
Abstract
Cerebral hypoperfusion is a key factor for determining the outcome after subarachnoid hemorrhage (SAH). A subset of SAH patients develop neurogenic stress cardiomyopathy (NSC), but it is unclear to what extent cerebral hypoperfusion is influenced by cardiac dysfunction after SAH. The aims of this study were to examine the association between cardiac function and cerebral perfusion in a murine model of SAH and to identify electrocardiographic and echocardiographic signs indicative of NSC. We quantified cortical perfusion by laser SPECKLE contrast imaging, and myocardial function by serial high-frequency ultrasound imaging, for up to 7 days after experimental SAH induction in mice by endovascular filament perforation. Cortical perfusion decreased significantly whereas cardiac output and left ventricular ejection fraction increased significantly shortly post-SAH. Transient pathological ECG and echocardiographic abnormalities, indicating NSC (right bundle branch block, reduced left ventricular contractility), were observed up to 3 h post-SAH in a subset of model animals. Cerebral perfusion improved over time after SAH and correlated significantly with left ventricular end-diastolic volume at 3, 24, and 72 h. The murine SAH model is appropriate to experimentally investigate NSC. We conclude that in addition to cerebrovascular dysfunction, cardiac dysfunction may significantly influence cerebral perfusion, with LVEDV presenting a potential parameter for risk stratification.
Similar content being viewed by others


Introduction
Spontaneous subarachnoid hemorrhage (SAH), a type of hemorrhagic stroke, is caused primarily by rupture of an intracranial aneurysm1,2. There are two major phases of brain injury after SAH, early and delayed, with both shared and distinct causative factors. Early brain injury (EBI) occurs as an immediate result of bleeding and associated transient global cerebral ischemia1,3. Alternatively, delayed cerebral ischemia (DCI), occurring mostly within 21 days after SAH onset, demonstrates a multifactorial pathogenesis, including vasospasms of microvessels and macrovessels, microthrombosis, and cortical spreading depressions3,4. Clinically, DCI manifests as new neurological deficits related to cerebral ischemia5. Cerebral hypoperfusion is central to the development of both EBI and DCI3 and, therefore, may be a promising target for therapeutic intervention to improve the neurological outcome.
SAH impairs cardiac function by disrupting the neuro-cardiac axis, leading to neurogenic stress cardiomyopathy (NSC) of variable severity [reviewed in6,7]. Under physiological conditions, cerebral autoregulation attenuates the effects of cardiac function on cerebral perfusion. However, cerebral autoregulation is disrupted after SAH5,8. Further, several studies have shown that decreased cardiac output (CO) can induce both acute9,10 and chronic cerebral hypoperfusion11,12, and that impaired left ventricular (LV) function is associated with cognitive decline13,14. Presumably, impaired LV function leads to cerebral hypoperfusion after SAH. This assumption is supported by a clinical study that found an association between cardiac dysfunction and cerebral hypoperfusion during the first 24 h after hospital admission in patients with SAH15. Based on these clinical findings5,6,7,8,9,10,11,12,13,14,15, improving cardiac function appears essential for mitigating cerebral hypoperfusion and optimal neurological outcome after SAH. Nevertheless, this approach has never been studied. Rather, current treatment guidelines focus on the importance of cerebral perfusion pressure (CPP) and the role of the cerebral vasculature in cerebral perfusion16,17,18,19.
Murine models are an increasingly important research tool to study SAH pathophysiology. Similar to the human disease, rodents develop cerebral hypoperfusion after experimental induction of SAH20,21,22. Histological studies have reported myocardial injury 7 days after SAH induction in mice23,24,25, suggesting that murine models are suitable for studies on the neuro-cardiac axis. Cerebral hypoperfusion after SAH is pivotal to both EBI and DCI pathophysiology26,27,28, so most experimental studies have focused on the cerebral vasculature’s role in dysregulating cerebral circulation29,30,31,32. However, the link between post-stroke cardiac dysfunction and cerebral hypoperfusion has also not been established in experimental studies.
Therefore, we investigated the association between cerebral perfusion and cardiac function in a murine endovascular filament perforation model of SAH. The primary purpose was to analyze the correlation between cardiac function and cerebral cortical perfusion after SAH induction. We also examined electrocardiographic (ECG) and echocardiographic signs indicative of NSC to facilitate future interventional studies for improving CO and cerebral perfusion following SAH.
Methods
Ethics approval
The animal experiments were approved by the responsible animal care committee (Landesuntersuchungsamt Rheinland-Pfalz) and conducted in accordance with the German Animal Welfare Act (TierSchG). All applicable international, national, and institutional guidelines for the care and use of animals were followed. The study was carried out in compliance with the ARRIVE guidelines.
Animals and housing conditions
Female C57BL/6N mice (Janvier, Saint-Berthevin Cedex, France; aged 11–12 weeks) were used for all experiments. As described in our previous study22, the mice were housed under controlled environmental conditions (12-h light–dark cycles, 23 ± 1 °C, 55% ± 5% relative humidity) with free access to water and food (Altromin, Lage, Germany). Body weight was measured daily during the experimental duration.
Study design
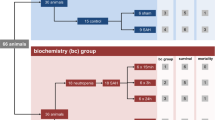
Twenty mice were randomized to receive either SAH (13 animals) or sham surgery (7 animals). Cerebral cortical blood flow and intracranial pressure (ICP) were determined before surgery and 15 min, 3 h, 24 h, 72 h, and 7 days after SAH induction or sham surgery. ECG and echocardiography were performed at baseline on the day before surgery, and immediately after each post-surgical cerebral cortical blood flow measurement. Mice were sacrificed after the last echocardiography exam, 7 days post-surgery. Non-invasive measurements of arterial blood pressure were acquired on the day before surgery, and on days 1, 3, and 7 after surgery, using the Coda mouse tail-cuff system (Kent-Scientific, Torrington, CT, USA). The study design is shown in Fig. 1.

Study concept. (A) Study design. Mice were randomized to sham surgery and experimental subarachnoid hemorrhage (SAH) groups and subjected to serial measures of cerebral perfusion and cardiac function at the indicated times. (B) Schematic illustration of the SPECKLE imaging method for measuring cerebral cortical perfusion. (C) Illustration of high-frequency small animal transthoracic echocardiography for assessing cardiac output (CO), stroke volume, left ventricular ejection fraction (LVEF), and left ventricular end-diastolic volume (LVEDV).
Murine model of SAH
SAH was induced by endovascular filament perforation, under isoflurane anesthesia (Abbvie, Wiesbaden, Germany) with continuous ICP monitoring21,22,32,33. Anesthesia was induced with 4% isoflurane for 1 min and maintained with 2% isoflurane. For analgesia, Buprenorphin (Indivior, Slough, Berkshire, UK; 0.1 mg/kg body weight) was subcutaneously injected before surgery and then three times per day until the end of the experiment on post-surgical day 7. Body temperature was maintained at 36 °C during surgery using a heating pad (Physiotemp Instruments LLC, Clifton, NJ, USA).
For SAH induction, the anaesthetized mice were first placed in the prone position, and an ICP probe (Codman, Johnson & Johnson, Raynham, MA, USA) was placed in the right frontal region through a burr hole. After surgical preparation, a filament (Prolene 5.0; Ethicon, Norderstedt, Germany) was inserted into the left external carotid artery and intracranially advanced through the internal carotid artery21,22,32,33. A sharp rise in ICP was considered as an indicator of successful endovascular perforation. The ICP probe was removed at the end of the procedure. Surgical wounds were closed using Prolene 6.0 sutures (Ethicon). The same procedure was conducted in the sham group, but without advancing the endovascular filament into the internal carotid artery. After surgery, the animals were kept in an incubator, heated to 36 °C (IC8000; Draeger, Luebeck, Germany) for 1 h, to prevent hypothermia.
Determination of cerebral cortical perfusion with laser SPECKLE contrast imaging
All measurements were performed under anesthesia (2% isoflurane) while maintaining the body temperature at 36 °C using a heating pad (Physitemp Instruments LLC). Cerebral cortical perfusion was determined, using a laser perfusion imager (MoorFLPI-2-blood flow imager; Cologne, Germany), and analyzed with Moor review software (moorFLPI Full-Field Laser Perfusion Imager Review Version 4.0) as described21,22. Briefly, following a midline incision to expose the calvaria, 60 transcalvarian perfusion images were recorded at one picture per second. The wound was then closed with Prolene 6.0 sutures. A mean image was calculated from the 60 perfusion images. The mean flux values were determined by evaluating a 3-mm diameter region of interest (ROI) over the perfusion territory of the left middle cerebral artery. Changes in perfusion are expressed relative to the preoperative flux value. This method is illustrated in Fig. 1.
High-frequency small animal echocardiography and ECG recording
Cardiac function was assessed by transthoracic echocardiography. Serial examinations were performed under isoflurane anesthesia (1.2–1.5%) using the Vevo3100 high-resolution imaging system (VisualSonics, FujiFilm, Toronto, Canada) equipped with a 38 MHz (MZ 400) linear array transducer. Images were acquired at above 200 frames/s. The one-lead ECG and breathing rate were monitored throughout, and the mouse’s body temperature was kept stable during anesthesia using a heating system within the handling platform. Brightness (B)-mode and Motion (M)-mode movies of the parasternal long axis (PLAX) and parasternal short axis (SAX, mid ventricular) were acquired. Post-acquisition analysis was performed using VevoLab Software (VisualSonics). Left ventricular ejection fraction (LVEF), left ventricular end-diastolic volume (LVEDV), CO, and stroke volume were measured as LV function indices.
Statistical analysis
GraphPad Prism software (La Jolla, CA, USA) was used for correlation analyses, and SigmaPlot version 12.5 (Systat Software, San Jose, CA, USA) was used for descriptive statistics and statistical power analyses. Data are presented as mean ± standard error of the mean (SEM). The data were tested for normal distribution using the Kolmogorow-Smirnow normality test. To compare variables between 2 groups, group means (sham vs. SAH) were compared by Mann–Whitney U test in case of non-normal distribution and Student’s t-test in case of normal distribution. A p < 0.05 (two-tailed) was considered statistically significant. Association strengths were measured by Pearson’s correlation coefficients in case of normal distribution and by Spearman’s correlation coefficients in case of non-normal distribution, again with a p < 0.05 considered significant.
Group sizes were calculated based on the mortality rates in our previous experimental SAH studies21,22 and statistical power analysis with alpha of 0.05, power of 0.8, and assuming an r value of 0.85.
Results
Mortality and body weight
13 animals were randomized to SAH induction and seven to sham surgery. Two SAH animals and one sham animal died within 3 h after surgery, and one SAH animal died within 24 h after surgery. These animals were excluded from the evaluation. Of the remaining 10 SAH animals, one showed subdural hematoma over the left hemisphere, which precluded measuring cerebral cortical perfusion. Therefore, this animal was also excluded from the evaluation, leaving nine SAH and six sham animals. One further SAH animal was found dead on day 3 before the perfusion measurement. Another three SAH animals and three sham animals died on day 4 after surgery. Accordingly, statistical analyses were performed on 9 SAH group animals for the time points until 24 h post-surgery, 8 animals for the 72 h time point, and on 5 animals at 7 days post-SAH. In the sham group, statistical analyses were performed on 6 animals for the time points until 72 h post-surgery and on 3 animals at 7 days post-surgery. In the sham group, cerebral perfusion in 1 animal at 7 days and in 2 animals at 72 h post-surgery, and the cardiac parameters in 1 animal at the time points before surgery and 15 min post-surgery could not be evaluated due to technical reasons.
The body weights were similar between SAH and sham animals at all measurement points post-surgery but differed slightly (~ 5%) at baseline (before surgery: 20.0 ± 0.5 g vs. 18.9 ± 0.1 g, p < 0.05; day 1: 18.5 ± 0.3 vs. 18.1 ± 0.4 g; day 3: 17.1 ± 0.7 vs. 15.8 ± 0.5 g; day 7: 19.1 ± 0.4 vs. 19.0 ± 1.0 g).
Elevated ICP and cerebral hypoperfusion after SAH
Mice were randomized to either SAH induction or sham surgery. Cerebral perfusion was determined along with ICP, ECG, and echocardiography at baseline and 15 min, 3 h, 24 h, 72 h, and 7 days after SAH or sham surgery. The study protocol is illustrated in Fig. 1.
We first examined whether SAH induces a reduction in cerebral perfusion. Similar to previous reports20,21,22,29,30,31, SAH induction led to a rapid and significant increase in ICP after endovascular perforation (SAH: 71.3 ± 9.7, sham 11.3 ± 2.5 mmHg, p < 0.001), which normalized slowly. Induction of SAH also caused a significant decrease in cortical perfusion, which recovered to near baseline by 24 h, again consistent with previous studies20,21,22,29,30,31. Mean arterial pressure (MAP) decreased after surgery, but the difference between SAH and sham animals did not reach statistical significance. The detailed time courses of these physiological parameters are shown in the Fig. 2.

Comparisons of cerebral and cardiovascular parameters between SAH and sham groups. (A)–(H) Cerebral and cardiovascular parameters were measured from SAH and sham animals before surgery and 15 min, 3 h, 24 h, 72 h, and 7 days post-surgery. SAH: n = 9 for the time points before surgery and 15 min, 3 h, 24 h postop.; n = 8 at 72 h postop.; n = 5 at 7 days postop.; Sham: cerebral perfusion: n = 4 at 72 h and n = 2 at 7 days post-surgery; heart reate, stroke volume, cardiac output, LVEF, LVEDV: n = 5 for the time points before surgery and 15 min post-surgery; in all other cases: n = 6 for the time points before surgery and 15 min, 3 h, 24 h, and 72 h post-surgery; n = 3 for 7 days post-surgery. Data are presented as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001 (Mann–Whitney U test or Student's t-test as appropriate). arb: arbitrary; ICP: intracranial pressure; LVEDV: left ventricular end-diastolic volume; LVEF: left ventricular ejection fraction; MAP: mean arterial pressure; SAH: subarachnoid hemorrhage; SEM: standard error of the mean.
Pathophysiological features of NSC after SAH
We then investigated whether SAH animals showed echocardiographic or electrocardiographic signs of NSC. We observed abnormal echocardiographic or electrocardiographic changes in only a subset of SAH model mice, in accord with clinical observations6,7,34,35. In two of ten SAH group animals, LVEF was reduced substantially at 3 h post-SAH despite an overall increase in LVEF after SAH compared to sham (Fig. 3 and supplementary material (video)). Three of ten SAH group animals exhibited a right bundle branch block at 15 min and 3 h post-surgery. In one animal, this conduction block was clearly associated with a wall-motion abnormality. None of these pathological findings were present at baseline, confirming SAH-dependent induction, and all abnormal ECG or echocardiographic findings recovered by 24 h post-surgery.

ECG and echocardiographic features of neurogenic stress cardiomyopathy (NSC) in mice subjected to experimental subarachnoid hemorrhage (SAH). (A) Echocardiographic findings from one of two SAH group animals in which the left ventricular function was markedly reduced at 3 h post-surgery (in contrast to the overall changes in the SAH group but consistent with emergence of NSC in a subset of SAH patients). Recovery was complete by 72 h post-surgery. A video of the echocardiography can be found in the supplementary material. (B) An example of abnormal electrocardiographic findings (right bundle branch block) observed in three mice after SAH induction. Recovery was complete at 24 h post-SAH.
We also investigated whether there was an overall reduction in LV function among the SAH group animals compared to the sham controls. In the SAH group, stroke volume and LVEF were elevated for up to 72 h post-surgery, with a significant group difference at 15 min, 24 h, and 72 h for LVEF, while CO was elevated at 15 min, 24 h, and 72 h, with a significant group difference at 15 min. Conversely, LVEF was significantly higher in sham animals at 7 days post-surgery. Similarly, LVEDV was higher in the sham group at 24 h and 72 h post-surgery, which reached the level of significance at 24 h.
Taken together, our data show overall enhanced LV function until 72 h post-surgery and reduced cardiac contractility at 7 days post-surgery in the SAH group compared to sham animals (Fig. 2). However, a few animals showed impaired LV systolic function resembling severe human NSC at 15 min and 3 h post-SAH.
Cardiac function correlates with cerebral perfusion after SAH
Cardiac output reportedly influences cerebral perfusion and function9,10,11,13, so we next tested whether LV function impacts post-SAH cerebral perfusion. To this end, we examined the correlations of cardiac parameters, determined by high-frequency ultrasound, with cerebral cortical perfusion determined by SPECKLE imaging. LVEDV correlated significantly with cerebral perfusion at 3 h, 24 h, and 72 h post-SAH, which was not the case in sham animals. This indicates a link between diastolic filling and cerebral perfusion at these time points. There was a trend for correlation of cardiac stroke volume and CO with cerebral perfusion at 72 h after cranial bleeding, which, however, did not reach statistical significance. This reveals a link between systolic left ventricular output and cerebral perfusion at this time point. Alternatively, there was no significant correlation between cerebral perfusion and LVEF (Figs. 4, 5, and supplementary material (Figs. E1–E4)).

Correlations of LVEDV with cerebral perfusion after SAH. (A)–(F) Correlations of left ventricular end-diastolic volume (LVEDV) with cerebral perfusion at the indicated times post-surgery. Extended analyses can be found in the supplementary material. n = 9 for the time points before surgery and 15 min, 3 h, 24 h postop.; n = 8 at 72 h postop.; n = 5 at 7 days postop. Correlation is expressed by Pearson’s or Spearman’s correlation coefficient (r) as appropriate, with p < 0.05 considered statistically significant.

Correlations of LVEDV with cerebral perfusion in sham animals. (A)–(E) Correlations of left ventricular end-diastolic volume (LVEDV) with cerebral perfusion at the indicated times post-surgery. (F) For 7 days postop., a correlation analysis was not performed due to the case number. n = 5 for the time points before surgery and 15 min post-surgery; n = 6 for 3 h and 24 h postop.; n = 4 at 72 h and n = 2 at 7 days postop. Correlation is expressed by Pearson’s or Spearman’s correlation coefficient (r) as appropriate, with p < 0.05 considered statistically significant.
Discussion
To the best of our knowledge, this is the first study to analyze the relationship between cardiac function and cerebral perfusion in a murine model of SAH. The most important findings of our study are: (i) Overall, SAH enhances LV contractility in the early period after SAH but with marked inter-individual variability, (ii) only a specific subset of animals developed echocardiographic and ECG signs of NSC, a pattern similar to SAH patients6,7,34,35, and (iii) LVEDV correlated significantly with cerebral perfusion at defined time points post-SAH, and there was a non-significant trend for correlation of cardiac output and stroke volume with cerebral perfusion. Taken together, these data show a relation between cerebral perfusion and cardiac function and therefore indicate that, in addition to cerebrovascular dysfunction, cardiac dysfunction may significantly influence cerebral perfusion, which presumably affects neurological outcome after SAH.
A substantial minority of SAH patients develop NSC, and post-SAH alterations in cardiac function have been linked to NSC development6,7,34,35,36. NSC symptoms range from shortness of breath to pulmonary edema, life-threatening cardiac arrhythmias, and congestive heart failure, and may be paralleled by increased biomarkers of myocardial injury, greater filling pressures, and abnormal ECG findings6,7. In a prospective clinical study, reduced LV function was observed in 15% and regional wall-motion abnormalities (RWMAs) without reduced LV function in 13% of 173 SAH patients in the first days after cranial bleeding34. Another prospective study reported pathological echocardiography findings in 17% of 63 SAH patients35. Neurogenic stress cardiomyopathy most likely results from excessive catecholamine release induced by the SAH, which leads to myocardial sympathetic overstimulation, excessive myocardial contraction, and ATP consumption, ensuing cardiomyocyte death, and finally cardiac inflammation6,7,24,37,38,39,40. NSC development has been linked to DCI and worse neurological outcome6,7,35,36.
Recent clinical research indicates that the degree of cerebral hypoperfusion during the first hours after SAH determines EBI severity, DCI occurrence, and neurological outcome26,27,28. Based on our experimental data, we suggest that cardiac dysfunction contributes directly to cerebral hypoperfusion and cerebral injury after SAH. This is supported by a clinical study that found an association between cardiac dysfunction and cerebral hypoperfusion during the first 24 h after hospital admission in SAH patients15. The similar patterns of cardiovascular and cerebrovascular dysfunction following both experimental SAH and clinical cases further validate this notion and suggest that our murine SAH model is well suited to study NSC and cardiac pathogenesis after SAH as well as to investigate novel therapeutic approaches.
We found a significant positive correlation between LVEDV and cerebral perfusion between 3 and 72 h post-SAH, as well as markedly lower LVEDV among individual animals. One possible reason for low LVEDV in some individuals (those showing the most severe hypoperfusion) could be a lack of intravascular volume, resulting in low preload and afterload. However, there were no obvious differences in body weight loss or intraoperative blood loss between animals with low and high LVEDV, making it unlikely that hypovolemia can account for poor post-SAH LVEDV. A clinical multicenter observational study, using pulse contour analysis, found that the global end-diastolic volume index (GEDI) was significantly lower on the 2 days preceding DCI in a group of SAH patients41. However, this apparent lack of intravascular volume was not caused by differences in fluid balance compared to patients without DCI. The strong correlation between LVEDV and cerebral perfusion at multiple early time points suggest that reduction in GEDI may directly contribute to DCI.
Another potential reason for reduced LVEDV is diastolic dysfunction. A clinical study observed diastolic dysfunction in 71% of 223 SAH patients, and diastolic dysfunction was associated with pulmonary edema42. However, a link between diastolic dysfunction and cerebral hypoperfusion had not been investigated. Therefore, we assessed the E / A relation, but found no difference between animals showing low or high LVEDV at time points with significant correlations between LVEDV and cerebral perfusion (3, 24, and 72 h post-SAH). Furthermore, there was no significant correlation between E/A and cerebral perfusion among SAH group animals or a significant difference in E/A between SAH and sham groups (supplementary material, Figs. E5, E6). These findings would argue against diastolic dysfunction accounting for low LVEDV and associated cerebral hypoperfusion; however, considering the sample sizes of our experimental study we cannot eliminate a contribution from diastolic dysfunction.
Finally, we want to address the limitations of our study. Firstly, only 9 SAH and 6 sham (control) animals entered the final analysis. Group sizes further decreased over time leaving 5 animals in the SAH group and 3 animals in the control group on day 7. Although statistical analysis revealed significant results, the small sample sizes need to be pointed out as a limitation of this study. Furthermore—because the measurements of perfusion and cardiac function were only performed at defined timepoints and not continuously—we cannot exclude that the lack of significance in several measures at the later time points is because the most affected mice were the ones that died. Secondly, a correlation between parameters of cardiac function and cerebral perfusion does not prove a causal relationship. We can furthermore not rule out that cerebral perfusion has an impact on cardiac function rather than cardiac function on cerebral perfusion. Nevertheless, our study demonstrates that a rodent model and a new experimental setting is available to experimentally investigate NCS and the relation of cardiac function and cerebral perfusion, and to potentially develop new therapeutic strategies to either prevent or treat NSC after SAH. Thirdly, the animals underwent repeated surgeries for imaging of cortical perfusion and ICP. Although the surgical trauma by the single measures was low and anesthesia times were short, stress imposed by the repeated surgeries as well as pharmacological effects of the anesthetic agents may present confounders. It should be noted, however, that the longitudinal nature of the study, which is made possible by the experimental setting used here, is a strength.
In summary, we show, to the best of our knowledge for the first time, that cerebral cortical hypoperfusion correlates with cardiac function after experimental SAH in mice. We conclude that in addition to cerebrovascular dysfunction, cardiac dysfunction may significantly influence cerebral perfusion, with LVEDV presenting a potential parameter for risk stratification. This finding may be of substantial clinical significance since it implies that both mitigation of cardiac dysfunction and suppression of cerebrovascular abnormalities, such as local cortical micro-vasospasm, are required to restore cerebral perfusion, which presumably is critical to obtain optimal neurological outcome. Our study confirms clinical approaches focusing on monitoring and optimizing cardiac function in patients suffering from SAH.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
Macdonald, R. L. & Schweizer, T. A. Spontaneous subarachnoid haemorrhage. Lancet 389, 655–666. https://doi.org/10.1016/s0140-6736(16)30668-7 (2017).
Lawton, M. T. & Vates, G. E. Subarachnoid hemorrhage. N. Engl. J. Med. 377, 257–266. https://doi.org/10.1056/NEJMcp1605827 (2017).
van Lieshout, J. H. et al. An introduction to the pathophysiology of aneurysmal subarachnoid hemorrhage. Neurosurg. Rev. 41, 917–930. https://doi.org/10.1007/s10143-017-0827-y (2018).
Macdonald, R. L. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 10, 44–58. https://doi.org/10.1038/nrneurol.2013.246 (2014).
Francoeur, C. L. & Mayer, S. A. Management of delayed cerebral ischemia after subarachnoid hemorrhage. Crit. Care 20, 277. https://doi.org/10.1186/s13054-016-1447-6 (2016).
Lee, V. H., Oh, J. K., Mulvagh, S. L. & Wijdicks, E. F. Mechanisms in neurogenic stress cardiomyopathy after aneurysmal subarachnoid hemorrhage. Neurocrit. Care 5, 243–249. https://doi.org/10.1385/ncc:5:3:243 (2006).
Chen, Z. et al. Brain–heart interaction: Cardiac complications after stroke. Circ. Res. 121, 451–468. https://doi.org/10.1161/circresaha.117.311170 (2017).
Budohoski, K. P. et al. Clinical relevance of cerebral autoregulation following subarachnoid haemorrhage. Nat. Rev. Neurol. 9, 152–163. https://doi.org/10.1038/nrneurol.2013.11 (2013).
Levine, B. D., Giller, C. A., Lane, L. D., Buckey, J. C. & Blomqvist, C. G. Cerebral versus systemic hemodynamics during graded orthostatic stress in humans. Circulation 90, 298–306. https://doi.org/10.1161/01.cir.90.1.298 (1994).
van Lieshout, J. J., Pott, F., Madsen, P. L., van Goudoever, J. & Secher, N. H. Muscle tensing during standing: Effects on cerebral tissue oxygenation and cerebral artery blood velocity. Stroke 32, 1546–1551. https://doi.org/10.1161/01.str.32.7.1546 (2001).
Gruhn, N. et al. Cerebral blood flow in patients with chronic heart failure before and after heart transplantation. Stroke 32, 2530–2533. https://doi.org/10.1161/hs1101.098360 (2001).
Massaro, A. R., Dutra, A. P., Almeida, D. R., Diniz, R. V. & Malheiros, S. M. Transcranial Doppler assessment of cerebral blood flow: Effect of cardiac transplantation. Neurology 66, 124–126. https://doi.org/10.1212/01.wnl.0000191397.57244.91 (2006).
van den Hurk, K. et al. Heart failure and cognitive function in the general population: The Hoorn Study. Eur. J. Heart Fail. 13, 1362–1369. https://doi.org/10.1093/eurjhf/hfr138 (2011).
Eggermont, L. H. et al. Cardiac disease and cognitive impairment: A systematic review. Heart 98, 1334–1340. https://doi.org/10.1136/heartjnl-2012-301682 (2012).
Cremers, C. H. et al. Relationship between cardiac dysfunction and cerebral perfusion in patients with aneurysmal subarachnoid hemorrhage. Neurocrit. Care 24, 202–206. https://doi.org/10.1007/s12028-015-0188-8 (2016).
Diringer, M. N. et al. Critical care management of patients following aneurysmal subarachnoid hemorrhage: Recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit. Care 15, 211–240. https://doi.org/10.1007/s12028-011-9605-9 (2011).
Connolly, E. S. Jr. et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: A guideline for healthcare professionals from the American Heart Association/american Stroke Association. Stroke 43, 1711–1737. https://doi.org/10.1161/STR.0b013e3182587839 (2012).
Gathier, C. S. et al. Effects of induced hypertension on cerebral perfusion in delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: A randomized clinical trial. Stroke 46, 3277–3281. https://doi.org/10.1161/strokeaha.115.010537 (2015).
Gathier, C. S. et al. Induced hypertension for delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: A randomized clinical trial. Stroke 49, 76–83. https://doi.org/10.1161/strokeaha.117.017956 (2018).
Westermaier, T., Jauss, A., Eriskat, J., Kunze, E. & Roosen, K. Time-course of cerebral perfusion and tissue oxygenation in the first 6 h after experimental subarachnoid hemorrhage in rats. J. Cereb. Blood Flow Metab. 29, 771–779. https://doi.org/10.1038/jcbfm.2008.169 (2009).
Neulen, A. et al. Large vessel vasospasm is not associated with cerebral cortical hypoperfusion in a murine model of subarachnoid hemorrhage. Transl. Stroke Res. https://doi.org/10.1007/s12975-018-0647-6 (2018).
Neulen, A. et al. Neutrophils mediate early cerebral cortical hypoperfusion in a murine model of subarachnoid haemorrhage. Sci. Rep. 9, 8460. https://doi.org/10.1038/s41598-019-44906-9 (2019).
Burch, G. E., Sun, S. C., Colcolough, H. L., DePasquale, N. P. & Sohal, R. S. Acute myocardial lesions; following experimentally-induced intracranial hemorrhage in mice: A histological and histochemical study. Arch. Pathol. 84, 517–521 (1967).
McNair, J. L., Clower, B. R. & Sanford, R. A. The effect of reserpine pretreatment on myocardial damage associated with simulated intracranial hemorrhage in mice. Eur. J. Pharmacol. 9, 1–6 (1970).
Hawkins, W. E. & Clower, B. R. Myocardial damage after head trauma and simulated intracranial haemorrhage in mice: The role of the autonomic nervous system. Cardiovasc. Res. 5, 524–529. https://doi.org/10.1093/cvr/5.4.524 (1971).
van der Schaaf, I. et al. CT after subarachnoid hemorrhage: Relation of cerebral perfusion to delayed cerebral ischemia. Neurology 66, 1533–1538. https://doi.org/10.1212/01.wnl.0000216272.67895.d3 (2006).
Schubert, G. A., Seiz, M., Hegewald, A. A., Manville, J. & Thome, C. Acute hypoperfusion immediately after subarachnoid hemorrhage: A xenon contrast-enhanced CT study. J. Neurotrauma 26, 2225–2231. https://doi.org/10.1089/neu.2009.0924 (2009).
Etminan, N. et al. Early perfusion computerized tomography imaging as a radiographic surrogate for delayed cerebral ischemia and functional outcome after subarachnoid hemorrhage. Stroke 44, 1260–1266. https://doi.org/10.1161/strokeaha.111.675975 (2013).
Westermaier, T., Jauss, A., Eriskat, J., Kunze, E. & Roosen, K. Acute vasoconstriction: Decrease and recovery of cerebral blood flow after various intensities of experimental subarachnoid hemorrhage in rats. J. Neurosurg. 110, 996–1002. https://doi.org/10.3171/2008.8.Jns08591 (2009).
Friedrich, B., Muller, F., Feiler, S., Scholler, K. & Plesnila, N. Experimental subarachnoid hemorrhage causes early and long-lasting microarterial constriction and microthrombosis: An in-vivo microscopy study. J. Cereb. Blood Flow Metab. 32, 447–455. https://doi.org/10.1038/jcbfm.2011.154 (2012).
Liu, H. et al. Microvasospasms after experimental subarachnoid hemorrhage do not depend on endothelin a receptors. Stroke 49, 693–699. https://doi.org/10.1161/strokeaha.117.020028 (2018).
Neulen, A. et al. A segmentation-based volumetric approach to localize and quantify cerebral vasospasm based on tomographic imaging data. PLoS ONE 12, e0172010. https://doi.org/10.1371/journal.pone.0172010 (2017).
Luh, C. et al. The contractile apparatus is essential for the integrity of the blood–brain barrier after experimental subarachnoid hemorrhage. Transl. Stroke Res. https://doi.org/10.1007/s12975-018-0677-0 (2018).
Banki, N. et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J. Neurosurg. 105, 15–20. https://doi.org/10.3171/jns.2006.105.1.15 (2006).
Kilbourn, K. J., Ching, G., Silverman, D. I., McCullough, L. & Brown, R. J. Clinical outcomes after neurogenic stress induced cardiomyopathy in aneurysmal sub-arachnoid hemorrhage: A prospective cohort study. Clin. Neurol. Neurosurg. 128, 4–9. https://doi.org/10.1016/j.clineuro.2014.10.017 (2015).
van der Bilt, I. A. et al. Impact of cardiac complications on outcome after aneurysmal subarachnoid hemorrhage: A meta-analysis. Neurology 72, 635–642. https://doi.org/10.1212/01.wnl.0000342471.07290.07 (2009).
Nguyen, H. & Zaroff, J. G. Neurogenic stunned myocardium. Curr. Neurol. Neurosci. Rep. 9, 486–491 (2009).
Salem, R. et al. Subarachnoid hemorrhage induces an early and reversible cardiac injury associated with catecholamine release: One-week follow-up study. Crit. Care 18, 558. https://doi.org/10.1186/s13054-014-0558-1 (2014).
Masuda, T. et al. Sympathetic nervous activity and myocardial damage immediately after subarachnoid hemorrhage in a unique animal model. Stroke 33, 1671–1676 (2002).
Novitzky, D., Wicomb, W. N., Cooper, D. K., Rose, A. G. & Reichart, B. Prevention of myocardial injury during brain death by total cardiac sympathectomy in the Chacma baboon. Ann. Thorac. Surg. 41, 520–524 (1986).
Watanabe, A. et al. Global end-diastolic volume is associated with the occurrence of delayed cerebral ischemia and pulmonary edema after subarachnoid hemorrhage. Shock 38, 480–485. https://doi.org/10.1097/SHK.0b013e31826a3813 (2012).
Kopelnik, A. et al. Prevalence and implications of diastolic dysfunction after subarachnoid hemorrhage. Neurocrit. Care 3, 132–138. https://doi.org/10.1385/ncc:3:2:132 (2005).
Acknowledgements
The authors would like to thank Stefan Kindel (Mainz, Germany) for preparing the illustrations in Fig. 1. This manuscript contains results that are part of the doctoral thesis of E. Holzbach, presented to the Medical Faculty of the Johannes Gutenberg-University of Mainz (Mainz, Germany). A preliminary version of this study was presented as an abstract at the 45th annual meeting on intracranial pressure, cerebral perfusion, and hydrocephalus of the German Society of Neurosurgery, and at the 71st Annual Meeting of the German Society of Neurosurgery. The authors would like to thank Enago (http://www.enago.com) for the English language review.
Funding
Open Access funding enabled and organized by Projekt DEAL.. PW, MM and SHK were supported by the German Federal Ministry for Education and Research (BMBF 01EO1503) and by the Boehringer Ingelheim Foundation “Novel and neglected cardiovascular risk factors: molecular mechanisms and therapeutic implications”. The work was supported by a Large Instrumentation Grant of the German Research Foundation (DFG INST 371/47-1 FUGG).
Author information
Authors and Affiliations
Contributions
Study design and concept: A.N., M.M., S.H.K., S.C.T.; Experiments: A.N., M.M., T.P., W.S.R. and E.H.; Data analysis: A.N., M.M., M.K., E.H., S.C.T.; Manuscript writing: A.N., M.M., S.H.K., S.C.T.; Revision of the manuscript: A.N., M.M., M.K., T.P., E.H., P.W., S.H.K., F.R., S.C.T.; Academic supervision: P.W., F.R., S.C.T.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary Video 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Neulen, A., Molitor, M., Kosterhon, M. et al. Correlation of cardiac function and cerebral perfusion in a murine model of subarachnoid hemorrhage. Sci Rep 11, 3317 (2021). https://doi.org/10.1038/s41598-021-82583-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-82583-9
This article is cited by
-
Blood pressure and outcome after aneurysmal subarachnoid hemorrhage
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
