
Abstract
We report a method to generate a 3D motor neuron model with segregated and directed axonal outgrowth. iPSC-derived motor neurons are cultured in extracellular matrix gel in a microfluidic platform. Neurons extend their axons into an adjacent layer of gel, whereas dendrites and soma remain predominantly in the somal compartment, as verified by immunofluorescent staining. Axonal outgrowth could be precisely quantified and was shown to respond to the chemotherapeutic drug vincristine in a highly reproducible dose-dependent manner. The model was shown susceptible to excitotoxicity upon exposure with excess glutamate and showed formation of stress granules upon excess glutamate or sodium arsenite exposure, mimicking processes common in motor neuron diseases. Importantly, outgrowing axons could be attracted and repelled through a gradient of axonal guidance cues, such as semaphorins. The platform comprises 40 chips arranged underneath a microtiter plate providing both throughput and compatibility to standard laboratory equipment. The model will thus prove ideal for studying axonal biology and disease, drug discovery and regenerative medicine.
Similar content being viewed by others


Introduction
Motor neurons project their axons to innervate other neurons and smooth- and skeletal muscle, allowing muscles in the body to contract. Impaired motor neuron function due to nerve injury or disease, affects voluntary movement and can cause paralysis. Although peripheral nerves are able to regenerate after injury, complete recovery does not occur1,2,3. Disease is another cause of disrupted motor neuron and axon function. In amyotrophic lateral sclerosis (ALS), upper and lower motor neurons undergo progressive degeneration leading to muscle denervation, paralysis and death within 3–5 years after diagnosis4,5. Although exact causes are still unknown, aberrances in axonal stability, axonal transport, and axon growth dynamics have been reported amongst other hallmarks5,6,7,8,9,10. Accordingly, there is an increased research interest in studying axonal repair after peripheral nerve injury, as well as in studying axonal pathology in ALS. In vitro models that are relevant in their physiological behavior and allow spatially segregated interrogation of motor neuron somata and axons, are critical in advancing our understanding of peripheral nerve injury and disease, and in developing novel curing or regenerating therapies.
The introduction of induced pluripotent stem cell (iPSC) technology and subsequent differentiation into human motor neurons provides a unique opportunity to study motor neuron disease and nerve regeneration mechanisms. In particular, the generation of iPSC-derived motor neurons from patients carrying specific mutations is a powerful tool for in vitro disease modeling. Most studies have focused on iPSC-derived lower motor neurons to study ALS. Lower motor neurons from familial ALS patients carrying superoxide dismutase 1 (SOD1) mutations showed various disease phenotypes compared to healthy controls, including reduced survival, abnormal mitochondrial mobility and endoplasmic reticulum (ER) stress11,12. Lower motor neurons derived from patients carrying a mutation in the fused in sarcoma (FUS) gene showed cytoplasmic mislocalization of FUS and accumulation into SGs, in addition to aberrant axonal transport of mitochondria and ER vesicles13,14. Relevant disease phenotypes were also observed using iPSC technology for other common mutations in familial ALS, such as chromosome 9 open reading frame 72 (C9ORF72) and TAR DNA binding protein 43 (TDP-43) mutations. An extensive overview of this topic is provided elsewhere15.
Despite recent developments in iPSC technology, progress in dissecting ALS disease mechanisms, or in developing efficient therapies, is often hampered by a lack of relevant and scalable in vitro platforms that can rapidly be used to study axonal pathology. Two-dimensional culture platforms have been used extensively to model neurite outgrowth and regeneration, reviewed by Al-Ali et al.16. Low-density neuronal cultures allow for assessment of morphology and neurite outgrowth of individual cells. These cultures can be used to assess the effects of genetic mutations and effects of neurotoxic and neuroprotective compounds. Alternatively, stripe assays and spot assays can be employed. These 2D assays make use of control substances and test substances applied in stripes or spots, after which neurons are plated and neurite outgrowth onto the stripes or into the spots is detected via microscopy17,18. High-density neuronal cultures do not allow for assessment of morphology and neurite outgrowth of single cells. However, they can still provide information on overall outgrowth in a system, e.g. via scratch assays, in which cells and neurites are removed or damaged mechanically and recovery can be observed by microscopy19.
While traditional 2D cultures such as the ones described above have shown invaluable for studying neurite outgrowth, it has been reported that neuronal cultures in 3D display increased survival rates and distinct gene expression profiles as compared to 2D monolayers20. More importantly, traditional cultures do not allow for directed neurite outgrowth and separation of cell somata and neurites, which is of major interest in studying motor axon biology. Microfluidic systems are emerging as novel tools that counterbalance important drawbacks of commonly used in vitro culture setups. Microfluidic systems provide excellent spatial control of cells and matrices21, and are routinely used to study axon outgrowth. Drawing inspiration from early work by Robert Campenot22, Taylor and colleagues developed a microfluidic chip that allowed assessment of neurite outgrowth in a two-dimensional setting by employing microgrooves23. A similar setup was employed by many others to generate compartmentalized neuronal cultures that segregate cell somata and axons24,25,26,27,28,29,30. Others have developed three-dimensional microfluidic cultures for the same purpose, showing compartmentalized somata and axon outgrowth and connection to skeletal muscle31,32. Although these platforms were successfully used to study axonal injury and regeneration, they are typically implemented as a single chip system and not compatible with automation, and therefore not suited for routine experimentation or high-throughput screening. In addition, most chips are fabricated only by the research groups that developed them, and use a silicon rubber as prototyping material33. This material typically leads to high absorption of hydrophobic compounds. Moreover, the fabrication of most chips is cumbersome and hampers routine adoption by other laboratories.
Here we report a method to generate a compartmentalized neurite outgrowth model in a 3D fashion, using a high-throughput microfluidic platform that is made out of materials that are biocompatible and non-absorbing34. iPSC-derived motor neuron progenitors were differentiated into cholinergic lower motor neurons in 3D in an extracellular matrix gel. While somata and dendrites stayed predominantly within the somal compartment, axons extended into an adjacent layer of gel, resulting in spatial segregation of axons from somata and dendrites. Axon outgrowth and stability were assessed in response to the chemotherapeutic drug vincristine, which is known to bind tubulin monomers and disrupt microtubule networks35. In addition, cultures were exposed to high concentrations of glutamate or to sodium arsenite, mimicking excitotoxicity and SG formation seen in ALS pathology. Finally, axon attraction and repulsion through gradients of chemotrophic molecules was studied, which is of interest for regenerative approaches. The OrganoPlate platform used comprises 40 chips underneath a microtiter plate, enabling both throughput and compatibility of the platform. We envision the use of this model in studying motor neuron disease- and regeneration-related processes that may aid the discovery of novel therapeutic directions.
Results
Culture of motor neuron progenitors in the OrganoPlate
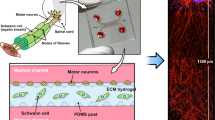
The OrganoPlate 3-lane (Fig. 1A) is a microfluidic platform comprising 40 tissue culture chips which can be used for the generation of miniaturized tissues34. It employs a microtiter plate design, making it compatible with standard laboratory equipment. A schematic depiction of a single chip is shown in Fig. 1B. Within a chip, the microfluidic lanes are patterned by small pressure barriers termed phaseguides36. Phaseguides cause a change in capillary pressure subsequently preventing fluids to overflow into the adjacent lane, thereby patterning them without the use of artificial membranes. We leveraged the OrganoPlate 3-lane to generate a segregated motor neuron neurite outgrowth model in which developing neurites are separated from cell somata (Fig. 1B). Matrigel-growth factor reduced (GFR) was dispensed in the middle lane, after which motor neuron progenitors (MNPs) embedded in matrigel-GFR were added to the top lane. Motor neuron differentiation medium was then added to the top and bottom lane to differentiate the MNPs into mature motor neurons. In this set-up, MNP differentiation was accompanied by neurite extension into the adjacent microfluidic lane. A schematic overview of the procedure is shown in Fig. 1C,D.

Motor neuron progenitors form 3D networks in the OrganoPlate. (A) Image of an OrganoPlate 3-lane. The OrganoPlate is a microfluidic platform based on a 384-well plate design containing 40 individual tissue culture chips. The image is copyright of MIMETAS BV. (B) Each tissue culture chip contains 3 microfluidic lanes that join in the center of the chip. The 3-lane architecture in the chips’ center is assured by phaseguides that enable patterning of extracellular matrix gel. Inlet and outlet wells allow access to each lane: top inlet (TI) and top outlet (TO) wells allow access to top lane; middle inlet (MI) and outlet (MO) wells to middle lane; and bottom inlet (BI) and outlet (BO) wells to the bottom lane. Matrigel-GFR without cells is added to the middle lane. Following gelation, motor neuron progenitors embedded in matrigel-GFR are seeded in the top lane. (C) Schematic overview of cell seeding procedure. Matrigel-GFR is seeded in the middle lane and MNPs embedded in matrigel-GFR are seeded in the top lane (panel 1). MNPs form networks in the top lane (panel 2), after which neurites protrude into the middle and eventually bottom lane (panel 3). Blue color indicates presence of gel, red color indicates presence of medium. (D) Schematic overview of the spatial segregation of motor neuron somata (top lane) and axons (middle and bottom lanes) in OrganoPlate 3-lane cultures.
Characterization of motor neuron differentiation and neurite outgrowth in the OrganoPlate
To characterize the cells in the OrganoPlate, we performed immunostaining and qPCR analyses. MNPs were differentiated in 3D in the OrganoPlate for 17 days and immunostained to confirm the presence of motor neuron markers SMI32, islet-1 (ISL1), and choline acetyltransferase (CHAT) (Fig. 2A). To evaluate the success of the differentiation, gene expression of the 17-day differentiated motor neurons was compared to non-differentiated MNPs and iPSCs. MNPs and differentiated motor neurons showed expression of motor neuron markers ISL1, CHAT, NFH, and SLC18A3, encoding the vesicular acetylcholine transporter (VACHT) protein, mostly at similar levels. This indicates that the MNPs used in this study already express many genes associated with motor neurons. However, the differentiated motor neurons showed reduced expression of progenitor markers OLIG2, SOX2, and PAX6 compared to the MNPs, indicating further differentiation and maturation (Fig. 2B, Supplementary Fig. S1). The expression of the glial marker S100β was low and did not increase substantially over time. To characterize neurite outgrowth, cultures were immunostained for the somatodendritic marker microtubule-associated protein 2 (MAP2) and the axonal marker microtubule-associated protein TAU (MAPT)/TAU. Neurites penetrating the middle lane were mostly TAU-positive, while only some neurites contained MAP2 (Fig. 2C). These results indicated that the neurites protruding the adjacent lane were predominantly axonal.

Progenitors differentiate into motor neurons and extend their axons into an adjacent gel lane. (A) MNPs were differentiated in the OrganoPlate for 17 days and immunostained for motor neuron markers SMI-32, ISL1, and CHAT. Maximum projection images of neuronal networks in the top lane are shown, the scale bar is 200 µm. (B) mRNA expression levels of induced pluripotent stem cells (iPSC), motor neuron progenitors (MNP) and motor neurons differentiated for 17 days (MN d17) in the OrganoPlate. Expression of markers of neural stem cells (NANOG, SOX2, PAX6), motor neuron progenitors (OLIG2), motor neurons (ISL1, CHAT, RBFOX3, VACHT, NFH), mature neurons (SYN1/SYNAPSIN, MAP2) and glial cells (S100β) was evaluated. Heatmap shows fold-change values as compared to expression in iPSCs, and the graphic representation was created using GraphPad Prism, version 8.3.1 (https://www.graphpad.com/scientific-software/prism/). (C) Motor neurons (day 17) were immunostained for dendritic marker MAP2 (red) and axonal marker TAU (green). Neurites protruding into the middle lane are mostly TAU-positive and MAP2-negative, indicating a predominantly axonal nature. Maximum projection images, the scale bar is 200 µm.
Finally, we assessed spontaneous calcium fluctuations as an indication of electrophysiological activity. The calcium-sensitive dye Cal-520 AM was loaded into the cells, after which intracellular calcium fluctuations were detected using high-speed imaging (Supplementary Video S1). The videos show spontaneous calcium fluctuations confirming that the neurons are electrophysiologically active. Overall, these data show that differentiated motor neuron progenitors are mature, electrophysiologically active, and send their axons into the middle lane of the tissue culture chip.
Neuronal network formation and quantification of axon outgrowth
Motor neurons send their axons over long distances to connect with their target cells, and changes in motor axon projections are a hallmark of motor neuron disease. Thus, as axon outgrowth and morphology are important parameters, we investigated our ability to quantify axon outgrowth in the OrganoPlate. MNPs seeded in the OrganoPlate formed neuronal networks in the top lane only during the first 6 days, after which they started to protrude their processes into the middle lane (Fig. 3A). This network became increasingly complex over time, where axons eventually penetrated into the bottom lane (Fig. 3A, Supplementary Fig. S2). Interestingly, the addition of 10 ng/mL brain-derived neurotrophic factor BDNF to the bottom lane increased cell migration into the middle lane (Supplementary Fig. S2). Moreover, upon filling the middle lane with a collagen-I ECM, axon outgrowth was substantially reduced (Supplementary Fig. S3). These observations indicate that specific molecular changes affect the behavior of motor neurons and axons in the OrganoPlate. To quantify the outgrowth, the raw signal from the middle lane (region of interest, ROI) was converted into a binary image where black signal indicates the presence of live cells and processes. The black signal was then skeletonized, after which total axon length was extracted (Fig. 3B). As expected, total axon outgrowth increased over time (Fig. 3C). Axon outgrowth was significantly increased at day 14 (P = 0.0003) and 17 (P < 0.0001) when compared to day 6. For situations of abundant outgrowth, an alternative quantification approach may be employed where black signal is quantified directly as a percentage of the total ROI measured (Supplementary Fig. S4). The first approach, in which axons are skeletonized, offers the advantage of quantifying axon length in micrometers. However, both quantification approaches yielded comparable results. To finally demonstrate the 3-dimensionality of the cultures, Z-slices from a single tissue culture chip were made and used for the generation of a 3D reconstruction (Fig. 3D, Supplementary Video S2). Taken together, these results show that motor neurons in the OrganoPlate increasingly extend their axons into adjacent lanes, which can be reliably quantified using different approaches.

Characterization of 3D axon outgrowth. (A) Motor neuron axon outgrowth over time, visualized using calcein-AM dye. Dashed lines indicate region of interest (ROI) used for axon outgrowth quantification. Maximum projection images, the scale bar is 140 µm. (B) Quantification of axon outgrowth in the middle lane at day 17. Green-fluorescent signal of calcein-labeled axons was converted into a binary image, and subsequently converted into a skeletonized image that allows quantification of neurite length. (C) Quantification of motor neuron axon outgrowth over time using the procedure depicted in b. Graph shows mean neurite length ± SEM, n = 10, N = 2. *** (P < 0.001), **** (P < 0.0001); Kruskall–Wallis test. The graphic representation was created using GraphPad Prism, version 8.3.1 (https://www.graphpad.com/scientific-software/prism/). (D) 3D reconstruction of neuronal network and axonal outgrowth rendered from a confocal Z-stack, visualized using a calcein-AM dye at day 17. A side view is shown in panel i. Panel ii shows a top view. Panels i and ii were created using Imaris Viewer (https://imaris.oxinst.com/imaris-viewer). Panel iii depicts the Z-axis of the culture, in which structures at different depths within the chip are depicted using a color gradient to visualize the 3D nature of the culture. Deeper structures are depicted in blue, more superficial structures are depicted in green, followed by yellow and orange. Panel iii was created in ImageJ76.
Robust modelling of disease-relevant phenotypes for screening
We next assessed the robustness of the outgrowth between chips and found highly similar outgrowth in 40 chips of a complete OrganoPlate 3-lane (Fig. 4A,B). As axon outgrowth was robust, we then proceeded with exposing our cultures to various concentrations of vincristine. Vincristine binds tubulin monomers and prevents their incorporation in the microtubule network, halting axon outgrowth and destabilizing developing axons. Vincristine concentrations of 0.01 nM and lower did not notably affect axon outgrowth, while concentrations of 10 nM and higher almost completely disrupted axons in the middle and bottom lane (Fig. 4C,D). In the OrganoPlate, vincristine is thus shown to have a dynamic range between 0.1 and 10 nM. In concordance, the IC50 of vincristine exposure was determined to be 0.5039 nM. To further characterize these experiments, LDH release was measured as an indicator of cytotoxicity. Vincristine substantially affected axon outgrowth, while only at the highest concentration tested vincristine may lead to increased LDH activity (P = 0.065; Supplementary Fig. S5). These results indicated that our treatment can specifically target axon outgrowth and stability.

Robust 3D axonal outgrowth model allows assessment of neurite disrupting compounds. (A) Montage of a complete OrganoPlate containing 40 chips, each showing 3D axonal outgrowth at day 17 of culture visualized by calcein-AM dye. Maximum projection images, the scale bar is 200 µm. (B) Quantification of axonal outgrowth shown in (A), n = 40. Graph shows mean ± SD. (C) Motor neurons (day 17) were exposed to various concentrations of vincristine for 4 days. Cultures were visualized with calcein-AM dye. Maximum projection images, the scale bar is 200 µm. (D) Quantification of axonal outgrowth depicted in (C). Dose–response curve showing the effect of vincristine exposure on axonal outgrowth. Graph shows mean values ± SEM and a 95% confidence interval (dotted lines) around the curve. n = 5. The graphic representations in (B) and (D) were created using GraphPad Prism, version 8.3.1 (https://www.graphpad.com/scientific-software/prism/).
Glutamate toxicity is a hallmark of diseases such as ALS (Taylor et al., 2016). To examine whether we could model this pathological situation in the OrganoPlate culture, we exposed cultures to 5 mM glutamate for 7 days (day 10–17), in line with a previous study32. Excess glutamate significantly reduced axon outgrowth as compared to control treatment (P = 0.041; Fig. 5A,B). Exposure to sodium arsenite is another commonly used treatment for studying neuronal vulnerability in ALS and other diseases, as patient cells may respond differently to cellular stress37,38,39. Short-term exposure to sodium arsenite (3 h) did not significantly reduce axon outgrowth (Fig. 5A,B), but did induce formation of SGs, as did treatment with excess glutamate (Fig. 5C). SGs are accumulations of proteins and RNAs, mostly consisting of stalled translation initiation complexes, that form in a reversible manner upon cellular stress. Treatment with glutamate or sodium arsenite induced SG formation as shown by G3BP1 immunostainings. G3BP1 is a major component of SGs and shows a diffuse expression pattern in control conditions. Following both treatments, more granular patterns were found that are indicative of SG formation (Fig. 5C). We confirmed that the SGs are formed in neurons rather than non-neuronal cells, as results from immunostaining (data not shown) and qPCR analysis only showed sporadic presence of glial cells in our cultures. The low mRNA levels of s100β are presented in Supplementary Fig. S1. As expected, FUS remained mostly nuclear in all conditions (Fig. 5C), indicating that the incorporation of FUS in SGs requires ALS mutations in the FUS gene or other additional insults. Overall, these data show that the OrganoPlate motor neuron cultures can be used to study disease-relevant phenotypes, such as axon degeneration and SG formation.

Excitotoxic conditions induce reduced axon outgrowth and formation of stress granules. (A) Motor neurons (day 17) were exposed to 5 mM glutamate for 7 days or 0.05 mM sodium arsenite for 3 h and labeled with calcein-AM dye. The scale bar is 200 µm. (B) Quantification of the axon outgrowth depicted in (A). Graph shows mean ± SEM. n = 2–4, *(P < 0.05); Kruskall-Wallis test. The graphic representation was created using GraphPad Prism, version 8.3.1 (https://www.graphpad.com/scientific-software/prism/). (C) Motor neurons (day 17) grown in an OrganoPlate 2-lane were exposed to 5 mM glutamate for 7 days or 0.05 mM sodium arsenite for 3 h. Cells were immunostained for the stress granule marker G3BP1 (green) and for FUS (red). Maximum projection images, the scale bar is 20 µm.
Axon outgrowth can be directed in the OrganoPlate
The observation that axons extend towards the bottom lane, demonstrates that the OrganoPlate setup can be used for investigating directed human motor axonal outgrowth. This is of interest for studying motor axon biology or for devising regenerative strategies. Previously, gradient formation in the OrganoPlate has been reported to be stable for up to 6 days40. We now verify this for our assay setup. Figure 6A shows a gradient of 150 kDa fluorescein isothiocyanate (FITC)-Dextran molecules in our culture setup comprising neurons in matrigel. The gradient stays stable for at least 72 h. The stability of the gradients is longer than the time interval between medium changes (2–3 days), indicating that gradients can be maintained over the entire culture period (Fig. 6A).

Attraction and repulsion of axons. (A) Gradient formation is visualized by perfusion of a fluorescein isocyanate (FITC)-Dextran (150 kDa) molecule through the bottom lane of a tissue culture chip. Images were taken at 24 h, 48 h, and 72 h after perfusion, showing stable gradient formation in the OrganoPlate. (B) Motor neurons (day 17) were cultured with either DMEM/F12 or motor neuron differentiation medium in the bottom lane to assess axon attraction. Cultures are visualized using calein-AM dye. Maximum projection images, the scale bar is 200 µm. (C) Quantification of axon outgrowth shown in (B) shows a significant (P < 0.0001) increase in outgrowth upon perfusion of the bottom lane with motor neuron differentiation medium. Graph shows mean ± SEM, n = 20–29, N = 2; unpaired t test. (D) Motor neurons (day 17) were cultured with either semaphorin 3F (SEMA3F) or 3A (SEMA3A) in the bottom lane to assess axon repulsion. Cultures are visualized using calein-AM dye. Maximum projection images, the scale bar is 200 µm. Dashed lines indicate the region of interest for axon outgrowth quantification in (E). (E) Quantification of the axon outgrowth depicted in (D). Axons were significantly less prevalent in the bottom microfluidic lane when perfused with SEMA3F (1 µg/mL, P = 0.001; 0.1 µg/mL, P = 0.0003) or SEMA3A (1 µg/mL, P = 0.001). Graph shows mean ± SEM, n = 5, *(P < 0.05), ** (P < 0.01), or *** (P < 0.001); Brown-Forsythe and Welch test. The graphic representations in (B) and (D) were created using GraphPad Prism, version 8.3.1 (https://www.graphpad.com/scientific-software/prism/).
We then attempted to use these gradients to direct axonal outgrowth. Interestingly, perfusion of the bottom lane with motor neuron differentiation medium caused significantly increased extension of axons into this lane as compared to perfusion with DMEM/F12 (P < 0.0001; Fig. 6B,C). In contrast, perfusion of the axonal repellents SEMA3A or SEMA3F41,42,43 in the bottom lane inhibited axonal outgrowth. The results show that SEMA3A (1 µg/mL) prevented axons from extending towards the edge of the gel region, as compared to the vehicle control (P = 0.0003; Fig. 6D,E). Application of SEMA3F induced a dose-dependent decrease in axons extending to the edge of the gel region, with concentrations of 1 µg/mL (P = 0.001) and 0.1 µg/mL (P = 0.0003) having statistically significant effects, when compared to the vehicle control. The edge of the gel extends into the lower perfusion region as a consequence of meniscus stretching during phaseguide-assisted patterning44. For this reason, quantification in Fig. 6C,E is based on a region of interest taken in the lower perfusion channel.
In conclusion, gradients formed in the OrganoPlate can be used to attract and repel motor axons. Moreover, gradient-based studies may also be extended to toxicity studies, by solely perfusing the bottom lane with the toxin. We demonstrated this with vincristine, where axons in the middle lane were again significantly affected using vincristine concentrations of 1 nM and higher (P < 0.0001, Supplementary Fig. S6). However, it is important to note that the steepness of the gradient may be altered due to the small molecular size of vincristine. Together, the results suggest that the OrganoPlate setup can be used for investigating directed human motor axon outgrowth, which is of interest for advancing our knowledge in motor axon biology or for devising regenerative strategies.
Discussion
Here, we present a robust 3D model for studying human motor neurons and their axons in a perfused microfluidic platform. The platform enables segregated neurite outgrowth from motor neuron somata. Importantly, outgrowing neurites were demonstrated to be predominantly axonal. Outgrowth was shown to be highly reproducible and could be quantified by standard image analysis techniques. Moreover, axon outgrowth and stability were affected by a compound acting on the microtubule network. Furthermore, we could reproduce disease-relevant phenomena such as excitotoxicity and stress granule formation, while axons could also be attracted and repelled, demonstrating the ability to direct motor axonal outgrowth.
Segregation of axons from cell somata is of crucial importance for studying motor neuron diseases such as ALS. ALS is characterized by prominent axonal changes, eventually leading to axon degeneration and target de-innervation. Although changes such as altered axonal transport and cytoskeletal abnormalities have been reported45,46, early pathological changes in motor axons in ALS remain poorly characterized. However, they provide important therapeutic targets due to their early occurrence in the disease process. It is expected that high-throughput platforms can be used to identify reagents that target axon degeneration, which will accelerate drug development for ALS and other motor neuron diseases. To demonstrate the applicability of our model for compound testing, we exposed our cultures to a concentration range of vincristine. Vincristine is a chemotherapeutic agent which belongs to the vinca-alkaloids and primarily inhibits and disrupts microtubule networks by binding α- and β-tubulin monomers35. We show that vincristine caused a dose-dependent decrease in the axon density in the middle lane. Lack of clear changes in LDH activity indicates that axons may be affected in the absence of robust effects on cell viability in general. This observation not only confirms that changes in axon integrity can be modeled in our platform, but also that it is suitable for generating dose–response curves for specific compounds.
In our study, glutamate exposure led to a decrease in axon outgrowth and to a qualitative increase in the number of SGs. These results indicate that ALS-relevant phenotypes, such as glutamate toxicity and SG formation, can be captured in the OrganoPlate platform. It is thought that in ALS patients, motor neurons are exposed to pathological levels of extracellular glutamate, causing excitotoxicity47. Furthermore, motor neurons of most ALS patients show pathological protein aggregates that contain the RNA-binding protein TDP-4348,49,50. Other proteins such as the RNA binding protein FUS also localize in protein aggregates, but in a smaller group of patients51,52. Both TDP-43 and FUS play important roles in RNA processing and predominantly localize in the nucleus of healthy motor neurons. However, FUS and TDP-43 become more cytosolic in motor neurons of ALS patients, where they influence the assembly and dynamics of SGs53. SGs form when cells are exposed to adverse environmental conditions, such as heat shock, viral infection and oxidative stress54, and are believed to contribute to the formation of insoluble protein aggregates53,55. Changes in SG formation and dynamics are therefore thought to contribute to ALS pathology37. The mechanisms with which this happens may be studied more quantitatively in future studies.
The OrganoPlate may also be used to identify cues to enhance motor nerve regeneration following peripheral nerve or spinal cord injury. Motor axons often fail to successfully repair severed motor axon projections and no therapeutic interventions are available to stimulate this process. Since molecular gradients are known to influence axon outgrowth in vivo, we first demonstrated our ability to generate gradients in the OrganoPlate. We then attempted to utilize gradients to provide outgrowing axons with chemotrophic cues. Perfusion of motor neuron differentiation medium in the bottom lane increased axon outgrowth as compared to basal medium. This may either be explained by gradient formation of growth factors which influence the rate with which axons protrude into the middle lane, while it cannot be excluded that, upon perfusion of the bottom lane with basal medium, dilution of the differentiation medium contributes to the reduced axonal outgrowth.
Van Duinen et al.40 showed that a gradient could be maintained for many hours in the OrganoPlate. They demonstrated this is in a cell-free setup in which a gel was symmetrically perfused and in which one perfusion channel acted as a source and the other perfusion channel as a sink. Here we confirmed these results for our own assay setup. A crucial difference, however, is that our assay comprises neurons in matrigel that were inserted in the top lane. It should be expected that matrigel impedes perfusion flow in the top channel, thereby reducing the sink capacity. Our results show that the gradient is stable over prolonged periods, nonetheless. This may be explained by the interstitial flow through the matrigel, or by neuronal remodeling of the ECM which subsequently causes some level of perfusion flow.
We then perfused the bottom lane with semaphorins, which were expected to form directional repulsive cues. The molecular weight of these semaphorins upon dimerization (200–250 kDa) is higher than that of the FITC-dextran molecule (150 kDa) which has been used to confirm gradient formation. This implies that the semaphorin gradient is of similar or of lower steepness as compared to the gradient that we found for FITC-dextran. We prevented axons from protruding into the bottom microfluidic lane by providing SEMA3A or SEMA3F to this lane. Secreted class 3 semaphorins can act as axonal repellents via the Neuropilin receptor41,43,56,57. These results show that axons can be attracted and repelled in vitro in 3D in a segregated manner. Quantification was performed on the lower lane, where axons mostly bundle together at the interface of gel region with the perfusion lane. This causes some loss in spatial resolution as axons are overlapping in this region. We show, however, that this approach is still useful for capturing differences in axonal outgrowth. Such studies could employ the platform for studying the mechanism-of-action and function of various axon guidance cues as well as to search for reagents that could neutralize repulsive gradients, known to inhibit axon regeneration in vivo58, or to boost axon growth across sites of injury.
Evidently, motor axons do not grow in isolation but are surrounded by other cell types such as astrocytes and microglia in the central nervous system and Schwann cells in the peripheral nervous system, where they form synapses with skeletal muscle cells. The physiological relevance of our model can therefore be enhanced through the incorporation of these other cell types, which have shown to influence axon regeneration59,60,61, as well as ALS pathology62,63,64,65. The neuromuscular junction is of special interest, as it is affected at early stages in ALS66. In addition, the use of a chemically defined synthetic gel as an alternative to matrigel-GFR may be considered to further reduce the presence of growth factors in the ECM gel and to reduce possible batch-to-batch variation67.
The work described here employed motor neurons derived from a healthy donor and induced common motor neuron disease phenotypes by means of chemical triggers, such as excess glutamate and sodium arsenite. Although these techniques are widely used to study motor neuron diseases in vitro, a more complex approach can further improve the understanding of ALS disease mechanisms. Since diseases such as ALS are highly heterogeneous, it is likely that personalized treatments need to be developed for individual patients or specific patient groups. The compatibility of our platform with stem cell technology is beneficial for developing such personalized medicine approaches, allowing patient-derived material to be used68,69. Using the 3D neurite outgrowth model presented here, one can study differences between healthy and patient-derived motor neurons and their response to insults in a high-throughput ECM-embedded setting. Neurite outgrowth can be analyzed in 2D maximum projection images to assess high-level differences between conditions, as was done in this study, or in 3D at higher magnification to study individual somas and axons. Finally, the model may allow the development of other axon-based read-outs in the OrganoPlate. For example, axonal transport is widely disturbed in ALS and could be studied in 3D in our model14,45,70. As axonal pathology is not limited to motor neuron disease71,72,73, it will be interesting to assess whether growth of other neuronal populations in the OrganoPlate also results in axonal enrichment.
An important feature of the assay described here is that neurite outgrowth is 3D and not guided by mechanical cues such as microgrooves. In addition, particular attention was paid to the robustness and reproducibility of the assay, to allow smooth adoption by other laboratories. A crucial aspect is that all ingredients necessary for the assay are commercially available. The throughput of 40 chips per OrganoPlate, as well as its compatibility with standard microscopy and automation equipment, provides a good basis for routine experimentation. We thus foresee that the assay will be picked up by a wide audience.
In conclusion, we have established a motor neuron model with directed and segregated axon outgrowth, which can be used to study motor axons in 3D. We demonstrate the versatility of the model by showing changes in axon outgrowth upon exposure to various neurotoxins and signaling molecules. The platform will allow us to obtain further insight into motor axon biology and disease.
Materials and methods
Cell culture
iPSC-derived motor neuron progenitors (MNPs) (Axol, ax0078) were expanded in T75 flasks (734-2705, Corning, NY, USA) coated with 1:100 matrigel-GFR (Corning, 356341). MNPs were cultured in a chemically defined motor neuron progenitor expansion medium previously described by Du et al.74. The medium consists of equal parts DMEM/F12 with Glutamax (Gibco, 31331-028) and Neurobasal (Gibco, 21103-049) medium, supplemented with 0.5 × N2 supplement (Thermo Fisher, 17502-048), 0.5X B27 supplement (Thermo Fisher, 12587-010), 1% penicillin/streptomycin (Sigma, P4333), 0.1 mM ascorbic acid (Sigma, 49752), 3 µM CHIR99021 (Axon Medchem, 1386), 2 µM DMH-1 (Sigma, D8946), 2 µM SB431542 (Sigma, S4317), 0.5 µM purmorphamine (Enzo Life Sciences, ALX-420-045-M001), 0.5 mM VPA (Sigma, P4543-10G), and 0.1 µM retinoic acid (Sigma, R2625). Medium was refreshed three times a week. Cells were cultured up to passage 10 in a humidified incubator at 37 °C and 5% CO2 before seeding in the OrganoPlate. Cell detachment was performed by washing the cells 1 × using Phosphate Buffered Saline (PBS), after which cells were incubated for 5 min using accutase (Sigma, A6964).
Culture of iPSC-derived MNPs in the OrganoPlate
iPSC-derived MNPs were seeded in an OrganoPlate 3-lane (MIMETAS, 4004-400B) for neurite outgrowth experiments. Each tissue culture chip consists of three microfluidic lanes of 400 µm × 220 µm (w × h) each44. Before seeding the MNPs, matrigel-GFR was thawed for a minimum of 3 h on ice. On the day of seeding, 50 µL of PBS was added to the observation windows to prevent the ECM from drying out and to increase optical clarity. Then, 1.1–1.4 µL of matrigel-GFR was dispensed into each middle inlet and the plate was incubated for 3.5 min at 37 °C, 5% CO2, and 11.5 more at room temperature (RT). Alternatively, 1.7 µL of a collagen-I based ECM was disposed into the middle inlet, as previously described44, but with a 15-min incubation time. MNPs were then resuspended to a cell density of 30,000 cells/µL using matrigel-GFR, and 2.0 µL of this cell suspension was added to the top inlet. The plate was then again transferred to an incubator (37 °C, 5% CO2) for 15 min, after which a motor neuron differentiation medium was added to the top and bottom in- and outlet (50 µL to each well). The motor neuron differentiation medium used in this study has previously been described74 and consists of equal parts DMEM/F12 and Neurobasal medium, supplemented with 0.5 × N2 supplement, 0.5× B27 supplement, 1% penicillin/streptomycin, 0.1 mM ascorbic acid, 0.5 µM purmorphamine, and 0.1 µM retinoic acid. After day 6 of differentiation, 0.1 µM Compound E (Bio-Connect, AG-CR1-0081-C250) was added to the differentiation medium for the remaining culture time. The plate was then transferred to an incubator and placed on a rocking platform (7° inclination, 8-min interval), to initiate bidirectional perfusion flow.
For calcium imaging experiments and for the immunostainings following glutamate and sodium arsenite exposure, cells were seeded in an OrganoPlate 2-lane (MIMETAS, 9605-400B) with 200 µm × 200 µm (w × h) microfluidic lanes. Cells were detached and pelleted as described above. The OrganoPlate 2-lane was subsequently seeded as described previously75. Medium was added to the gel inlet, perfusion inlet and the perfusion outlet (50 µL to each well).
All read-outs were conducted on day 17, as the differentiation method used in this study was previously reported to yield mature motor neurons after 16 days of differentiation74. At day 17, the cultures in this study showed abundant and robust neurite outgrowth that could be quantified.
Assessment of gradient formation and stability
Directly following MNP seeding to an OrganoPlate 3-lane, motor neuron differentiation medium supplemented with 0.5 mg/mL 150 kDa FITC-Dextran (Sigma, 48946) was perfused through the bottom lane (50 µL per inlet and outlet). Identical volumes of motor neuron differentiation medium without dye were again added to the top inlet and outlet. Images of the corresponding wavelength were then taken using an ImageXpress Micro XLS microscope (Molecular Devices, Sunnyvale, CA, USA) using a 4 × objective. The plate was placed back onto the rocking platform following each imaging session. Pictures were taken after 24, 48 and 72 h to assess the stability of the gradient.
Calcein labeling of live motor neurons
To visualize live cells, including neurites, the medium was supplemented with 0.5 µg/mL calcein-AM (Thermo Fisher, C3099). Here, 50 µL of calcein-AM-supplemented medium was added to the top in- and outlets, while 25 µL was added to all middle and bottom in- and outlets. The plate was then placed back onto the rocking platform for 3 h. Images were captured using an ImageXpress Micro XLS-c microscope. Using a 10 × objective, Z-stacks with 3 µm spacing were captured and a maximum projection was saved per tissue culture chip. 3D reconstructions were rendered from a confocal Z-stack using Imaris Viewer (version: Viewer, Bitplane, Zürich, Switzerland, https://imaris.oxinst.com/imaris-viewer). ImageJ76 was used to depict the height of the cells in the Z-stack, rendering a 2D image.
Neurite outgrowth quantification
Maximum projections were processed in ImageJ76 to quantify neurite outgrowth. The center of the middle lane of the chips was selected as the region of interest for the data shown in Figs. 1, 2, 3, 4 and 5, as this lane features the 3D axonal outgrowth of the model. In short, calcein signal from the middle lane was isolated and subsequently converted into a binary image using the Huang threshold ImageJ plugin. The binary image was then skeletonized using the skeletonization ImageJ plugin, after which the total neurite length could be extracted from the skeletonized image. For more complex neurite networks, we used a slightly different quantification approach. Here, a binary image was generated in an identical manner. The proportion of the binary image containing signal (and thus neurites) was calculated. After Fig. 2, neurite outgrowth is referred to as axonal outgrowth. Extracted values were then compared across conditions and these were plotted in GraphPad Prism, version 8.3.1 (GraphPad Software, San Diego, CA, USA, https://www.graphpad.com/scientific-software/prism/). The skeletonization approach was used in Fig. 2 and Supplementary Fig. S3 only.
RNA isolation, cDNA synthesis and qPCR and data analysis
2D cultures of proliferating MNPs (Axol) and OrganoPlate cultures of 17-day differentiated motor neurons (from MNPs, Axol) were lysed using TRIzol (Thermo Fisher, 15596026). For the differentiated neurons, all contents of 40 OrganoPlate chips were lysed and pooled into one sample. Because the iPSCs from the same donor were not commercially available, iPSCs from a different donor were used as a reference and have previously been characterized more extensively, annotated as iPSC177,78. Samples were stored at − 20 °C before RNA extraction. RNA was extracted and cDNA prepared as previously described79. Primers were intron-spanning and designed with PrimerBLAST (NCBI). The expression fold changes were determined (2−ΔΔCT) and normalized with the geomean of the three reference genes GAPDH, BETA-ACTIN and TBPQ (see primer sequences in Table 1). Graphical representations were created in GraphPad Prism, version 8.3.1 (GraphPad Software, San Diego, CA, USA, https://www.graphpad.com/scientific-software/prism/).
Immunocytochemistry
At day 17 following MNP seeding, cultures were fixed using 4% paraformaldehyde (PFA; Sigma, P6148) in PBS. The following steps were performed as described previously44, with minor modifications. Cells were washed with PBS, blocked using 2% BSA, 0.1% Triton X-100 and either 5% goat serum or 1% normal donkey serum (NDS) when staining for CHAT. Moreover, 1% Triton X-100 was used in the blocking buffers for G3BP1 and FUS stainings only. Antibodies were diluted in a blocking buffer containing 2% BSA, 1% Triton X-100 and 1% NDS or 5% goat serum and incubated for 2–3 day and DNA was labeled using Hoechst (Thermo Fisher, H3570). Images were subsequently captured with an ImageXpress Micro XLS microscope. Using a 10 × objective, Z-stacks were captured with 3 µm spacing and a maximum projection was saved per tissue culture chip. For Fig. 5, images were captured using a 40 × objective. Immunostainings were processed using ImageJ76. Immunostainings for ISL1 and CHAT, MAP2 and TAU were despeckled (ImageJ plugin) during analysis to remove background signal. All antibodies, buffers, and antibody dilutions are listed in Table 2.
Calcium imaging
Cells cultured in an OrganoPlate 2-lane were incubated with Cal-520 AM to measure spontaneous calcium fluctuations. Motor neuron differentiation medium was supplemented with 200 µM Cal-520 AM and 1:250 pluronic F-120 (Thermo Fisher, P3000MP). 30 µL of solution was added to the medium inlet and 20 µL to the medium outlet. Cells were incubated under low perfusion (2°, 8 min) in 37 °C, 5% CO2 conditions for 90 min, followed by 30 min at RT in the dark. Images were captured at 2.08 Hz using an ImageXpress Micro XLS microscope (4 × magnification) and were subsequently processed in ImageJ to allow the visualization of the calcium fluctuations. Calcium recordings were corrected for bleaching (exponential fit) using a bleach correction plugin in ImageJ76. Recordings depict changes in intracellular calcium concentrations over time. Red depicts large changes in intracellular fluorescence and blue depicts small changes.
Vincristine exposure
MNPs cultured in the OrganoPlate 3-lane were exposed to vincristine (Sigma, V8879) from day 13 to day 17. A medium change was performed on day 15. On day 17, a calcein labeling experiment was performed while the old medium was sampled to assess cell viability using a lactate dehydrogenase (LDH) activity assay. The results for the axonal outgrowth quantification were plotted in a dose–response curve. Here, the ROI involved the bottom lane. The dotted lines interconnected the 95% confidence intervals for each concentration. The graph was created in GraphPad Prism, version 8.3.1 (GraphPad Software, San Diego, CA, USA, https://www.graphpad.com/scientific-software/prism/).
LDH activity assay
Lactate dehydrogenase (LDH) activity was analyzed using an LDH activity kit (Sigma, MAK066). LDH reduces NAD to NADH, which can be detected in a spectrophotometric multiwell plate reader. The assay was performed according to manufacturer’s protocol and has been previously described80. In short, at day 17, medium from the top microfluidic lane was sampled, and used for the LDH activity assay. Measurements were done after 1 min and every 2 min following the initial measurement. Absorbance was measured at 450 nm for a total duration of 50 min.
Excitotoxicity assay in the OrganoPlate
MNPs were exposed to high glutamate from day 10 to 17. Glutamic acid (Sigma, G1251) was freshly dissolved in motor neuron differentiation medium to a concentration of 5 mM before performing a medium change. On day 17, cells were exposed to 0.05 mM sodium arsenite (Merck Milipore, 1062771000) for 3 h as a positive control for SG formation. Following exposure, MNPs in the OrganoPlate 3-lane were stained using calcein-AM, while the OrganoPlate 2-lane was fixated using 4% paraformaldehyde.
Semaphorin exposure
Directly following seeding, motor neuron differentiation medium was supplemented with 0, 10, 100 or 1000 µg/mL semaphorin 3A (SEMA3A, Peprotech, 15017H) or semaphorin 3F (SEMA3F, R&D, 9878S3025) and perfused throughout the bottom lane (50 µL/well). Regular differentiation medium was added to the top lane (50 µL/well). Medium was changed every 2–3 days and calcein labeling was performed at day 17. Axonal outgrowth was quantified using a similar approach as described earlier, with the exception that the outgrowth in the bottom lane was quantified.
Statistical design and statistical analyses
Data was analyzed using GraphPad Prism, version 8.3.1 (GraphPad Software, San Diego, CA, USA, https://www.graphpad.com/scientific-software/prism/). Data normality was assessed using the Kolmogoriv-Smirnov, Shapiro–Wilk, D’Agostino–Pearson omnibus and the Anderson–Darling tests. Equality of variances was assessed using the Brown–Forsythe and Bartlett’s tests, or using the F-test when performing a t-test. In case the assumptions were not violated, unpaired t-tests or one-way ANOVAs were performed. For one-way ANOVAs, Tukey’s multiple comparisons tests were performed. In case the assumption of equality of variances was violated, the Brown-Forsythe and Welch test was performed with a Dunnett T3 multiple comparisons test. When data was not normally distributed, the nonparametric Kruskall–Willis with Dunn’s multiple comparisons test was performed. For multiple comparisons tests, all groups were compared with all groups, unless stated otherwise. Statistical significance is indicated by one or more asterisks. *(P < 0.05), **(P < 0.01), ***(P < 0.001), or ****(P < 0.0001). Following multiple comparisons tests, p-values adjusted for these comparisons are reported. The total number of technical replicates is depicted as n, while the number of independent experiments is depicted as N.
Data availability
The datasets generated during in this study are available from the corresponding authors on reasonable request.
References
Gomes-Osman, J., Cortes, M., Guest, J. & Pascual-Leone, A. A systematic review of experimental strategies aimed at improving motor function after acute and chronic spinal cord injury. J. Neurotrauma 33, 425–438 (2016).
McCreedy, D. A. & Sakiyama-Elbert, S. E. Combination therapies in the CNS: Engineering the environment. Neurosci. Lett. 519, 115–121 (2012).
Palispis, W. A. & Gupta, R. Surgical repair in humans after traumatic nerve injury provides limited functional neural regeneration in adults. Exp. Neurol. 290, 106–114 (2017).
Nijssen, J., Aguila, J., Hoogstraaten, R., Kee, N. & Hedlund, E. Axon-seq decodes the motor axon transcriptome and its modulation in response to ALS. Stem Cell Rep. 11, 1565–1578 (2018).
Osking, Z. et al. ALS-linked SOD1 mutants enhance neurite outgrowth and branching in adult motor neurons. iScience 11, 294–304 (2019).
Swarup, V., Audet, J. N., Phaneuf, D., Kriz, J. & Julien, J. P. Abnormal regenerative responses and impaired axonal outgrowth after nerve crush in TDP-43 transgenic mouse models of amyotrophic lateral sclerosis. J. Neurosci. 32, 18186–18195 (2012).
Wang, Z. B., Zhang, X. & Li, X. J. Recapitulation of spinal motor neuron-specific disease phenotypes in a human cell model of spinal muscular atrophy. Cell Res. 23, 378–393 (2013).
Comley, L. H., Nijssen, J., Frost-Nylen, J. & Hedlund, E. Cross-disease comparison of amyotrophic lateral sclerosis and spinal muscular atrophy reveals conservation of selective vulnerability but differential neuromuscular junction pathology. J. Comp. Neurol. 524, 1424–1442 (2016).
Xu, C. C., Denton, K. R., Wang, Z. B., Zhang, X. & Li, X. J. Abnormal mitochondrial transport and morphology as early pathological changes in human models of spinal muscular atrophy. DMM Dis. Model. Mech. 9, 39–49 (2016).
Pandya, V. A. & Patani, R. Decoding the relationship between ageing and amyotrophic lateral sclerosis: A cellular perspective. Brain 143, 1057–1072 (2020).
Kiskinis, E. et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795 (2014).
Bhinge, A., Namboori, S. C., Zhang, X., VanDongen, A. M. J. & Stanton, L. W. Genetic correction of SOD1 mutant iPSCs reveals ERK and JNK activated AP1 as a driver of neurodegeneration in amyotrophic lateral sclerosis. Stem Cell Rep. 8, 856–869 (2017).
Higelin, J. et al. FUS mislocalization and vulnerability to DNA damage in ALS patients derived hiPSCs and aging motoneurons. Front. Cell. Neurosci. 10, 290 (2016).
Guo, W. et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 8, 1–14 (2017).
Hawrot, J., Imhof, S. & Wainger, B. J. Modeling cell-autonomous motor neuron phenotypes in ALS using iPSCs. Neurobiol. Dis. 134, 104680 (2020).
Al-Ali, H., Beckerman, S. R., Bixby, J. L. & Lemmon, V. P. In vitro models of axon regeneration. Exp. Neurol. 287, 423–434 (2017).
Knöll, B., Weinl, C., Nordheim, A. & Bonhoeffer, F. Stripe assay to examine axonal guidance and cell migration. Nat. Protoc. 2, 1216–1224 (2007).
Yu, P., Wang, H., Katagiri, Y. & Geller, H. M. An in vitro model of reactive astrogliosis and its effect on neuronal growth. Methods Mol. Biol. 814, 327–340 (2012).
Zou, Y. et al. Gene-silencing screen for mammalian axon regeneration identifies Inpp5f (Sac2) as an endogenous suppressor of repair after spinal cord injury. J. Neurosci. 35, 10429–10439 (2015).
Brännvall, K. et al. Enhanced neuronal differentiation in a three-dimensional collagen-hyaluronan matrix. J. Neurosci. Res. 85, 2138–2146 (2007).
van Duinen, V., Trietsch, S. J., Joore, J., Vulto, P. & Hankemeier, T. Microfluidic 3D cell culture: From tools to tissue models. Curr. Opin. Biotechnol. 35, 118–126 (2015).
Campenot, R. B. Local control of neurite development by nerve growth factor. Proc. Natl. Acad. Sci. U. S. A. 74, 4516–4519 (1977).
Taylor, A. M. et al. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2, 599–605 (2005).
Park, J. W., Vahidi, B., Taylor, A. M., Rhee, S. W. & Jeon, N. L. Microfluidic culture platform for neuroscience research. Nat. Protoc. 1, 2128–2136 (2006).
Kim, Y. T., Karthikeyan, K., Chirvi, S. & Davé, D. P. Neuro-optical microfluidic platform to study injury and regeneration of single axons. Lab Chip 9, 2576–2581 (2009).
Hellman, A. N. et al. Examination of axonal injury and regeneration in micropatterned neuronal culture using pulsed laser microbeam dissection. Lab Chip 10, 2083–2092 (2010).
Hosmane, S. et al. Valve-based microfluidic compression platform: Single axon injury and regrowth. Lab Chip 11, 3888–3895 (2011).
Kim, H. J. et al. Integrated microfluidics platforms for investigating injury and regeneration of CNS axons. Ann. Biomed. Eng. 40, 1268–1276 (2012).
Dollé, J. P., Morrison, B., Schloss, R. S. & Yarmush, M. L. An organotypic uniaxial strain model using microfluidics. Lab Chip 13, 432–442 (2013).
van de Wijdeven, R. et al. Structuring a multi-nodal neural network in vitro within a novel design microfluidic chip. Biomed. Microdevices 20, 9 (2018).
Uzel, S.G.M. et al. Microfluidic device for the formation of optically excitable, three-dimensional, compartmentalized motor units. Sci. Adv. 2, e1501429 (2016).
Osaki, T., Uzel, S. G. M. & Kamm, R. D. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 4, 1–16 (2018).
Wong, I. & Ho, C. M. Surface molecular property modifications for poly(dimethylsiloxane) (PDMS) based microfluidic devices. Microfluid. Nanofluidics 7, 291–306 (2009).
Trietsch, S. J., Israëls, G. D., Joore, J., Hankemeier, T. & Vulto, P. Microfluidic titer plate for stratified 3D cell culture. Lab Chip 13, 3548–3554 (2013).
Van Helleputte, L. et al. Inhibition of histone deacetylase 6 (HDAC6) protects against vincristine-induced peripheral neuropathies and inhibits tumor growth. Neurobiol. Dis. 111, 59–69 (2018).
Vulto, P. et al. Phaseguides: A paradigm shift in microfluidic priming and emptying. Lab Chip 11, 1596–1602 (2011).
Marrone, L. et al. Isogenic FUS-eGFP iPSC reporter lines enable quantification of FUS stress granule pathology that is rescued by drugs inducing autophagy. Stem Cell Rep. 10, 375–389 (2018).
Mackenzie, I. R. et al. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95, 808–816 (2017).
Gal, J. et al. ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 132, 563–576 (2016).
van Duinen, V. et al. Perfused 3D angiogenic sprouting in a high-throughput in vitro platform. Angiogenesis 22, 157–165 (2019).
Pasterkamp, R. J. Getting neural circuits into shape with semaphorins. Nat. Rev. Neurosci. 13, 605–618 (2012).
Masuda, T. & Taniguchi, M. Contribution of semaphorins to the formation of the peripheral nervous system in higher vertebrates. Cell Adhes. Migr. 10, 593–603 (2016).
Pond, A., Roche, F. K. & Letourneau, P. C. Temporal regulation of neuropilin-1 expression and sensitivity to semaphorin 3A in NGF- and NT3-responsive chick sensory neurons. J. Neurobiol. 51, 43–53 (2002).
Trietsch, S. J. et al. Membrane-free culture and real-time barrier integrity assessment of perfused intestinal epithelium tubes. Nat. Commun. 8, 262 (2017).
Bilsland, L. G. et al. Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. U. S. A. 107, 20523–20528 (2010).
Wu, C. H. et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503 (2012).
Blasco, H., Mavel, S., Corcia, P. & Gordon, P. H. The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 21, 3551–3575 (2014).
Blokhuis, A. M., Groen, E. J. N., Koppers, M., Van Den Berg, L. H. & Pasterkamp, R. J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 777–794 (2013).
Ling, S. C., Polymenidou, M. & Cleveland, D. W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 79, 416–438 (2013).
Neumann, M. Molecular neuropathology of TDP-43 proteinopathies. Int. J. Mol. Sci. 10, 232–246 (2009).
Deng, H. X. et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 67, 739–748 (2010).
Guerrero, E. N. et al. TDP-43/FUS in motor neuron disease: Complexity and challenges. Prog. Neurobiol. 145–146, 78–97 (2016).
Baradaran-Heravi, Y., Van Broeckhoven, C. & van der Zee, J. Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol. Dis. 134, 104639 (2020).
Anderson, P. & Kedersha, N. Stress granules: The tao of RNA triage. Trends Biochem. Sci. 33, 141–150 (2008).
Scotter, E. L., Chen, H. J. & Shaw, C. E. TDP-43 proteinopathy and ALS: Insights into disease mechanisms and therapeutic targets. Neurotherapeutics 12, 352–363 (2015).
Huber, A. B. et al. Distinct roles for secreted semaphorin signaling in spinal motor axon guidance. Neuron 48, 949–964 (2005).
McCormick, A. M., Jarmusik, N. A. & Leipzig, N. D. Co-immobilization of semaphorin3A and nerve growth factor to guide and pattern axons. Acta Biomater. 28, 33–44 (2015).
Giger, R. J., Hollis, E. R. & Tuszynski, M. H. Guidance molecules in axon regeneration. Cold Spring Harb. Perspect. Biol. 2, 1–22 (2010).
Buss, A. et al. Growth-modulating molecules are associated with invading Schwann cells and not astrocytes in human traumatic spinal cord injury. Brain 130, 940–953 (2007).
Falnikar, A., Li, K. & Lepore, A. C. Therapeutically targeting astrocytes with stem and progenitor cell transplantation following traumatic spinal cord injury. Brain Res. 1619, 91–103 (2015).
Kroner, A. & Rosas Almanza, J. Role of microglia in spinal cord injury. Neurosci. Lett. 709, 134370 (2019).
Brites, D. & Vaz, A. R. Microglia centered pathogenesis in ALS: Insights in cell interconnectivity. Front. Cell. Neurosci. 8, 1–24 (2014).
Haidet-Phillips, A. M. et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828 (2011).
Picchiarelli, G. et al. FUS-mediated regulation of acetylcholine receptor transcription at neuromuscular junctions is compromised in amyotrophic lateral sclerosis. Nat. Neurosci. 22, 1793–1805 (2019).
Rentzos, M. et al. Alterations of T cell subsets in ALS: A systemic immune activation?. Acta Neurol. Scand. 125, 260–264 (2012).
Mora, G. & Chiò, A. Disorders of upper and lower motor neurons. In Prognosis of Neurological Diseases (eds. Sghirlanzoni, A., Lauria, G. & Chiapparini, L.) 261–272 (Springer, Milan, 2015).
Caliari, S. R. & Burdick, J. A. A practical guide to hydrogels for cell culture. Nat. Methods 13, 405–414 (2016).
Kane, K. I. W. et al. Automated microfluidic cell culture of stem cell derived dopaminergic neurons. Sci. Rep. 9, 1–12 (2019).
Schutgens, F. et al. Tubuloids derived from human adult kidney and urine for personalized disease modeling. Nat. Biotechnol. 37, 303–313 (2019).
Alami, N. H. et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543 (2014).
Saha, A. R. et al. Parkinson’s disease α-synuclein mutations exhibit defective axonal transport in cultured neurons. J. Cell Sci. 117, 1017–1024 (2004).
Smith, K. D. B., Kallhoff, V., Zheng, H. & Pautler, R. G. In vivo axonal transport rates decrease in a mouse model of Alzheimer’s disease. Neuroimage 35, 1401–1408 (2007).
Wang, Z. X., Tan, L. & Yu, J. T. Axonal transport defects in Alzheimer’s disease. Mol. Neurobiol. 51, 1309–1321 (2015).
Du, Z. W. et al. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 6, 1–9 (2015).
Wevers, N. R. et al. High-throughput compound evaluation on 3D networks of neurons and glia in a microfluidic platform. Sci. Rep. 6, 1–10 (2016).
Schindelin, J. et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Harschnitz, O. et al. Autoantibody pathogenicity in a multifocal motor neuropathy induced pluripotent stem cell-derived model. Ann. Neurol. 80, 71–88 (2016).
Ormel, P.R. et al. Microglia innately develop within cerebral organoids. Nat. Commun. 9, 1–14 (2018).
Melief, J. et al. Characterizing primary human microglia: A comparative study with myeloid subsets and culture models. Glia 64, 1857–1868 (2016).
Vormann, M. K. et al. Nephrotoxicity and kidney transport assessment on 3D perfused proximal tubules. AAPS J. 20, 1–11 (2018).
Acknowledgements
The authors thank Frederik Schavemaker for his contribution to developing artist impressions of the OrganoPlate and Thomas Olivier for his assistance with microscopy and data analysis. This work was supported by the Stichting ALS Nederland (ALS-on-a-chip, awarded to PV and RJP; TOTALS, awarded to RJP).
Author information
Authors and Affiliations
Contributions
X.M.S.: methodology, investigation, formal analysis, visualization, writing-original draft preparation. S.P.-V.: methodology, investigation, formal analysis, writing-original draft preparation, supervision. J.C.D.: investigation. P.V.: conceptualization, writing-review & editing, supervision, funding acquisition. N.R.W.: methodology, visualization, writing-original draft preparation, review & editing, supervision. R.J.P.: conceptualization, resources, writing-review & editing, supervision, funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
SPV and RJP declare no conflict of interest. XMS, JCD, PV, and NRW are employees of MIMETAS BV and PV is shareholder of that same company. The OrganoPlate is a registered trademark of MIMETAS BV.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Spijkers, X.M., Pasteuning-Vuhman, S., Dorleijn, J.C. et al. A directional 3D neurite outgrowth model for studying motor axon biology and disease. Sci Rep 11, 2080 (2021). https://doi.org/10.1038/s41598-021-81335-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-81335-z
This article is cited by
-
Local production of reactive oxygen species drives vincristine-induced axon degeneration
Cell Death & Disease (2023)
-
Organ-on-a-chip models for development of cancer immunotherapies
Cancer Immunology, Immunotherapy (2023)
-
Human organs-on-chips for disease modelling, drug development and personalized medicine
Nature Reviews Genetics (2022)
-
Multi-scale Fabrication Techniques of Collagen Hydrogel for Developing Physiological 3D In vitro Barrier Model
International Journal of Precision Engineering and Manufacturing (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
