
Abstract
Kochia, a major weed species, has evolved resistance to four herbicide modes of action. Herbicide resistance appears to spread quickly, which could result in diminished standing genetic variation, reducing the ability of populations to adapt further. Here we used double digest restriction enzyme associated sequencing to determine the level of gene flow among kochia populations and whether selection for glyphosate resistance reduces genetic variation. Canadian Prairie populations show little to no genetic differentiation (FST = 0.01) and no correlation between genetic and geographic distance (r2 = − 0.02 p = 0.56), indicating high gene flow and no population structure. There is some evidence that kochia populations are genetically depauperate compared to other weed species, but genetic diversity did not differ between glyphosate susceptible and resistant populations or individuals. The inbreeding coefficients suggest there are 23% fewer heterozygotes in these populations than expected with random mating, and no variation was found within the chloroplast. These results indicate that any alleles for herbicide resistance can be expected to spread quickly through kochia populations, but there is little evidence this spread will reduce the species’ genetic variation or limit the species’ ability to respond to further selection pressure.
Similar content being viewed by others


Introduction
A key area of interest in evolutionary biology is understanding the consequences of selection for genetic diversity and the future ability of populations to adapt. One potential consequence of strong selection is a reduction in the raw material, the genetic variation, available for selection1. At the individual level, this reduction can be detected in persistent genomic signatures of selective sweeps such as those associated with human evolution (reviewed by2,3), animal e.g.4,5 and crop domestication e.g.6,7. The evolution of herbicide resistance (HR) in agricultural weeds results from strong selection pressure. The spread of these resistance alleles through populations provides a growing number of study systems for understanding the consequences of selective sweeps at the individual and population level8,9,10. For practical reasons, weed managers are interested in understanding whether or not the standing variation for other herbicide modes of action is likely to be lost from populations following this selection. This could delay the evolution of additional HR within the population resulting from the time expected for new mutations conferring this resistance to arise in the population9,11,12,13,14. However, even when selection pressure is strong, loss in genetic variation depend on multiple factors, including the genetic basis of the resistance, mating system, population size, spatial or temporal variation in selection pressure, and gene flow9.
Kochia (Bassia scoparia (L.) A.J. Scott syn. Kochia scoparia (L.) Schrad.) is native to Europe and Asia and introduced to Canada, the United States, Africa, and South America. It is an annual noted for early germination, tolerance of arid and saline conditions, and a tumble weed habit. Kochia is wind pollinated and produces a large amount of pollen. However, it is also self-compatible, and as a result, the species is likely predominately outcrossing, but with high levels of variablity15. It has North American herbarium collections dating from the 1880s (reviewed by15). Kochia was found to be the fastest spreading alien species in the western USA from 1880 to 198016. It causes significant yield losses (30–60%) in crops such as winter wheat and sugar beet15, a problem exacerbated by the evolution of multiple herbicide resistance17.
The species has evolved resistance to four herbicide modes of action18. This includes resistance to photosystem II inhibitors18, acetolactate synthase (ALS) inhibitors19,20, synthetic auxins21 and glyphosate22,23,24. Individuals with multiple herbicide resistance to all four modes of action have been detected in Kansas17, while individuals combining ALS, glyphosate (GR) and synthetic auxin resistance were documented in Alberta, Canada in 201725. Kochia populations with ALS inhibitor resistance were first detected in Canada in 198826 and the point mutations conferring this resistance have become nearly ubiquitous throughout the Prairie provinces in less than 20 years27. Glyphosate resistance was first detected in Kansas in 200722 and was widespread in the USA’s Great Plains and confirmed in all three Canadian Prairie provinces by 201328,29,30. This suggests that glyphosate resistance arose de novo in Texas after 33 years of glyphosate use31 and 11 years of intensified glyphosate use following the introduction of glyphosate resistant crops31,32. This resistance appears to have then spread through populations, rather than being a common variant in the species’ standing variation or emerging repeatedly de novo across the range. The glyphosate resistance (GR) mechanism described for kochia is increased copy number and expression of the 5-enolpyruvylshikimate-3-phosphate synthase (EPSPS) enzyme, which is inhibited by glyphosate in susceptible plants33. EPSPS copy number correlates with glyphosate resistance, with four or more copies resulting in resistance17,24. Inheritance of increased copy number follows a single locus Mendelian pattern, as the gene copies are in a tandem array on a single chromosome34. At this time, no other mechanisms of glyphosate resistance for kochia have been described.
While the spread of herbicide resistance genes, via pollen and seed, depends on the interconnectivity of populations, the selection of novel herbicide resistance from standing variation in a population depends on variation being available. In addition to selection for multiple HRs, often which have resulted from changes at a single locus (ALS, EPSPS, and photosystem II inhibitors), kochia’s history as an introduced species may limit available variation as only a subset of the variation available within a species is expected to be introduced to a new region35,36,37. Here we used double digested restriction enzyme associated markers and 26 populations from the Canadian Prairies to understand the current genetic variation in kochia populations and how selection for glyphosate resistance may have changed this variation. Specifically, we investigated three questions: (1) what is the level of gene flow and inbreeding among the kochia populations? (2) what is the current level of genetic diversity in these populations? and (3) do populations where high EPSPS copy number (EPSPSCN) has been introduced or individuals with high EPSPSCN show evidence of reduced diversity?
Results
No individual sampled from a susceptible population had increased EPSPSCN relative to ALS, while resistant populations were generally a mixture of individuals with and without increased EPSPSCN. In one population, identified as 10% resistant in the initial screening of 100 individuals29, none of the 12 individuals sampled had increased EPSPSCN (Table 1), as a result, this pair was excluded from comparisons of resistant and susceptible populations.
In total, after following the STACKS pipelines38 for SNP discovery and genotyping, 360 (94%) and 362 (95%) of individuals had sufficient coverage in the de novo and reference based pipelines, respectively. However, the sibling sets were excluded in the majority of analyses, resulting in the inclusion of 89 high EPSPS individuals and 206 low EPSPS individuals (Table 1). For the reference based pipeline, the dataset included 3248 variable (polymorphic) loci with 10.6% missing data, while the de novo pipeline had 3173 variable loci with 11.2% missing data. Overall, 1.29% of nucleotides examined were polymorphic across alleles and nucleotide diversity (π) was 0.0036. Most consensus reference loci (99.6% of 6041 polymorphic and fixed loci) and 83.9% (of the 5626 polymorphic and fixed) of the de novo loci mapped to the genome (Bowtie239). In total, 3440 loci were identified by both pipelines. Population analyses were run on all four data sets, but statistical results were very similar (Supplementary Table S2) and only the reference based pipeline’s results are presented. The minor allele frequency (MAF) in the reference-based set of loci averaged 0.19.
Population structure and gene flow
Overall population differentiation (FST) was very low at 0.01, and pair-wise FST values ranged from 0 to 0.07 (Fig. 1) and did not correlate with geographic distance (Mantel test, r2 = − 0.02 p = 0.57). AMOVAs indicated the majority of molecular variation (79.8%) was attributed to individuals, with less than 1% of the variation explained by differences among populations and no variation explained by population status (resistant or susceptible) (Table 2). A principal components analysis (PCA) explained 23.6% and 11.2% of the variance on the first and second axes, respectively, but showed no clustering by province, population or EPSPS type (Fig. 2). Different runs of find.clusters assigned the lowest Bayesian information criterion to different numbers of clusters. However, six clusters were selected with the lowest BIC for both the reference and the de novo loci sets. These groups did not correspond to population, province or EPSPS status (Supplementary Fig. S1), nor did they correspond to clusters or regions within the PCA. When the full sibling groups were included, the optimal number of groups ranged from eight to eleven, but full siblings were not assigned to the same group (Supplementary Fig. S2). The analysis produced by fineRADStructure indicated little population structure with a diffuse pattern of co-ancestry levels averaged by population. Groups of individuals with higher co-ancestry were mixed by province, population and EPSPS status (Supplementary Fig. S3). The overall FIS was calculated as 0.23, but ranged from 0 to 0.42 within populations (Table 1). The proportion of alleles shared by two individuals averaged 75% (range 64–98%), the proportion of polymorphic nucleotide sites (Pn) averaged 2.73 × 10–3 (range 1.02–4.58 × 10–3), and the average proportion of loci showing variation within individuals (HL) was 13% (range 5% to 23%).

Between population heat map with higher values as more intensely coloured with Nei’s genetic distance (DST; ranged from 0.01 to 0.05) below the diagonal and FST values (ranged from 0 to 0.08) above the diagonal. Values that were not considered statistically different from 0 (bootstrap p-value < 0.05) are coded in black.

Principal components analysis (PCA) using SNPs from 3248 variable loci with first and second axes account for 23.6% and 11.2% of the variation, respectively, but showing no clustering by EPSPS:ALS ratio (size), population (colour), or province (shape) indicating little to no population structure in this species.
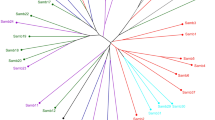
The unweighted pair group method with arithmetic mean dendrograms included groupings mixed by province, population and EPSPS status (Fig. 3). While some clustering of individuals with increased EPSPSCN was apparent, for example, a small cluster of high EPSPSCN individuals from Alberta, Saskatchewan and Manitoba populations (e.g. Fig. 3 at 10 o’ clock), others were scattered through the tree likely reflecting the high rates of gene flow rather than multiple independent origins.

UPGMA tree based on Prevosti’s Genetic distance (see scale at top of tree) for individuals from kochia populations in Alberta, Saskatchewan and Manitoba. Population of origin is represented by the coloured blocks at the tips, while EPSPS:ALS ratio is represented by the size of the bar. Red branches belong to individuals with an EPSPS:ALS ratios of 4 or greater, which are considered resistant to glyphosate, and blue dots indicate nodes with 80 or greater bootstrap support.
The number of migrants among the populations (Nm) was calculated as 16.4 for comparison to40.
Population statistics and genetic diversity by EPSPS status
AMOVAs indicated that neither population type (Table 2A) nor individual EPSPSCN status within resistant populations (Table 2B) explained genetic variation. Neither the proportion of polymorphic nucleotide sites (Pn) nor the proportion of variable loci (HL) differed by population type or individual EPSPS status (Table 3). Nucleotide diversity estimates were 0.0034 and 0.0035 for individuals with low and high EPSPSCN, respectively. BayseScan indicated that no loci showed evidence of selection when coded by individual or population’s EPSPS status.
Alignment to chloroplast
Thirty-six de novo consensus loci aligned to the chloroplast, but all were fixed across individuals. This may indicate that too few regions were used to detect variation, that markers fell in invariant regions, or that there is little variation in the chloroplast. Previous attempts in our laboratory to find variability in rbcL, matK, trnL-F, psbA-H2, pshH-psbB and atp-rbcL yielded only a pair of SNPs across these populations (unpublished data), which may indicate a lack of variation.
Discussion
Here, we determined that these 26 kochia populations, sampled from across the Canadian Prairie provinces, showed high levels of gene flow. This was indicated by: (1) the very low levels of genetic differentiation (Table 1, Fig. 1); (2) individuals harbouring the majority of genetic variation (Table 2); and (3) the absence of population structure (Figs. 1, 2, 3). This estimate of genetic differentiation for kochia (FST 0.01) is lower than the moderate level41 reported for 13 North Dakota and Minnesota populations based on 45 microsatellites (GST = 0.0940). While we note that calculating the number of migrants from FST has been criticized as underlying assumptions are likely to be violated42,43, our estimate would be six times higher than that of40, which suggests a higher level of connection between these populations. The level of genetic differentiation observed here for kochia is also lower than the average genetic differentiation reported for outcrossing wind-pollinated species (GST value 0.101) based on allozymes44. Other weedy species with similarly low FST include the wind-pollinated grasses Apera spica-venti L. (GST = 0.01, 0.024 for Canadian and European populations, respectively, from allozymes45) and Alopecurus myosuroides Huds. (FST = 0.023 from AFLPs46), as well as the outcrossing and self-incompatible Rosa rugosa Thub. (FST = 0.045 from microsatellites47). Populations of GR plants with similar genetic differentiation include the outcrossing and self-incompatible Lolium perenne ssp. multiflorium (Lam.) Husnot (FST = 0.006–0.088 from microsatellites48); and some population pairs of the obligate outcrossing species Amaranthus palmeri S. Watson in the USA (e.g. FST = 0.052 for R-S pairs from Arizona from SNPs49).
We observed an overall inbreeding coefficient (FIS) of 0.23, indicating a 23% higher level of homozygosity than expected by random mating. As material used here was grown from openly pollinated seed, this could indicate that inbreeding is occurring in many populations. Given the low level of genetic differentiation between populations, these high inbreeding coefficients are unusual; plants are generally expected to have either low or high values for both FST and FIS, as self-pollination increases divergence while outcrossing reduces divergence. For example, for two species with low FST values mentioned above, R. rugosa and A. palmeri, the FIS values were estimated at 0.04347 and 0.01649, respectively. In contrast, central European populations of Amaranthus retroflexus L. had inbreeding coefficients similar to the higher values found here at 0.382, but FST was 0.27 indicating strong population differentiation (allozymes50). However, a similar relationship to that observed here was reported for L. perenne ssp. multiflorium, which had FIS estimates ranging from 0.396 to 0.517 despite low values for genetic differentiation48. The authors suggested that this could result from genetic bottlenecks caused by glyphosate selection, but noted that FIS values did not correlate with the frequency of GR plants. Similar processes may be contributing to the high FIS values estimated in this study, since both high and low ESPSCN populations have been subject to selection pressure.
Currently, it is difficult to evaluate kochia’s level of genetic diversity compared to other species, as it is unclear what levels should be expected in weedy or outcrossing plants. This challenge results from the variety of molecular markers used over the last 50 years and the variety of information presented by SNP studies. Genetic diversity (HE) in these populations averaged 0.28, lower than the previous report for kochia of 0.35 (Nei’s gene diversity51 or h in40). While this may represent a reduction in genetic diversity, these populations are further north along kochia’s invasion path and may have had lower initial genetic diversity. Alternatively, this difference may be the result of using different genetic markers. Kochia’s genetic diversity is higher than the average of 0.16 for outcrossing wind-pollinated species from allozyme studies44. However, the percentage of variable loci (variable vs. variable and fixed loci) was 53%, similar to the 51% of variable allozyme loci in outcrossing species52. It is lower than genetic diversity of 0.678–0.824 estimated for GR populations of L. perenne ssp. multiflora48, but slightly higher than estimates for 42 GR populations of the highly self-pollinating Conyza canadensis (L.) Cronq. (microsatellites; 0.21 (0–0.45)53). Unfortunately, few other studies using approaches such as ddRADseq to identify SNPs report the number of loci that were identified, but that were invariant. Our estimate is higher than that for two species of bee-pollinated perennial Rhododendron in Japan, where 23% (144 loci of 675) of loci were variable; this was the only other study we located that reported this information54. Similarly, kochia’s nucleotide diversity (all sites) was 0.0036, which is lower than estimates of 0.0047 from AFLPS for A. myosuroides46, but higher than the nucleotide diversity estimated overall for core and invasive populations of Mercurialis annua L. (0.0021; SNPs47). With increasing numbers of GBS studies examining genetic diversity, we anticipate that sufficient context to evaluate the potential for a particular weed to adapt from standing variation will soon be available14.
Whether kochia populations are more or less diverse than expected, there were no differences associated with high EPSPSCN within population or individuals. Similarly, population genetics parameters (e.g. FST, Pn, HL) did not differ between high EPSPSCN populations or individuals and their low EPSPSCN counterparts. The bootstrapped confidence intervals of the inbreeding coefficient for low and high EPSPSCN individuals did not overlap (high EPSPSCN individuals FIS = 0.29, low FIS = 0.24), but random permutation tests indicated no statistical significance (p = 0.057). As a result, if kochia populations are depauperate or rich in comparison to expectations, this is the case whether or not the individuals or populations have high EPSPSCN, indicating that we have no evidence that selection for EPSPSCN has altered genetic diversity in these populations. This study sampled at an early stage in the spread of high EPSPSCN individuals across the Prairie Provinces (i.e. prior to fixation); sampling kochia populations after fixation of GR or after the spread of additional HR genes would provide additional insights.
The high gene flow among populations suggests that kochia on the Canadian Prairies, and perhaps beyond, could be considered a single population. Estimates from tracking tumbling kochia suggest that approximately 90% of seeds are dispersed over the first kilometer55, leaving 10%, potentially 3000 seeds15, to be dispersed over greater distances. This strong dispersal likely contributes to high connectivity, though human-mediated seed movement is also likely a factor. As a result, beneficial alleles, such as those for herbicide resistance, can be expected to spread rapidly through the species’ range. Further, any selection for a suitable combination of beneficial alleles and genetic background will have all genetic material available in the species to select from when introduced into one area56,57. This is congruent with the speed at which ALS mutations spread through the Prairies27 and suggests that high EPSPSCN and auxinic resistance are likely to spread as rapidly as ALS resistance. This prediction is supported by Canadian Prairie random surveys showing a rapid increase in incidence of GR (5 to 50% of populations) and auxinic-resistant kochia (0 to 18%) in a 5-year period25. With the refinement of our understanding of kochia’s genome and the generation of a chromosome level assembly for the species, it will become possible to use this data to look at the signatures of selection near the EPSPS gene and determine whether this event is associated with a hard or soft sweep and better explore the origin of GR resistance in this species and its consequences2, 58.
A potential consequence of this high gene flow is that evolving locally adapted ecotypes would require extremely strong selection in kochia. However, the evolution of locally adapted ecotypes has been considered a key feature of successful invasions59,60. Based on this study, we expect the spread of GR will result in little change in kochia’s capacity to evolve additional herbicide resistance from standing variation. Swift, comprehensive, and ongoing action would be needed to curtail the spread of herbicide resistance genes from points of evolution in kochia populations. The species will need to be managed as a whole, as there are no smaller, individually controllable units require coordination and cooperation among producers and levels of government36. Future work expanding the geographic coverage of sampling, and investigating the genetic variation of these populations as GR and auxinic resistance spread, would provide further insights.
Materials and methods
Plant material
Plant material was from bulk-collected seed from population pairs where high EPSPSCN individuals had been detected (resistant) and from where they had not (susceptible) in relatively close geographic proximity. These populations were sampled and identified during surveys to determine the extent of GR kochia in Alberta (2011, 2012), Saskatchewan (2013) and Manitoba (2013) (Fig. 4)28,29,30. In these surveys, populations were considered resistant if they had individuals not controlled by glyphosate at 900 g ae/ha in greenhouse screens28,29,30. We extracted DNA from twelve individuals from four, seven and two pairs from Alberta, Saskatchewan and Manitoba, respectively. We also used six groups of reciprocally related progeny, sibling plants resulting from reciprocal controlled crosses between high and low EPSPSCN individuals from within populations in Alberta and Saskatchewan61. In total, this included 312 individuals from 26 populations and 72 progeny. The maps of the locations of these populations were made in QGIS Desktop 2.18.1562 with layers available and downloaded from Natural Earth (https://www.naturalearthdata.com/downloads/).

Locations of the kochia populations sampled in Alberta, Saskatchewan and Manitoba, Canada with north toward the top of the figure. Populations where glyphosate resistant individuals were detected in screens by Hall et al.28 and Beckie et al.29 are filled and contain a “R” in their label while those with no resistance detected in these screens are shown as empty and include an “S” in their labels.
DNA sequencing and analysis
DNA extraction
Seed were germinated and grown in the greenhouse at Agriculture and Agri-Food Canada’s Ottawa Research and Development Centre. Young leaves were collected and DNA extracted using FastDNA kit (MP BioMedicals, USA). All material from the greenhouses and residual debris following seed cleaning were autoclaved before disposal.
Quantitative PCR
Quantitative PCR was used to determine relative EPSPSCN compared to ALS following the method described in33. Specifically, two replicates were averaged to determine the EPSPS:ALS ratio. We measured the DNA concentrations of samples using a NanoDrop ND-1000 and 8000 Spectrophotometers (Thermo Scientific, Wilmington, DE, USA), corrected their concentration to 5 ng/μL and conducted Quantitative PCR (qPCR) using an Eppendorf Mastercycler ep cycler. The specific primers for EPSPS: 5′ GGCCAAAAGGGCAATCGTGGAG 3′ and 5′CATTGCCGTTCCCGCGTT TCC 3′63, and ALS ALS890F: 5′AGCCTGTGTTGTATGTGGGA 3′ and ALS999R: 5′ AGCGCCCAAACCCATTAAAG 3′61 were used and produced products of 102 and 110 bp, respectively. BioRad strip wells containing 10 μL ABI Power Sybr Green MM (2X) (Life Technologies, Hercules, CA, USA), 0.5 μL of the appropriate forward and reverse primer (5 μM), 10 ng of gDNA, and 7 μL of dH2O were used for the qPCR reactions33. Cycle parameters were initial denaturing at 95 °C for 15 min, followed by 95 °C for 30 s and annealing and extension at 60 °C for 60 s, for a total of 40 cycles. The ALS reference gene was used to standardize the EPSPSCN using the equation R = 2−ΔCTsample−ΔCTcalibrator to produce the estimate of the ratio between EPSPS and ALS33.
Double digested restriction enzyme associated markers
Double digested restriction enzyme associated marker library preparation and sequencing were completed at the University of Georgia using a 3DRAD based protocol. The enzymes used to generate the markers were HindIII (A|AGCTT) and NdeI (C|ATATG), and the project was designed to result in 300 million paired end reads for each of four plates.
Population genetics
Distances between the populations were calculated from their GPS coordinates using the Geographic Distance Matrix Generator v1.2.364, and the population map was created in QGIS 2.18.2562 with data from Natural Earth (https://www.naturalearthdata.com).
Data were analyzed using STACKS v1.4438 and custom R (3.4.3 “Kite-Eating Tree”) scripts65. STACKS (process_radtags) was used to demultiplex and filter data. In total, 1.6 billion reads were received and 1 billion were retained for an average of 1.5 million reads per individual. Stacks parameters were determined using a subset of samples as recommended66 and both the de novo and reference pipelines were followed. The parameters used were (M = n =) 5, (m =) 3, the minimum minor allele frequency allowed was 0.05, the maximum observed heterozygosity allowed was 0.7, and a random SNP was used from each locus. For the reference based pipeline, tags were aligned to kochia’s draft genome67 using Bowtie 2 version 2.1.039. Average coverage was 20.2 × for the reference pipeline and 15.8 × for the de novo pipeline. Individuals with less than 10 × coverage were excluded. Additionally, we removed loci with more than four alleles identified within full sibling groups, loci with 3 or fewer individuals represented in a population, and individuals with fewer than 60% of the loci. To identify loci associated with the chloroplast, consensus loci sequences were aligned to an assembly of kochia’s chloroplast67.
Population genetics parameters were calculated in R65. Observed heterozygosity (HO), within-population gene diversity (HS), overall gene diversity (HT), bootstrapped estimates of the inbreeding coefficient (FIS) with confidence intervals, and levels of genetic differentiation among populations (FST) were all calculated by hierfstat68. Mantel tests were conducted with ade469. Bootstrapped values for FST, associated p-values and Nei’s genetic distance (DST) were calculated with StAMPP70. BayeScan71 (version 2.1), used with default parameters except an increased prior of 300, produced estimates of FST with upper and lower limits for each population. The R packages boa72 and coda73 were used to assess BayeScan results and model convergence. Allelic richness estimates were generated by PopGenReports74. Unweighted pair group method with arithmetic mean trees were calculated using poppr75, while AMOVAs were calculated with 1000 permutations by poppr.manova with the “ade4” method69. The proportion of shared alleles among groups and k-means clustering were estimated (testing k = 1 to 40) with adegenet76. The program fineRADStructure was used to further investigate clustering77. A custom R script processed STACKS’ haplotype files containing all SNPs for each loci in order to calculate the proportion of polymorphic nucleotide sites (Pn) and the percentage of heterozygous loci (HL). The function oneway_test from coin78 estimated the p-value for comparing low and high EPSPSCN individuals or populations using 100,000 permutations. The proportion of shared alleles was calculated by adegenet76. Following40, the number of migrants were estimated with Nm = 0.25((1 − FST)/FST)79.
The packages ape80, gdata81, pegas82, phytools83, reshape84, Hmisc85 and vcfR86 were used for data handling and manipulation, while ggplot87 and colorspace88 were used for plotting.
Data availability
Data for this study will be made available by request to the corresponding author.
References
Wright, S. The roles of mutation, inbreeding, crossbreeding and selection in evolution. Proc. Sixth Int. Congr. Genet. 1, 356–366 (1932).
Rees, J. S., Castellano, S. & Andrés, A. M. The genomics of human local adaptation. Trends Genet. 36, 415–428. https://doi.org/10.1016/j.tig.2020.03.006 (2020).
Fan, S., Hansen, M. E. B., Lo, Y. & Tishkoff, S. A. Going global by adapting local: A review of recent human adaptation. Science 80, 354 (2016).
Marsden, C. D. et al. Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc. Natl. Acad. Sci. U.S.A. 113, 152–157. https://doi.org/10.1073/pnas.1512501113 (2016).
Oget, C., Servin, B. & Palhière, I. Genetic diversity analysis of French goat populations reveals selective sweeps involved in their differentiation. Anim. Genet. 50, 54–63. https://doi.org/10.1111/age.12752 (2019).
Kui, L. et al. Identification of selective sweeps in the domesticated table and wine grape (Vitis vinifera L.). Front Plant Sci. 11, 1–11. https://doi.org/10.3389/fpls.2020.00572 (2020).
Wegary, D. et al. Molecular diversity and selective sweeps in maize inbred lines adapted to African highlands. Sci. Rep. 9, 1–15. https://doi.org/10.1038/s41598-019-49861-z (2019).
Baker, H. G. The evolution of weeds. Annu. Rev. Ecol. Syst. 5, 1–24. https://doi.org/10.1146/annurev.es.05.110174.000245 (1974).
Kreiner, J. M., Stinchcombe, J. R. & Wright, S. I. Population genomics of herbicide resistance: Adaptation via evolutionary rescue. Annu. Rev. Plant Biol. 69, 611–635. https://doi.org/10.1146/annurev-arplant-042817-040038 (2018).
Baucom, R. S. Evolutionary and ecological insights from herbicide-resistant weeds: What have we learned about plant adaptation, and what is left to uncover?. New Phytol. https://doi.org/10.1111/nph.15723 (2019).
Hawkins, N. J., Bass, C., Dixon, A. & Neve, P. The evolutionary origins of pesticide resistance. Biol. Rev. 94, 135–155. https://doi.org/10.1111/brv.12440 (2019).
Orr, H. A. & Unckless, R. L. The population genetics of evolutionary rescue. PLoS Genet. 10, 1–9. https://doi.org/10.1371/journal.pgen.1004551 (2014).
Hermisson, J. & Pennings, P. S. Soft sweeps: Molecular population genetics of adaptation from standing genetic variation. Genetics 169, 2335–2352. https://doi.org/10.1534/genetics.104.036947 (2005).
Barrett, R. D. H. & Schluter, D. Adaptation from standing genetic variation. Trends Ecol. Evol. 23, 39–44. https://doi.org/10.1016/j.tree.2007.09.008 (2007).
Friesen, L. F., Beckie, H. J., Warwick, S. I. & Van Acker, R. C. The biology of Canadian weeds. 138. Kochia scoparia (L.) Schrad. Can. J. Plant Sci. 89, 141–167. https://doi.org/10.4141/CJPS08057 (2009).
Forcella, F. Final distribution is related to rate of spread in alien weeds. Weed Res. 25, 181–191. https://doi.org/10.1111/j.1365-3180.1985.tb00634.x (1985).
Varanasi, V. K. et al. Field-evolved resistance to four modes of action of herbicides in a single kochia (Kochia scoparia L. Schrad.) population. Pest Manage. Sci. 71, 1207–1212. https://doi.org/10.1002/ps.4034 (2015).
Heap, I. The International Survey of Herbicide Resistant Weeds (2019). www.weedscience.org. Accessed 4 Jan 2018.
Saari, L. L., Cotterman, J. C. & Primiani, M. M. Mechanism of sulfonylurea herbicide resistance in the weed Kochia scoparia. Plant Physiol. 93, 55–61 (1990).
Primiani, M. M., Erman, J. C. C. O. T. & Saari, L. L. Resistance of Kochia (Kochia scoparia) to sulfonylurea and imidazolinone herbicides. Weed Technol. 4, 169–172 (1990).
Nandula, V. K. & Manthey, F. A. Response of Kochia (Kochia scoparia) inbreds to 2,4-D and dicamba. Weed Technol. 16, 50–54. https://doi.org/10.1614/0890-037X(2002)016[0050:ROKKSI]2.0.CO;2 (2002).
Waite, J. et al. Differential Kochia (Kochia scoparia) populations response to glyphosate. Weed Sci. 61, 193–200. https://doi.org/10.1614/WS-D-12-00101.1 (2013).
Wiersma, A.T. Regional whole plant and molecular response of Kochia scoparia to glyphosate. M.S. thesis, Colorado State University, Fort Collins, CO (2012).
Godar, A. S., Stahlman, P. W., Jugulam, M. & Dille, J. A. Glyphosate-resistant kochia (Kochia scoparia) in Kansas: EPSPS gene copy number in relation to resistance levels. Weed Sci. 63, 587–595. https://doi.org/10.1614/WS-D-14-00145.1 (2015).
Beckie, H. J. et al. Triple-resistant kochia [Kochia scoparia (L.) Schrad.] in Alberta. Can. J. Plant Sci. 99, 281–285. https://doi.org/10.1139/cjps-2018-0256 (2019).
Morrisson, I. & Devine, M. Herbicide resistance in the Canadian prairie provinces : Five years after the fact. Phytoprotection 75, 5–16. https://doi.org/10.7202/706067ar (1994).
Beckie, H. J. et al. Occurrence and molecular characterization of acetolactate synthase (ALS) inhibitor–resistant Kochia (Kochia scoparia) in Western Canada. Weed Technol. 25, 170–175. https://doi.org/10.1614/WT-D-10-00067.1 (2011).
Hall, L. M. et al. Survey of glyphosate-resistant kochia ( Kochia scoparia L. Schrad.) in Alberta. Can. J. Plant Sci. 94, 127–130. https://doi.org/10.4141/cjps2013-204 (2014).
Beckie, H. J. et al. Glyphosate-resistant kochia (Kochia scoparia L. Schrad.) in Saskatchewan and Manitoba. Can. J. Plant Sci. 95, 345–349. https://doi.org/10.4141/CJPS-2014-328 (2015).
Beckie, H. J. et al. Glyphosate- and acetolactate synthase inhibitor–resistant kochia (Kochia scoparia) in Western Canada. Weed Sci. 61, 310–318. https://doi.org/10.1614/WS-D-12-00140.1 (2013).
Duke, S. O. The history and current status of glyphosate. Pest Manage. Sci. 74, 1027–1034. https://doi.org/10.1002/ps.4652 (2018).
Dill, G. M. Glyphosate-resistant crops: History, status and future. Pest Manage. Sci. 61, 219–224. https://doi.org/10.1002/ps.1008 (2005).
Wiersma, A. T. et al. Gene amplification of 5-enol-pyruvylshikimate-3-phosphate synthase in glyphosate-resistant Kochia scoparia. Planta 241, 463–474. https://doi.org/10.1007/s00425-014-2197-9 (2015).
Jugulam, M. et al. Tandem amplification of a chromosomal segment harboring EPSPS locus confers glyphosate resistance in Kochia scoparia. Plant Physiol. 166, 1200–1207. https://doi.org/10.1104/pp.114.242826 (2014).
Dlugosch, K. M. & Parker, I. M. Founding events in species invasions: Genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 17, 431–449. https://doi.org/10.1111/j.1365-294X.2007.03538.x (2008).
Sakai, A. K. et al. The population biology of invasive Species. Annu. Rev. Ecol. Syst. 32, 305–332 (2001).
Warwick, S. I. Allozyme and life history variation in five northwardly colonising North American weed species. Plant Syst. Evol. 169, 41–54 (1990).
Catchen, J. M. et al. Stacks: An analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140. https://doi.org/10.1111/mec.12354 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. https://doi.org/10.1038/nmeth.1923.Fast (2012).
Mengistu, L. W. & Messersmith, C. G. Genetic diversity of kochia. Weed Sci. 50, 498–503 (2002).
Hartl, D. L. & Clark, A. Principles of population genetics. Ecoscience 14(4), 544–554. https://doi.org/10.2980/1195-6860(2007)14[544b:POPG]2.0.CO;2 (2007).
Whitlock, M. C. & Mccauley, D. E. Indirect measures of gene flow and migration: F(ST) ≠ 1/(4Nm + 1). Heredity (Edinb.) 82, 117–125. https://doi.org/10.1038/sj.hdy.6884960 (1999).
Holsinger, K. E. & Weir, B. S. Genetics in geographically structured populations: Defining estimating and interpreting Fst. Nat. Rev. Genet. 10, 639–650. https://doi.org/10.1038/nrg2611 (2009).
Hamrick, J. L. & Godt, M. J. W. Effects of life history traits on genetic diversity in plant species. Philos. Trans. Biol. Sci. 351, 1291–1298. https://doi.org/10.1098/rstb.1996.0112 (1996).
Warwick, S. I., Thompson, B. K. & Black, L. D. Genetic variation in Canadian and European populations of the colonizing weed species Apera spica-venti. New Phytol. 106, 301–317 (1987).
Menchari, Y., Délye, C. & Le Corre, V. Genetic variation and population structure in black-grass (Alopecurus myosuroides Huds.), a successful, herbicide-resistant, annual grass weed of winter cereal fields. Mol. Ecol. 16, 3161–3172. https://doi.org/10.1111/j.1365-294X.2007.03390.x (2007).
Kelager, A., Pedersen, J. S. & Bruun, H. H. Multiple introductions and no loss of genetic diversity: Invasion history of Japanese Rose, Rosa rugosa, in Europe. Biol Invas. 15, 1125–1141. https://doi.org/10.1007/s10530-012-0356-0 (2013).
Karn, E. & Jasieniuk, M. Genetic diversity and structure of Lolium perenne ssp. multiflorum in California vineyards and orchards indicate potential for spread of herbicide resistance via gene flow. Evol. Appl. 10, 616–629. https://doi.org/10.1111/eva.12478 (2017).
Küpper, A. et al. Population genetic structure in glyphosate-resistant and -susceptible Palmer Amaranth (Amaranthus palmeri) populations using genotyping-by-sequencing (GBS). Front. Plant. Sci. 9, 29. https://doi.org/10.3389/fpls.2018.00029 (2018).
Mandák, B., Zákravský, P., Dostál, P. & Plačková, I. Population genetic structure of the noxious weed Amaranthus retroflexus in Central Europe. Flora Morphol. Distrib. Funct. Ecol. Plants 206, 697–703. https://doi.org/10.1016/j.flora.2011.01.010 (2011).
Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. U.S.A. 70, 3321–3323 (1973).
Hamrick, J. L., Linhart, Y. B. & Mitton, J. B. Relationships between life history characteristics and elecotrophoetically detectable genetic variation in plants. Annu. Rev. Ecol. Syst. 10, 173–200 (1979).
Okada, M. et al. Evolution and spread of glyphosate resistance in Conyza canadensis in California. Evol. Appl. 6, 761–777. https://doi.org/10.1111/eva.12061 (2013).
Yoichi, W. et al. Phylogeographic analysis suggests two origins for the riparian azalea Rhododendron indicum (L.) Sweet. Heredity (Edinb.) https://doi.org/10.1038/s41437-018-0064-3 (2018).
Beckie, H. J., Blackshaw, R. E., Hall, L. M. & Johnson, E. N. Pollen- and seed-mediated gene flow in Kochia (Kochia scoparia). Weed Sci. 64, 624–633. https://doi.org/10.1614/WS-D-16-00038.1 (2016).
Paris, M., Roux, F., Bérard, A. & Reboud, X. The effects of the genetic background on herbicide resistance fitness cost and its associated dominance in Arabidopsis thaliana. Heredity (Edinb.) 101, 499–506. https://doi.org/10.1038/hdy.2008.92 (2008).
Giacomini, D., Westra, P. & Ward, S. M. Impact of genetic background in fitness cost studies: An example from glyphosate-resistant Palmer Amaranth. Weed Sci. 62, 29–37. https://doi.org/10.1614/WS-D-13-00066.1 (2014).
Burke, M. K. How does adaptation sweep through the genome? Insights from long-term selection experiments. Proc. Biol. Sci. https://doi.org/10.1098/rspb.2012.0799 (2012).
González-Martínez, S. C., Ridout, K. & Pannell, J. R. Range expansion compromises adaptive evolution in an outcrossing plant. Curr. Biol. 27, 2544–2551. https://doi.org/10.1016/j.cub.2017.07.007 (2017).
Barrett, S. C. H. Genetic variation in weeds. In Biological Control of Weeds with Plant Pathogens (eds Walker, H. L. & Charudattan, R.) 73–98 (Springer, New York, 1982).
Martin, S. L. et al. Glyphosate resistance reduces kochia fitness: Comparison of segregating resistant and susceptible F2 populations. Plant Sci. https://doi.org/10.1016/j.plantsci.2017.04.010 (2017).
QGIS Development Team. QGIS Geographic Information System (2019).
Gaines, T. A. et al. Gene amplification confers glyphosate resistance in Amaranthus palmeri. Proc. Natl. Acad. Sci. U.S.A. 107, 1029–1034. https://doi.org/10.1073/pnas.0906649107 (2010).
Ersts, P. J. Geographic Distance Matrix Generator (2018).
R Core Team. R: A language and environment for statistical computing (2017).
Rochette, N. C. & Catchen, J. M. Deriving genotypes from RAD-seq short-read data using Stacks. Nat. Protoc. 12, 2640–2659. https://doi.org/10.1038/nprot.2017.123 (2017).
Patterson, E. L. et al. The draft genome of Kochia scoparia and the mechanism of glyphosate resistance via transposon-mediated EPSPS tandem gene duplication. bioRxiv https://doi.org/10.1101/600072 (2019).
Goudet, J. & Jombart, T. hierfstat: Estimation and Tests of Hierarchical F-Statistics, 58 (2015).
Dray, S. & Dafour, A. B. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 22, 1–20 (2007).
Pembleton, L. W., Cogan, N. O. I. & Forster, J. W. StAMPP: An R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Resour. 13, 946–952. https://doi.org/10.1111/1755-0998.12129%3e (2013).
Foll, M. & Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 180, 977–993. https://doi.org/10.1534/genetics.108.092221 (2008).
Smith, B. J. boa: An R package for MCMC Output convergence assessment and posterior inference. J. Stat. Softw. 21, 1–37 (2007).
Plummer, M., Best, N., Cowles, K. & Vines, K. CODA: Convergence diagnosis and output analysis for MCMC. R News 6, 45 (2006).
Adamack, A. T. & Gruber, B. PopGenReport: Simplifying basic population genetic analyses in R. Methods Ecol. Evol. 5, 384–387. https://doi.org/10.1111/2041-210X.12158 (2014).
Kamvar, Z. N., Tabima, J. F. & Grünwald, N. J. poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction v2.6.1. PeerJ 2, e281. https://doi.org/10.1093/biostatistics/1.4.465 (2018).
Jombart, T. & Ahmed, I. Adegenet 1.3–1: New tools for the analysis of genome-wide SNP data. Bioinformatics https://doi.org/10.1093/bioinformatics/btr521 (2011).
Malinsky, M., Trucchi, E., Lawson, D. J. & Falush, D. RADpainter and fineRADstructure: Population Inference from RADseq data. Mol. Biol. Evol. 35, 1284–1290. https://doi.org/10.1093/molbev/msy023 (2018).
Hothorn, T., Hornik, K., Van De Wiel, M.A., et al. Conditional Inference Procedures in a Permutation Test Framework: Package ‘Coin’, 98 (2017).
Slatkin, M. & Bartin, N. H. A comparison of three indirect methods for estimating average levels of gene flow. Evolution (N.Y.) 43, 1349–1368 (1989).
Paradis, E., Claude, J. & Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. https://doi.org/10.1093/biostatistics/1.4.465 (2004).
Warnes, G.R., et al. gdata: Various R programming tools for data manipulation (2017).
Paradis, E. pegas: An R package for population genetics with an integrated-modular approach. Bioinformatics 26, 419–420 (2010).
Revell, L. J. phytools: Phylogenetic tools for comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. https://doi.org/10.1111/j.2041-210X.2011.00169.x (2012).
Wickham, H. Flexibly reshape data. CRAN R Proj (2017).
Harrell, F.E. Hmisc: Harrell miscellaneous (2018).
Knaus, B. & Grünwald, N. vcfR: A package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 17, 44–53 (2017).
Wickham, H. & Chang, W. ggplot2 elegant graphics for data analysis (2015).
Ihaka, R., et al. Color space manipulation (2016).
Acknowledgements
We thank Dr. Tyler Smith for insightful discussions; Dr. Laforest for comments on the manuscript; Drs. Eric Patterson, Todd Gaines, and Pat Reeves for sharing genomic and chloroplast sequence data for kochia; and the greenhouse staff at Agriculture and Agri-Food Canada. Funding for this work was provided by Agriculture and Agri-Food Canada and the Alberta Crop Industry Development Fund.
Author information
Authors and Affiliations
Contributions
S.L.M., L.M.H. and H.J.B. conceived and designed this study. Material was collected by H.J.B. and L.M.H. DNA extraction and analysis was completed by L.B., W.W. and C.A.S. S.L.M. conducted data analysis and led the writing of the manuscript. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martin, S.L., Benedict, L., Wei, W. et al. High gene flow maintains genetic diversity following selection for high EPSPS copy number in the weed kochia (Amaranthaceae). Sci Rep 10, 18864 (2020). https://doi.org/10.1038/s41598-020-75345-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-75345-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
