G. Global genetic differentiation in a cosmopolitan pest of stored beans: Effects of geography, host-plant usage and anthropogenic factors. PLoS ONE 9, e106268 (2014).
Karsten, M., van Vuuren, B. J., Addison, P. & Terblanche, J. S. Deconstructing intercontinental invasion pathway hypotheses of the Mediterranean fruit fly (Ceratitis capitata) using a Bayesian inference approach: Are port interceptions and quarantine protocols successfully preventing new invasions?. Divers. Distrib. 21, 813–825 (2015).
Rodriguero, M. S. et al. Out of the forest: past and present range expansion of a parthenogenetic weevil pest, or how to colonize the world successfully. Ecol. Evol. 6, 5431–5445 (2016).
Kébé, K. et al. Global phylogeography of the insect pest Callosobruchus maculatus (Coleoptera: Bruchinae) relates to the history of its main host Vigna unguiculata. J. Biogeogr. 44, 2515–2526 (2017).
Lombaert, E. et al. Colonization history of the western corn rootworm (Diabrotica virgifera virgifera) in North America: insights from random forest ABC using microsatellite data. Biol. Invasions 20, 665–677 (2018).
Tuda, M., Ronn, J., Buranapanichpan, S., Wasano, N. & Arnqvist, G. Evolutionary diversification of the bean beetle genus Callosobruchus (Coleoptera: Bruchidae): Traits associated with stored-product pest status. Mol. Ecol. 15, 3541–3551 (2006).
Wei, S. J. et al. Population genetic structure and approximate Bayesian computation analyses reveal the southern origin and northward dispersal of the oriental fruit moth Grapholita molesta (Lepidoptera: Tortricidae) in its native range. Mol. Ecol. 24, 4094–4111 (2015).
Takano, S. et al. Unique clade of alphaproteobacterial endosymbionts induces complete cytoplasmic incompatibility in the coconut beetle. Proc. Natl. Acad. Sci. USA 114, 6110–6115 (2017).
Radcliffe, E. B. & Flanders, K. L. Biological control of alfalfa weevil in North America. Integr. Pest Manag. Rev. 3, 225–242 (1998).
Kuwata, R., Tokuda, M., Yamaguchi, D. & Yukawa, J. Coexistence of two mitochondrial DNA haplotypes in Japanese populations of Hypera postica (Col., Curculionidae). J. Appl. Entomol. 129, 191–197 (2005).
Skuhrovec, J. Host plants of weevils of the genus Hypera (Coleoptera: Curculionidae) occurring in the Czech Republic. Klapalekiana 41, 215–255 (2005).
Wood, K. A., Armbrust, E. J., Bartell, D. P. & Irwin, B. J. The literature of arthropods associated with alfalfa. V. A bibliography of the alfalfa weevil, Hypera postica (Gyllenhal), and the Egyptian alfalfa weevil, Hypera brunneipennis (Boheman) (Coleoptera: Curculionidae). Illinois Agricultural Experimental Station, Special Publication, 54 (1978).
Kimura, H., Okumura, M. & Yoshida, T. Emergence of and recent damage by the alfalfa weevil. Shokubutsu Boeki (Plant Protection) 42, 498–501 (in Japanese) (1988).
CAB International crop protection compendium. CAB International. http://www.cabicompendium.org/cpc/home.asp (2013).
Titus, E. G. On the life history of the alfalfa leaf-weevil. J. Econ. Entomol. 3, 459–470 (1910).
Wehrle, L. P. The discovery of an alfalfa weevil (Hypera brunneipennis Boheman) in Arizona. J. Econ. Entomol. 33, 119–121 (1940).
Poos, F. W. & Bissell, T. L. The alfalfa weevil in Maryland. J. Econ. Entomol. 46, 178–179 (1953).
Volker, K. C. & Simpson, R. G. Behavior of alfalfa weevil larvae affecting the establishment of Tetrastichus incertus in Colorado. Environ. Entomol. 4, 742–744 (1975).
Salt, G. & van den Bosch, R. The defense reactions of three species of Hypera (Coleoptera, Curculionidae) to an Ichneumon wasp. J. Invertebr. Pathol. 9, 164–177 (1967).
Maund, C. M. & Hsiao, T. H. Differential encapsulation of two Bathyplectes parasitoids among alfalfa weevil strains, Hypera postica (Gyllenhal). Can. Entomol. 123, 197–203 (1991).
Hsiao, T. H. Studies of interactions between alfalfa weevil strains, Wolbachia endosymbionts and parasitoids. In The ecology of agricultural pests: biochemical approaches (eds, Symondson, W. O. C. & Liddell, J. E.). 57–71 (Chapman & Hall, 1996).
Hsiao, T. H. & Stutz, J. M. Discrimination of alfalfa weevil strains by allozyme analysis. Entomol. Exp. Appl. 37, 113–121 (1985).
Erney, S. J., Pruess, K. P., Danielson, S. D. & Powers, T. O. Molecular differentiation of alfalfa weevil strains (Coleoptera: Curculionidae). Ann. Entomol. Soc. Am. 89, 804–811 (1996).
Böttger, J. A. A. Phylogenetic analysis of the alfalfa weevil complex (Coleoptera: Curculionidae) in North America. J. Econ. Entomol. 106, 426–436 (2013).
Iwase, S., Nakahira, K., Tuda, M., Kagoshima, K. & Takagi, M. Host-plant dependent population genetics of the invading weevil Hypera postica. Bull. Entomol. Res. 105, 92–100 (2015).
White, C. E., Armbrust, E. J. & Ashley, J. Cross-mating studies of eastern and western strains of alfalfa weevil. J. Econ. Entomol. 65, 85–89 (1972).
Iwase, S. & Tani, S. New haplotype and inter-strain reproductive compatibility of Wolbachia-uninfected alfalfa weevil, Hypera postica (Coleoptera: Curculionidae), in Japan. Entomol. Sci. 19, 72–76 (2016).
Werren, J. H. Biology of Wolbachia. Annu. Rev. Entomol. 42, 587–609 (1997).
LePage, D. P. et al. Prophage WO genes recapitulate and enhance Wolbachia-induced cytoplasmic incompatibility. Nature 543, 243–247 (2017).
Bailly-Bechet, M. et al. How long does Wolbachia remain on board?. Mol. Biol. Evol. 34, 1183–1193 (2017).
Hale, L. R. & Hoffmann, A. A. Mitochondrial DNA polymorphism and cytoplasmic incompatibility in natural populations of Drosophila simulans. Evolution 44, 1383–1386 (1990).
Ballard, J. W. O. & Kreitman, M. Unravelling selection in the mitochondrial genome of Drosophila. Genetics 138, 757–772 (1994).
Johnstone, R. A. & Hurst, G. D. D. Maternally inherited male-killing microorganisms may confound interpretation of mitochondrial DNA variability. Biol. J. Linn. Soc. 58, 453–470 (1996).
Jiggins, F. M. Male-killing Wolbachia and mitochondrial DNA: selective sweeps, hybrid introgression and parasite population dynamics. Genetics 164, 5–12 (2003).
Werren, J. H., Baldo, L. & Clark, M. E. Wolbachia: master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751 (2008).
Shoemaker, D. D., Dyer, K. A., Ahrens, M., McAbee, K. & Jaenike, J. Decreased diversity but increased substitution rate in host mtDNA as a consequence of Wolbachia endosymbiont infection. Genetics 168, 2049–2058 (2004).
Cariou, M., Duret, L. & Charlat, S. The global impact of Wolbachia on mitochondrial diversity and evolution. J. Evol. Biol. 30, 2204–2210 (2017).
Teixeira, L., Ferreira, A. & Ashburner, M. The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biol. 6, 2753–2763 (2008).
Brownlie, J. C. et al. Evidence for metabolic provisioning by a common invertebrate endosymbiont, Wolbachia pipientis, during periods of nutritional stress. PLoS Pathog. 5, e1000368 (2009).
Rand, D. M., Haney, R. A. & Fry, A. J. Cytonuclear coevolution: the genomics of cooperation. TRENDS Ecol. Evol. 19, 645–653 (2004).
Arnqvist, G. et al. The genetic architecture of metabolic rate: environment specific epistasis between mitochondrial and nuclear genes in an insect. Evolution 64, 3354–3363 (2010).
Blickenstaff, C. C. Partial intersterility of eastern and western US strains of the alfalfa weevil. Ann. Entomol. Soc. Am. 58, 523–526 (1965).
Hsiao, T. H. & Hsiao, C. Hybridization and cytoplasmic incompatibility among alfalfa weevil strains. Entomol. Exp. Appl. 37, 155–159 (1985).
Laven, H. Eradication of Culex pipiens fatigans through cytoplasmic incompatibility. Nature 216, 383–384 (1967).
Iwase, S. et al. Dynamics of infection with Wolbachia in Hypera postica (Coleoptera: Curculionidae) during invasion and establishment. Biol. Invasions 17, 3639–3648 (2015).
Sanaei, E. et al. Global genetic diversity, lineage distribution and Wolbachia infection of the alfalfa weevil Hypera postica (Coleoptera: Curculionidae). Ecol. Evol. 9, 9546–9563 (2019).
Ros, V. I. D., Fleming, V. M., Feil, E. J. & Breeuwer, J. A. J. How diverse is the genus Wolbachia? Multiple-gene sequencing reveals a putatively new Wolbachia supergroup recovered from spider mites (Acari: Tetranychidae). Appl. Environ. Microbiol. 75, 1036–1043 (2009).
Avise, J. C. Phylogeography: The history and formation of species (Harvard University Press, 2000).
Narita, S., Nomura, M., Kato, Y. & Fukatsu, T. Genetic structure of sibling butterfly species affected by Wolbachia infection sweep: evolutionary and biogeographical implications. Mol. Ecol. 15, 1095–1108 (2006).
Raychoudhury, R. et al. Phylogeography of Nasonia vitripennis (Hymenoptera) indicates a mitochondrial–Wolbachia sweep in North America. Heredity 104, 318–326 (2010).
Jäckel, R., Mora, D. & Dobler, S. Evidence for selective sweeps by Wolbachia infections: phylogeny of Altica leaf beetles and their reproductive parasites. Mol. Ecol. 22, 4241–4255 (2013).
Jiang, W. et al. Wolbachia infection status and genetic structure in natural populations of Polytremis nascens (Lepidoptera: Hesperiidae). Infect. Genet. Evol. 27, 202–211 (2014).
Jansen, V. A. A., Turelli, M. & Godfray, H. C. J. Stochastic spread of Wolbachia. Proc. R. Soc. Lond. B Biol. Sci. 275, 2769–2776 (2008).
Clancy, D. J. & Hoffmann, A. A. Environmental effects on cytoplasmic incompatibility and bacterial load in Wolbachia-infected Drosophila simulans. Entomol. Exp. Appl. 86, 13–24 (1998).
Bordenstein, S. R. & Bordenstein, S. R. Temperature affects the tripartite interactions between bacteriophage WO, Wolbachia, and cytoplasmic incompatibility. PLoS ONE 6, e29106 (2011).
Kamo, T. et al. Limited distribution of natural cyanamide in higher plants: Occurrence in Vicia villosa subsp varia, V. cracca, and Robinia pseudo-acacia. Phytochemistry 69, 1166–1172 (2008).
Megías, C., Cortes-Giraldo, I., Giron-Calle, J., Alaiz, M. & Vioque, J. Free amino acids, including canavanine, in the seeds from 32 Vicia species belonging to subgenus Vicilla. Biocatal. Agric. Biotechnol. 8, 126–129 (2016).
Rosenthal, G. A. & Dahlman, D. L. Incorporation of L-canavanine into proteins and the expression of its antimetabolic effects. J. Agric. Food Chem. 39, 987–990 (1991).
Kamo, T., Tokuoka. Y. & Miyazaki, M. Quantification of canavanine, 2-aminoethanol, and cyanamide in Aphis craccivora and its host plants, Robinia pseudoacacia and Vicia angustifolia: Effects of these compounds on larval survivorship of Harmonia axyridis. J. Chem. Ecol. 38, 1552–1560 (2012).
Hewitt, G. M. Post-glacial re-colonization of European biota. Biol. J. Linn. Soc. 68, 87–112 (1999).
Schmitt, T. Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Front. Zool. 4, 11 (2007).
Taberlet, P., Fumagalli, L., Wust-Saucy, A. G. & Cosson, J. F. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 7, 453–464 (1998).
Jordal, B. H. & Kambestad, M. DNA barcoding of bark and ambrosia beetles reveals excessive NUMTs and consistent east-west divergence across Palearctic forests. Mol. Ecol. Resour. 14, 7–17 (2013).
Quiros, C. F. & Bauchan, G. R. The genus Medicago and the origin of the Medicago sativa complex. In Alfalfa and alfalfa improvement (eds, Hanson, A. A., Barnes, D. K. & Hill, R. R.). 93–124 (American Society of Agronomy, Crop Science Society of America, Soil Science Society of America, 1988).
Small, E. Alfalfa and Relatives: Evolution and Classification of Medicago (NRC Research Press, 2011).
FAO Statistics Division. FAOSTAT: Crops and livestock products. http://www.fao.org/faostat/en/#data/TP (2017).
Simon, C. A. et al. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701 (1994).
Kim, C. G. et al. Pattern of morphological diversification in the Leptocarabus ground beetles (Coleoptera: Carabidae) as deduced from mitochondrial ND5 gene and nuclear 28S rDNA sequences. Mol. Biol. Evol. 17, 137–145 (2000).
Holden, P. R., Brookfield, J. F. Y. & Jones, P. Cloning and characterization of an ftsZ homologue from a bacterial symbiont of Drosophila melanogaster. Mol. Gen. Genet. 240, 213–220 (1993).
Kondo, N. I. et al. Wolbachia infections in world populations of bean beetles (Coleoptera: Chrysomelidae: Bruchinae) infesting cultivated and wild legumes. Zool. Sci. 28, 501–508 (2011).
Baldo, L. et al. Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl. Environ. Microbiol. 72, 7098–7110 (2006).
Huelsenbeck, J. P. & Ronquist, F. MrBayes: Bayesian inference of phylogeny. Biometrics 17, 754–755 (2001).
Nylander, J. A. A. MrAIC.pl. Program distributed by the author. Uppsala: Evolutionary Biology Centre, Uppsala University (2004).
Rambaut, A. & Drummond, A. J. Tracer v1.5, http://beast.bio.ed.ac.uk/ (2009).
Tajima, F. Statistical methods to test for nucleotide mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Fu, Y.-X. & Li, W.-H. Statistical tests of neutrality of mutations. Genetics 133, 693–709 (1993).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Yang, Z. PAML 4: A program package for phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Song, H., Buhay, J. E., Whiting, M. F. & Crandall, K. A. Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc. Natl. Acad. Sci. USA 105, 13486–13491 (2008).
Haran, J., Koutroumpa, F., Magnoux, E., Roques, A. & Roux, G. Ghost mtDNA haplotypes generated by fortuitous NUMTs can deeply disturb infra-specific genetic diversity and phylogeographic pattern. J. Zoolog. Syst. Evol. Res. 53, 109–115 (2015).
Clement, M., Snell, Q., Walker, P., Posada, D. & Crandall, K. TCS: Estimating gene genealogies. Parallel Distrib. Proces. Symp. Int. Proc. 2, 184 (2002).
Tajima, F. Evolutionary relationship of DNA sequences in finite populations. Genetics 105, 437–460 (1983).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Wright, S. Isolation by distance. Genetics 28, 114–138 (1943).
Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220 (1967).
Raymond, M. & Rousset, F. Genepop (version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 86, 248–249 (1995).
Nei, M. & Li, W. H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 76, 5269–5273 (1979).
Lemey, P., Rambaut, A., Drummond, A. J. & Suchard, M. A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 5, e1000520 (2009).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Bielejec, F. et al. SpreaD3: Interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 33, 2167–2169 (2016).
Acknowledgements
We thank Łukasz Kajtoch for information about the collection site in Poland, Tamás Németh and Zoltán György for guidance at the Hungarian Natural History Museum, Kumiko Kagoshima for assistance with PCR, and Anahí Espíndola for advice on phylogeographic methods.
Funding
Funding was provided by Japan Society for the Promotion of Science (Grant Nos. JP23405008, JP25430194, JP26304016, JP17H04612, JP18H02207 and JP19K06840), Kyushu University (Interdisciplinary Programs in Education and Projects in Research Development (25412)) and Ministerstvo Zemědělství (Grant No. RO0418).
Author information
Authors and Affiliations
Contributions
M.T., S.I., J.H. and J.S. conceived the idea, M.T., S.I. and JH. conducted molecular experiments, J.S., A.P., O.M., N.T., E.S., A.H.E. and K.M. performed morphological identification, M.T. and K.K. analyzed the data and M.T., J.H. and K.K. led the writing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tuda, M., Iwase, Si., Kébé, K. et al. Diversification, selective sweep, and body size in the invasive Palearctic alfalfa weevil infected with Wolbachia. Sci Rep 11, 9664 (2021). https://doi.org/10.1038/s41598-021-88770-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-88770-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
