
Abstract
Sweet potato leaf curl virus (SPLCV) causes yield losses in sweet potato cultivation. Diagnostic techniques such as serological detection have been developed because these plant viruses are difficult to treat. Serological assays have been used extensively with recombinant antibodies such as whole immunoglobulin or single-chain variable fragments (scFv). An scFv consists of variable heavy (VH) and variable light (VL) chains joined with a short, flexible peptide linker. An scFv can serve as a diagnostic application using various combinations of variable chains. Two SPLCV-specific scFv clones, F7 and G7, were screened by bio-panning process with a yeast cell which expressed coat protein (CP) of SPLCV. The scFv genes were subcloned and expressed in Escherichia coli. The binding affinity and characteristics of the expressed proteins were confirmed by enzyme-linked immunosorbent assay using SPLCV-infected plant leaves. Virus-specific scFv selection by a combination of yeast-surface display and scFv-phage display can be applied to detection of any virus.
Similar content being viewed by others


Introduction
The sweet potato (Ipomoea batatas L.) ranks among the world’s seven most important food crops, along with wheat, rice, maize, potato, barley, and cassava1,2. Because sweet potatoes propagate vegetatively, rather than through seeds, they are vulnerable to many diseases, including viruses3. Once infected with a virus, successive vegetative propagation can increase the intensity and incidence of a disease, resulting in uneconomical yields.
Geminiviruses have a twin icosahedral-particle morphology and their DNA consists of circular single-stranded genomes of approximately 3.0 kb4,5,6. Geminiviruses are classified into four genera, Mastrevirus, Begomovirus, Curtovirus and Topocuvirus, based on their insect vector7. The sweet potato leaf curl virus (SPLCV), a member of the genus Begomovirus, is transmitted by the whitefly (Bemisia tabaci Genn.), which is the only natural vector8. SPLCV, which causes symptoms including upward leaf curling in young stage (Fig. 1B), is responsible for declining yields around the world9,10.

(A) Healthy sweet potato leaves. (B) Symptomatic leaves from sweet potato leaf curl virus-infected sweet potato plants.
Efficient diagnosis and rapid treatment are important elements of any disease-control strategy. Several detection methods employing polymerase chain reaction (PCR) of SPLCV genomic DNA have been reported11,12,13. PCR is used widely to detect plant viruses in infected tissues14,15,16. However, PCR suffers from several disadvantages, such as the requirement for complete nucleic acid isolation and costly diagnostic equipment such as a thermocycler and UV transilluminator.
Serological diagnostic techniques are often preferred and provide standardization through simple and rapid analysis17. Antibodies are essential tools for serological diagnosis and their use is growing rapidly18,19. However, conventional serological techniques cannot be used because of difficulty obtaining target-specific antigens; expression of target proteins may not occur, making purification impossible20. Paradoxically, animals or animal cell cultures are required for plant virus diagnosis in plant virus laboratories.
Recombinant antibodies have proven useful for diagnostics and research19,21,22,23. The most commonly used form of recombinant antibodies is the single-chain variable fragment (scFv) which has a simple structure and low molecular weight24,25,26. An scFv consists of a variable heavy chain (VH) and a light chain (VL) of the antibody and is connected by a short polypeptide linker27. It is easily displayed on a phage, and a library can generate appropriate new recombinant antibodies without purification and special equipment28,29,30. It is possible to select an scFv with superior and specific affinity for a target antigen through bio-panning31,32. In addition, an scFv can be easily expressed in Escherichia coli, which grows rapidly in a simple and inexpensive medium and can express significant amounts of a desired protein23,33,34. Yeast-surface displays have recently emerged as a powerful platform for protein engineering35,36. Yeast cells can not only efficiently express 50,000 copies, but also take advantage of unique eukaryotic post-translational machinery such as glycosylation and disulfide isomerization37. For this reason, yeast-surface displays were chosen to express antigens in this study. Based on a combination of yeast-surface and scFv-phage display38, we screened SPLCV-specific scFv clones by bio-panning. In this work, a novel detection system for the SPLCV geminivirus is proposed. The application of phage- and yeast-surface displays to the selection of virus-specific scFv antibodies to SPLCV is also discussed.
Results
Virus antigen detection and selection of yeast cells expressing viral antigen
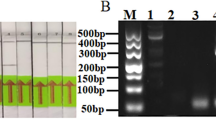
SPLCV is one of the monopartite begomoviruses in geminivirus family, and SPLCV has 6 ORFs for encoding different proteins for systemic movement (V1), cell-to-cell movement (V2), virus replication (C1), transcription activator (C2) or replication enhancer (C3), and symptom determinant (C4). V1 of SPLCV encodes coat protein which is the only structural protein of geminivirus particles. As a target antigen, V1 protein of SPLCV Haenam 1 strain was amplified by PCR and the products were visualized in the form of a slight single band product on 1% agarose gel containing ethidium bromide (Fig. 2A). The V1 sequence of SPLCV Haenam 1 strain consisted of 774 bp nucleotides. The amplified DNA fragments from the V1 of SPLCV was cloned into a pCTCON plasmid vector for yeast-surface display.

(A) PCR detection of target protein. The amplified sweet potato leaf curl virus (SPLCV) V1 gene was confirmed by 1% agarose gel electrophoresis. N lane is the no-template control and lanes 1–4 are amplified SPLCV V1 genes. (B) A schematic diagram of a plasmid (pCTCON-SPV1) for yeast-surface display used for antigen [SPLCV coat protein (CP)] expression. (C) A schematic diagram of a yeast-surface display. Antigen protein is expressed with yeast glycoprotein (Aga2) and multiple tags (HA, c-Myc). (D) FACS analysis of yeast cell lines displaying SPLCV V1. The x axis of each plot represents the cell count and the y-axis represents the intensity of the fluorescent signal of c-Myc-captured antibodies. The white graph is for the negative control cell, and gray histograms are for antigen-displaying induced cells. The x-mean values of antigen-displaying cells are shown at the bottom right of the plot. (E) Western blot analysis of the verification of antigen expression. The N lane is the negative control (EBY100), and lanes 1 and 2 are the induced samples (CP5, CP6). The anti-c-Myc antibody detected the tag fused to the antigen protein.
Yeast display has recently emerged as an alternative strategy, with one important advantage over phage display: the ability to precisely control selective parameters by FACS analysis. This technique can reliably quantify differences in antigen expression levels; antigens are fluorescently labeled with an antibody recognizing the C-terminal c-Myc tag encoded by the plasmid vector (Fig. 2B,C).
The surface display of SPLCV V1 by Saccharomyces cerevisiae also allows the detection of appropriately labeled antigen-antibody interactions by flow cytometry. The 6 colonies were evaluated by shifting degrees from wild-type yeast. The yeast expression cells grown in SDCAA media were used as controls. Representative flow cytometry histograms for selected clones are shown in Fig. 2D. The vertical axis indicates the cell number, and the horizontal axis indicates fluorescence. Similar Gaussian distribution patterns of all selected clones were observed. The mean value of the x axis (x-mean) was used to measure the degree of expression of SPLCV V1 as a statistic. Only one colony of SPLCV showed the shifting of the x-mean value from the control. The x-mean value of the control cell was 7.30 while the highest value of one colony was approximate three times higher (22.91). Two high-expression cell lines (CP 5 and CP 6) were analyzed by western blot to verify antigen expression (Fig. 2E). The anti-c-Myc antibody detected a C-terminal tag of target protein. The data showed a 44 kDa band corresponding to the expected size of SPLCV V1. No band was observed in EBY 100, which was used as the negative control.
Screening of scFvs by bio-panning
The antigen-binding affinity of randomly selected colonies was measured in each round to determine whether the affinity was higher by bio-panning rounds (Fig. 3A). OD450 was measured for screening of phage scFvs from randomly selected colonies. As the number of panning rounds increased, the binding affinity of positive libraries for positive antigens tended to increase. SPLCV-infected sweet potato leaves were used for the selection of antigen-specific scFv after bio-panning. SPLCV-infected sweet potato samples of leaves and phloem tissue39 were identified as shown in Fig. 3B. Both the sensitivity and the specificity used in the diagnosis were critical values. The negative samples (Healthy, TYLCV samples) had OD450 values of less than 0.20, and 15 phage scFv clones showed binding affinity with SPLCV- infected plants (Fig. 3C). The scFv DNA of selected clones was amplified by PCR and the nucleotide sequences were analyzed. The complementarity-determining regions (CDRs) were identified as shown in Table 1. The sequence was compared with the IgBLAST KABAT antibody sequence database40. Most of the scFv clones consisted of nonsense mutations or junk codons, and some clones could not be analyzed. A comparative analysis of VH and VL sequences showed significant differences in the CDRs, which are associated mainly with different biological activities41. Only two scFv clones (scFv “10” was named “G7”, and “12” was renamed “F7”) had complete sequences, including VH and VL chains.

(A) The bio-panning result with sweet potato leaf curl virus (SPLCV) displayed on the yeast cells as an antigen. Sixty randomly selected colonies were measured after each panning round. The collected phages were bound to the target antigen and quantitatively confirmed using HRP-conjugated anti-M13 antibodies at an OD of 450 nm. Data are presented as means ± SEMs. (B) PCR detection of SPLCV in different tissues (phloem tissues and leaves) of sweet potatoes. Lane N is a no-template control. Lane H is virus-free sweet potato samples. Lane P1 and P2 are amplified with phloem tissue of SPLCV-infected samples, and 1, 2 are amplified viral DNA from sweet potato leaf samples. (C) Binding of scFv antibodies, determined by ELISA against SPLCV-infected plant leaves. SPLCV-infected sweet potato leaves were coated onto 96-well microtiter plates. Each scFv was detected using HRP-conjugated anti-M13 antibodies and TMB substrate solution. ELISA readings (OD450) were collected after 30 min of incubation in a TMB substrate at 25 °C. Clone numbers 10 and 12 are the selected scFv used for expression.
Bacterial expression and characterization of anti-SPLCV scFv
The two scFv genes, F7 and G7, were subcloned into pET26b (+) (Fig. 4A) and pDEST-periHisMBP plasmids, respectively (Fig. 4B), and expressed in BL21 (DE3) pLysE. The expression test was performed under various IPTG concentrations (0, 0.1, 1, and 2 mM) and confirmed by western blot analysis (Fig. 4C). In the pET26b (+) plasmids, no soluble scFvs were expressed, and the plasmids were soluble in MBP fusion proteins. When the scFv protein was purified using Ni-NTA, the scFv did not bind well to the column. We therefore performed functional analysis using a filtrated medium in which scFv was expressed as soluble. To compare the binding affinity of different soluble scFv fragments, each protein was measured quantitatively by ELISA (Fig. 4D). Analysis of individual P values revealed a significant difference between negative and healthy samples (P < 0.0001, both) for F7 scFv. On the other hand, G7 scFv showed a slightly lower difference in the negative (P = 0.0049) and healthy (P = 0.0013) samples than did F7. Both scFv showed specific binding affinity to SPLCV sample.

(A) Schematic diagram of pET26b (+) scFv plasmid vector for expression of scFv. (B) Schematic diagram of scFv and MBP fusion protein expression plasmids. (C) Expression tests of anti-sweet potato leaf curl virus (SPLCV) scFv with various IPTG concentrations. Lane 1 shows non-induction, lanes 2–4 show induction with IPTG (0.5, 1, and 2 mM). E. coli (BL21 [DE3] pLysE) cells were induced at OD600 = 0.6, 26 °C for 6 h. (D) Quantitative analysis of ELISA results using a spectrophotometer and data are presented as means ± SEMs (**P < 0.01, ***P < 0.001, GraphPad Prism, GraphPad).
Avidity effects of bivalent scFv binding test
To increase antigen-binding affinity, bivalent F7 scFv was expressed and binding activity was confirmed. The bivalent scFv was expressed in BL21 (DE3) pLysE strain (Merck). The expression test was confirmed under various IPTG concentrations (0, 0.1, 1, and 1 mM). Expression of bivalent F7 scFv protein was confirmed by western blot (Fig. 5B).

Expression test and avidity effects of bivalent scFv. (A) Plasmid vector map for bivalent scFv. This construct was designed based on a MBP fusion expression plasmid, pDEST-periHisMBP64. The existing F7 gene (shown in yellow) and the E. coli codon-optimized F7 gene (shown in green) were ligated with a glycine-serine linker (shown in blue). The AflII and HindIII restriction enzyme sites that do not digest the backbone were added, so that the linker could be replaced. (B) Expression test of bivalent F7 scFv protein. The molecular weight of the expressed fusion proteins was 103.34 kDa in total and the molecular weight of each subunit was as follows: signal peptide (N-terminal signal peptide of MBP; 4.6 kDa) and MBP and polyhistidine tag (45.3 kDa). Lane 0 shows non-induction, lanes 1–3 show induction with IPTG (0.1, 1, and 2 mM). The induction was carried out at OD600 = 0.6, and overexpressed at 26 °C for 16 h. (C) Bivalent scFv (●) and monovalent scFv (■) were bonded with serial dilution in homogenized sweet potato leaf curl virus-infected leaf samples in coated ELISA wells. The 3D8 scFv (▲) was used as a negative control. Data are presented as means ± SEMs.
Different scFv formats containing only F7 scFv domains including monovalent and bivalent scFv were compared in ELISA (Fig. 5C). The monovalent F7 protein bound only at relatively high concentrations of more than 2 μg/mL. The bivalent F7 scFv bound 25 times as efficiently to SPLCV samples compared with the monovalent scFv.
Discussion
Generally, many of the conventional serological diagnosis tests by using polyclonal- or monoclonal antibodies are less specific and less sensitive than molecular diagnosis approaches. Therefore conventional serological diagnosis tests is not suitable for precise and accurate diagnosis of field samples but it is useful in serologically confirmatory testing and epidemiological studies42. To overcome the disadvantages associated with conventional serological diagnosis techniques, molecular diagnosis tests such as PCR, real-time PCR, recombinase polymerase amplification (RPA) and loop mediated isothermal amplification (LAMP) assays have been used for specific and sensitive detection of samples43,44,45. But still serological detection tests have many advantages to detect viruses easily and rapidly from many samples if suitable antibodies are provided. Recombinant scFv has been developed to overcome the limitations and drawbacks of conventional polyclonal- and/or monoclonal antibodies. In many review papers, recombinant scFv can provide us many advantages such as no animal immunization required, shorter periods for scFv production, much cheaper than conventional antibody for mass-production and easy maintaining the scFv as a gene in expression vectors46. Nowadays, scFv has a well-established protocol to produce a completely functional antigen-binding fragment in bacterial systems. In addition, the advances in scFv applications give us more efficient and generally applicable method to produce better scFv by an antibody engineering techniques.
To select an antigen-specific scFv with bio-panning, a large amount of antigen is required. We used yeast-surface display for this study because the system offers eukaryotic post-translational machinery such as disulfide isomerization and glycosylation37. In this study, we prepared a genetically engineered yeast cell that displays the SPLCV V1 as an antigen. Phage display has been used to develop target-specific recombinant antibodies30,47. Some advantages of phage display over conventional hybridoma techniques include shorter time, lower cost, and greater application23.
Based on a combination of yeast-surface display and scFv-phage display, we screened SPLCV-specific scFv clones by bio-panning as described in48,49. Two scFv clones were selected for SPLCV and these genes could be expressed in E. coli for mass production. The scFv was not expressed as soluble in E.coli with short fusion peptides such as a His tag, but was expressed in relatively large protein such as MBP. In addition, a small tag such as a His tag could be detected as an antibody in SDS-PAGE under denaturation conditions. However, the non-denatured protein was not purified through a Ni-NTA column. The His tag was likely not exposed to the outside due to the steric structure of the scFv proteins. The binding affinity for antigens and the properties of the expressed scFv clones were clearly identified by ELISA using SPLCV-infected plant leaves. The results showed that the expression of scFv in E. coli can induce reactivity and specificity of a recombinant antibody. Therefore SPLCV-specific scFv can be mass-produced easily and inexpensively in E. coli.
The design of a bivalent scFv-expressing vector enhanced the binding affinity of monovalent scFv protein50,51. The avidity effect of SPLCV-specific bivalent scFv-Fc proteins resulted in a 25-fold improvement in antigen binding compared with the monovalent scFv fragment. This recombinant scFv detection method with bivalent scFv will contribute to more efficient virus detection system development by enhancing scFv-antigen binding affinity. Then this method can detect virus from small piece of sweet potato samples compared the conventional serological virus detection method with monovalent scFv.
With these advantages, scFv could play an important role in SPLCV diagnosis. The main purpose of this study was screening specific antibodies for sweet potato viruses as a diagnostic method. Recombinant antibody fragments for specific antigens can be modified with genetic engineering to more specific and stable antibodies for serological methods of detection. In addition, a genetically engineered, recombinant scFv with higher efficiency can be produced, based on different scFv binding actions to the antigen.
Methods
Virus antigen DNA preparation
SPLCV Haenam 1 strain (GenBank No. HM754641)-infected sweet potato leaves (Fig. 1B) were homogenized in liquid nitrogen after sampling. The total genomic DNA of the sweet potato was extracted following a method described in52. A small amount of sweet potato (less than 1 g) was collected in a 1.5 mL microcentrifuge tube and 500 μL of Dellaporta extraction buffer (100 mM Tris, pH 8.0), 50 mM of ethylenediamine-tetraacetate EDTA, 500 mM of NaCl, ad 10 mM of β-mercaptoethanol (BME) were added. The microfuge tube was mixed vigorously and incubated for 10 min at 65 °C with 33 μL of 20% sodium dodecyl sulfate (SDS; w/v). Next, 160 μL of 5 M potassium acetate was added, mixed, and spun for 10 min at 15,000 g in a centrifuge. The 450 μL of supernatant was transferred to a new tube and the process was repeated until the supernatant was free of debris. Isopropanol (0.5 volumes) was added with a vortex and spun for 10 min at 15,000 g. When the supernatant was removed, all nucleic acids were in the bottom of the tube. The pellet was washed with 70% ethanol and spun repeatedly for 5 min at 9,000 g. After removal of the supernatant, the pellets were dried at room temperature for 30 min. Finally, the pellet was resuspended in 200 μL of RNase-treated TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). The extracted genomic DNA was purified and then stored at −86 °C.
SPLCV coat protein gene amplification
SPLCV coat protein (V1) was amplified by PCR with genomic DNA as a template. The forward primer 5′-CCTAGGATGACAGGGCGAATTCCC-3′ and reverse primer 5′-AGATCTATTATTGTGCGAATCATAGAAA-3′ were designed to amplify the coat protein. The amplification reaction was performed in PCR tubes containing 1 μL of the template, 10 pmol of each primer and 2 × PCR premix (Takara, Tokyo, Japan), for a total volume of 20 μL. PCR amplification was performed with a thermal cycler machine (T100; BioRad, California, USA) under the following conditions: 10 min at 96 °C for pre-denaturation, followed by thermal cycling for 35 cycles (30 s at 96 °C, 30 s at 55 °C, and 1 min at 72 °C), 10 min at 72 °C for the final extension and storage at 12 °C. The amplified product of the V1 gene (774 bp) was identified on 1.5% agarose gel containing ethidium bromide. The DNA was purified with a gel extraction kit (Macrogen, Seoul, Republic of Korea). Purified DNA was cloned into the TA-cloning vector pGEM®-T easy (Promega, Madison, WI, USA) and introduced into E. coli DH5α according to the manufacturer’s instruction. After transformation, a single colony was placed onto a Luria-Bertani (LB) agar (1.5% w/v) plate containing 50 μg/mL of ampicillin, 100 μg/mL of X-gal, and 1 mM of isopropylthio-β-D-galactoside (IPTG). The selected colony was cultured in 3 mL of LB broth with 50 μg/mL of ampicillin, and the plasmid was extracted with a plasmid mini-prep kit (Bioneer, Daejeon, Republic of Korea) after cell incubation. The plasmid sequence was analyzed by the Macrogen-sequencing service (Seoul, Republic of Korea) with T7 and SP6 primers and a basic local alignment search tool (BLAST) from the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov).
Subcloning for yeast surface display
The SPLCV V1 fragment was inserted into the pCTCON plasmid. The V1 gene in T-vector was digested with AvrII and BglII (Takara, Tokyo, Japan), and inserted between the NheI to BamHI sites of pCTCON according to the manufacturer’s instructions. The ligate was inserted into a DH5α-competent cell, and colony selection onto LB agar plate with ampicillin, colony culture selection in broth, mini-scale plasmid extraction, and sequencing analysis were performed.
Yeast transformation and yeast surface display
Yeast (Saccharomyces cerevisiae strain EBY100)-competent cell preparation was performed following a Clontech manual (Tokyo, Japan)53. Pre-culture was carried out by streaking cells onto a yeast-peptone-glucose (YPD) plate containing 2% dextrose (w/v) and letting them grow for 3 days at 30 °C. A single colony was inoculated with fresh YPD and cultured to an optical density at 600 nm (OD600) of 1.5 and diluted 10-fold in YPD and grown at 30 °C to an OD600 of 0.4–0.6. The cells were harvested and washed with distilled water. The yeast was resuspended in sterilized TE/LiAc buffer (100 mM lithium acetate, 100 mM Tris-HCl, 10 mM EDTA, pH 7.5). The plasmid DNA containing V1 and cell were mixed with carrier DNA (sheared salmon sperm DNA) and sterilized PEG/LiAc (40% of PEG 4000, 100 mM lithium acetate, 100 mM Tris-HCl, 10 mM EDTA, pH 7.5) in a fresh tube with vortexing and incubated at 30 °C for 30 min with shaking at 200 rpm. Dimethyl sulfoxide was added and the solution was incubated for 15 min in a 42 °C water bath and left on ice for 1 min. Cells were then harvested at 15,000 g for 5 s and resuspended in a 1X TE buffer and separated in synthetic defined media containing a dropout supplement (without tryptophan). A single colony was grown in SDCAA media overnight. For the seed culture, the cell was spun by centrifuge at 2,500 g for 15 min, and the media changed to SGCAA with 2% galactose and cultivated for 3 days.
Fluorescence-activated cell sorting analysis
SPLCV V1 protein-displaying yeast cells were analyzed by fluorescence-activated cell sorting (FACS) analysis. Cells (1.2 × 104) were rinsed with phosphate-buffered saline (PBS; pH 7.4) containing 0.1% (w/v) of bovine serum albumin (BSA), and then blocked in PBS with 0.1% BSA (PBS-B). After blocking, the cells were treated in dried-milk PBS (PBS-M)–diluted monoclonal anti-c-Myc antibody (1:500, produced in mice; Sigma, St. Louis, MO, USA) for an hour at 25 °C and washed with PBS-B. The cells were then incubated in PBS-B–diluted polyclonal anti-mouse immunoglobulin G (IgG) TRITC-conjugated antibodies (1:500; Abcam, Cambridge, MA, USA) for 2 h at 4 °C. Cells were washed with PBS-B, and analyzed on a Guava EasyCyte mini instrument (Merck, Darmstadt, Germany) and data were analyzed using Cytosoft software (version 4.2.1). Tomato yellow leaf curl virus (TYLCV) V1displaying yeast cells, cultivated in SDCAA were used as controls.
Yeast western blot analysis
Yeast media were spun and treated in a protein sample buffer (60 mM Tris-HCl, 25% glycerol, 2% SDS, 14.4 mM BME, 0.1% bromophenol blue, pH 6.8) for disulfide bond cleavage. Each supernatant was isolated by centrifuge and equal amounts were separated by 10% polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto a nitrocellulose membrane (GE healthcare, Chicago, IL, USA) with a Towbin buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, 20% [v/v] methanol) with a semi-dry transfer unit. The membranes were rinsed with Tris-buffered saline (TBS; pH 7.4) containing 0.1% (v/v) Tween-20 as a washing buffer (TBS-T). The membranes were blocked with TBS-T with 3% (w/v) skim milk (blocking buffer) and reacted with a blocking buffer-diluted anti-c-Myc monoclonal antibody (1:2000; Sigma, St. Louis, MO, USA) and anti-mouse IgG horseradish peroxidase (HRP)-conjugated antibodies (1:2000; Cell Signaling Technology, Danvers, MA, USA). The membrane-developing reaction was performed with enhanced chemiluminescence solution (GenDEPOT, Katy, TX, USA).
Bio-panning with phage-displayed scFv libraries
A higher initial antibody library size significantly increases the selectivity of high-affinity antigen-specific antibodies54,55,56. The human scFv library pRGA-scFv was provided by Prof. Myung-Hee Kwon (Department of Microbiology, Ajou University School of Medicine)57. The scFv gene fragments were inserted between the Sfi I and Not I restriction sites of a modified pCANTAB 5E phagemid vector. Titers of the propagated phage library were measured as kanamycin-resistant colony forming units (CFUs). Selection of anti-SPLCV phage scFvs by bio-panning was performed as described in48,49. Briefly, wells of 96-well plate (SPL Life Sciences, Pocheon, Korea) were coated with the yeast cells [blank (PBS), negative samples (TYLCV V1–displaying yeast cells) and positive samples (SPLCV V1–displaying yeast cells)] diluted to OD600 value of 0.6 with carbonate coating buffer (pH 9.6). After that, cover the plate with wet paper towel and incubate at 4 °C overnight. After a brief washing with TBS-T buffer (0.1% Tween 20 in TBS), all wells were blocked with blocking buffer (TBS, 0.1% Tween 20, 3% skim milk) for 2 h at 37 °C. For selection of phage scFv proteins, 108 mL/CFU recombinant phages in blocking buffer were added in blank wells, and incubated for 2 h at room temperature. Non-binding phages were transferred to negative-sample wells and incubated overnight at 4 °C. Collected non-binding phages were transferred to positive-sample wells and incubated for 1 h at room temperature. The positive-sample wells were washed vigorously with TBS-T 6 times. SPLCV-binding recombinant phages were eluted with 100 mM triethylamine and neutralized with 1 M Tris-HCl (pH 7.0). The eluted phages was titrated with XL1-blue (Agilent, Santa Clara, CA, USA) CFUs on an LB agar plate containing tetracycline (50 μg/mL) and carbenicillin (50 μg/mL). Three rounds of bio-panning were carried out, and phage titration was performed at every panning round.
Isolation and production of SPLCV-specific scFv
Randomly selected XL1-blue colonies containing individual clones from each bio-panning round were grown in 96-well mini tubes for 6 h at 37 °C and shaken with 2TY growth media containing 25 μg/mL tetracycline, 50 μg/mL carbenicillin, and 1% (w/v) glucose. After incubation, the cells were inoculated into new tubes with fresh 2TY growth media and incubated for 1 h at 37 °C with shaking. The M13KO7 helper phage was inoculated to cells at a multiplicity of infection of 50 and incubated 1 h at 37 °C without shaking. The soup was removed after centrifuging at 2,500 g, and resuspended in 2TY broth containing 25 μg/mL tetracycline, 50 μg/mL carbenicillin and 50 μg/mL kanamycin overnight at 30 °C.
Phage ELISA
To select putative antigen-binding phage scFv clones, a phage enzyme-linked immunosorbent assay (ELISA) assay was performed in 96-well microplates coated with sweet potato leaves (and healthy leaves as controls) in a general-extraction buffer (10 mM sodium sulfite, 2% (w/v) polyvinylpyrrolidone (MW 40,000), 0.2% (w/v) sodium azide, 2% (w/v) BSA, 2% (v/v) tween-20 with PBS, pH 7.4; GEB)58 overnight at 4 °C. Following rinsing with TBS-T 6 times, each phage clone culture’s supernatants were added to each antigen and control well and incubated for 2 h at room temperature. The wells were washed 3 times with TBS-T, and the plates were incubated with HRP-conjugated anti-M13 (1:1000; 11793, Sino Biological Inc., Beijing, China) antibodies at room temperature for 1 h. After washing 6 times with TBS-T, TMB substrate solution (Agdia) was added for 30 min. The enzymatic reaction was stopped by the addition of 1 M sulfuric acid. Absorbance was read at OD450 using a microplate spectrophotometer (BioTeK, Winooski, VT, USA).
Sequence analysis of selected SPLCV-specific phage scFv
Selected scFv clones by bio-panning were re-grown in 2TY media containing tetracycline and carbenicillin, and plasmid extraction was performed. Using these plasmids as a template, scFv sequences were amplified by PCR under the following conditions: 5 min at 96 °C for denaturation, followed by thermal cycling for 35 cycles (30 s at 96 °C, 1 min and 30 s at 55 °C and 1 min at 72 °C), and 5 min at 72 °C for the final extension. PCR was performed by 2X premix (Takara, Tokyo, Japan) using the primer sets pCANTAB5E-S1 5′-CAACGTGAAAAAATTATTATTCGC-3′ and pCANTAB5E-S6 5′-CATTTACTTAAAAGACATACTCC-3′. The amplified DNA was sent to Macrogen (Seoul, Republic of Korea) and analyzed with a DNA-standard sequencing service. Amino acid sequences were deduced from the obtained nucleotide data (ExPASy, https://web.expasy.org/translate). From the results of sequence analysis of screened scFv bound to each antigen, complementarity-determining regions (CDRs) of scFv were found with IgBlast (https://www.ncbi.nlm.nih.gov/igblast)40,59.
Bacterial expression of scFv
The selected scFv genes were cloned into a pET26b (+) (Merck) vector and expressed in E. coli. The genes of scFv were amplified by PCR using primers (Table 2) that added NcoI and EcoRI sites for the pET26b (+) plasmid to the 5′ and 3′ ends of the PCR product, respectively. The His-tag for pET26b (+) at the C-terminus was used for purification. The digested gene products were separated on 1% (w/v) agarose gels and the DNA was excised using a gel extraction kit (Bioneer). The digested PCR products and vectors were ligated overnight at 16 °C and inserted into E. coli strain BL21 (DE3) pLysE cells (Merck). To optimize the expression condition, 3 mL of exponential growth culture (with an OD600 of approximately 0.6) was induced at 26 °C overnight with 0.1, 1, and 2 mM IPTG in a shaking incubator. Large-scale expression of scFv genes in 200 mL of LB medium was induced by the addition of IPTG during 6 h of growth at 26 °C. The cells were harvested by centrifugation (2,500 g, 4 °C for 15 min) and the proteins were extracted from cell periplasm by cold osmotic shock60 and filtrated with a vacuum filter (Merck). For purification, supernatants of scFv expressed in pET26b(+) plasmids were applied to an nickel-nitrilotriacetic acid (Ni-NTA)61. 10% SDS-PAGE was performed to identify and separate each protein. Western blot analysis was carried out with primary anti-His–tag mouse monoclonal antibodies (1:5,000; R&D Systems, Minneapolis, MN, USA) and HRP-conjugated secondary anti-mouse IgG (Cell Signaling Technology) to detect scFv proteins.
Maltose-binding protein fusion expression
To improve protein expression, the scFv genes were subcloned into maltose-binding protein (MBP) encoding a plasmid vector, pDEST-periHisMBP (Addgene, 11086, Cambridge, MA, USA)62 using gateway cloning63. The scFv genes were amplified by PCR using primers (Table 3) in which the AttB1 and AttB2 sites were added to the 5′ and 3′ ends of the PCR product, respectively, for recombination site insertion. To produce entry clones, the BP reaction was carried out with BP Clonase™ II Enzyme mix (Invitrogen, Carlsbad, CA, USA) and pDONR221 (Invitrogen) as a donor plasmid. After addition of proteinase K, recombinant plasmids were transformed with DH5α and selected on the LB media containing kanamycin (50 μg/mL). Subcloning of an entry clone into a destination vector, pDEST-periHisMBP, was performed by LR Clonase™ II Enzyme mix (Invitrogen). After addition of proteinase K, the expression clones were transformed with DH5α and selected in an ampicillin (50 μg/mL) selection medium. The expression clone sequences were analyzed using a DNA sequencing service (Macrogen, Seoul, Republic of Korea). MBP-fusion scFvs were expressed in BL21 (DE3) pLysE cells (Merck). An expression test was then carried out as with the pET26b (+) clone.
Functional analysis of scFv proteins
To determine the binding activity of scFvs purified from E.coli, indirect ELISA was carried out. Briefly, sweet potato samples were coated on a 96-well microplate directly with a general extract buffer overnight at 4 °C. Following washing with TBS-T 6 times, the microplate was blocked with TBS-T containing 3% BSA solution and incubated for 2 h at room temperature. The wells were washed 3 times with TBS-T. The microplates were then coated with purified scFv proteins diluted in a blocking buffer at a concentration of 0.1 μg/μL for 1 h at room temperature. Following rinsing with TBS-T, anti-His tag mouse monoclonal antibodies (R&D Systems) and HRP-conjugated secondary anti-mouse IgG (Cell Signaling Technology) were applied to detect scFv proteins. After washing 6 times with TBS-T, TMB substrate solution (Agdia) was added for 30 min, and the enzymatic action was stopped with the addition of 1 M sulfuric acid. The absorbance was read at OD405 using a microplate spectrophotometer (Tecan Sunrise, Tecan, Switzerland).
Cloning and soluble expression of homodimer scFv
The synthesized gene was cloned into maltose-binding protein (MBP), encoding a plasmid vector, pDEST-periHisMBP (Addgene, 11086, Cambridge, MA, USA)62, by gateway cloning63. The BP reaction was carried out with BP Clonase™ II Enzyme mix (Invitrogen, Carlsbad, CA, USA) and pDONR221 (Invitrogen) as a donor plasmid. The proteinase K was added and, recombinant plasmids were transformed into DH5α and selected on the LB agar plates containing kanamycin (50 μg/mL). To produce expression clones, additional recombination was performed by LR Clonase™ II Enzyme mix (Invitrogen) with entry clones and pDEST-periHisMBP. After addition of proteinase K, the expression clones were transformed into DH5α and selected on LB agar plates containing ampicillin (50 μg/mL) selection medium. The expression clone, named pPHM-F7-di-scFv (Fig. 5A), was analyzed by a DNA sequencing service (Macrogen, Seoul, Republic of Korea). Bivalent F7 scFv protein was expressed in BL21 (DE3) pLysE cells (Merck). An expression test was then carried out as for the protein A fusion clone. For purification, the supernatant products of scFv expression were applied to a nickel-nitrilotriacetic (Ni-NTA) agarose column (Thermo Scientific, Waltham, MA, USA)64. SDS-PAGE (10%) was performed to identify and separate proteins. Western blot analysis was conducted as described by65 using primary anti-His tag mouse monoclonal antibodies (1:5000; R&D systems, Minneapolis, MN, USA) and HRP-conjugated secondary anti-mouse IgG (Cell Signaling Technology) to detect target proteins.
Functional analysis of bivalent scFv
To determine the binding activity of bivalent F7 scFv, indirect ELISA was carried out. Sweet potato samples were coated on a 96-well microplate directly with GEB at 4 °C for overnight in a humidified container. After washing six times with TBS-T, the microplate was blocked with TBS-T containing 3% BSA solution and incubated at room temperature for 2 h. The wells were rinsed three times with TBS-T. The microplates were then coated with purified bivalent scFv serially diluted in blocking buffer (five-fold dilution from 300 μg/mL) at room temperature for 1 hour. The serially diluted F7 scFv also used to compare. Each concentration of scFv protein was determined after calculation based on the values measured with a spectrophotometer with the molecular weight of the scFv protein and coefficient factor. We used estimates based on the values measured with a spectrophotometer to determine the protein concentration
Following rinsing with TBS-T, anti-His tag mouse monoclonal antibodies (1:1000; R&D systems) and HRP-conjugated anti-mouse IgG antibodies (1:1000; Cell Signaling Technology) were treated for detection. After six more rounds of washing, TMB substrate solution (Agdia, Evry, France) was added for 30 min, and the action was stopped by the addition of 1 M sulfuric acid. Absorbance was read at 405 nm against a reference wavelength of 620 nm (A450-A620) using a microplate spectrophotometer.
References
Woolfe, J. A. Sweet potato: an untapped food resource. (Cambridge University Press, 1992).
Pardina, P. R. et al. First report of Sweet potato leaf curl virus infecting sweet potato in Argentina. Australasian Plant Disease Notes 7, 157–160 (2012).
Frison, E. & Ng, S. Elimination of sweet potato virus disease agents by meristem tip culture. International Journal of Pest Management 27, 452–454 (1981).
Van Regenmortel, M. Guidelines to the demarcation of virus species. Arch. Virol. 142, 1505–1518 (1997).
Hanley-Bowdoin, L., Settlage, S. B., Orozco, B. M., Nagar, S. & Robertson, D. Geminiviruses: models for plant DNA replication, transcription, and cell cycle regulation. Critical Reviews in Plant Sciences 18, 71–106 (1999).
Varsani, A. et al. Establishment of three new genera in the family Geminiviridae: Becurtovirus, Eragrovirus and Turncurtovirus. Archives of virology 159, 2193–2203 (2014).
Fauquet, C. et al. Geminivirus strain demarcation and nomenclature. Archives of virology 153, 783–821 (2008).
Bedford, I. D., Briddon, R. W., Brown, J. K., Rosell, R. & Markham, P. G. Geminivirus transmission and biological characterisation of Bemisia tabaci (Gennadius) biotypes from different geographic regions. Annals of Applied Biology 125, 311–325 (1994).
Ling, K.-S., Jackson, D. M., Harrison, H., Simmons, A. M. & Pesic-VanEsbroeck, Z. Field evaluation of yield effects on the USA heirloom sweetpotato cultivars infected by Sweet potato leaf curl virus. Crop protection 29, 757–765 (2010).
Kwak, H.-R. et al. The current incidence of viral disease in Korean sweet potatoes and development of multiplex RT-PCR assays for simultaneous detection of eight sweet potato viruses. The plant pathology journal 30, 416 (2014).
Lotrakul, P., Valverde, R., Clark, C., Sim, J. & De La Torre, R. Detection of a geminivirus infecting sweet potato in the United States. Plant Disease 82, 1253–1257 (1998).
Lotrakul, P., Valverde, R., Clark, C., Hurtt, S. & Hoy, M. Sweetpotato leaf curl virus and related geminiviruses in sweetpotato. Acta horticulturae (2002).
Kwak, H.-R. et al. Virus disease incidences of sweet potatoes in Korea. The Plant Pathology Journal 22, 239–247 (2006).
Navas-Castillo, J., Dıaz, J., Sánchez-Campos, S. & Moriones, E. Improvement of the print-capture polymerase chain reaction procedure for efficient amplification of DNA virus genomes from plants and insect vectors. Journal of virological methods 75, 195–198 (1998).
Kim, J. et al. Seed transmission of Sweet potato leaf curl virus in sweet potato (Ipomoea batatas). Plant Pathology 64, 1284–1291 (2015).
Kil, E.-J. et al. Tomato yellow leaf curl virus (TYLCV-IL): a seed-transmissible geminivirus in tomatoes. Scientific reports 6, 19013 (2016).
Clark, M. F. & Adams, A. Characteristics of the microplate method of enzyme-linked immunosorbent assay for the detection of plant viruses. Journal of general virology 34, 475–483 (1977).
Koenig, R. & Paul, H. Variants of ELISA in plant virus diagnosis. Journal of Virological Methods 5, 113–125 (1982).
Nieri, P., Donadio, D., Rossi, S., Adinolfi, B. & Podesta, A. Antibodies for therapeutic uses and the evolution of biotechniques. Current medicinal chemistry 16, 753–779 (2009).
Voller, A., Bartlett, A., Bidwell, D., Clark, M. & Adams, A. The detection of viruses by enzyme-linked immunosorbent assay (ELISA). Journal of General Virology 33, 165–167 (1976).
Dübel, S. Recombinant therapeutic antibodies. Applied microbiology and biotechnology 74, 723–729 (2007).
Hagemeyer, C. E., von Zur Muhlen, C., von Elverfeldt, D. & Peter, K. Single-chain antibodies as diagnostic tools and therapeutic agents. Thrombosis and haemostasis 102, 1012–1019 (2009).
Kato, M. & Hanyu, Y. Construction of an scFv library by enzymatic assembly of VL and VH genes. Journal of immunological methods 396, 15–22 (2013).
Huston, J. S. et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proceedings of the National Academy of Sciences 85, 5879–5883 (1988).
Bird, R. E. et al. Single-chain antigen-binding proteins. Science 242, 423–426 (1988).
Ward, E. S., Güssow, D., Griffiths, A. D., Jones, P. T. & Winter, G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature 341, 544 (1989).
Huston, J. S. et al. In Methods in Enzymology Vol. 203 46–88 (Elsevier, 1991).
McCafferty, J., Griffiths, A. D., Winter, G. & Chiswell, D. J. Phage antibodies: filamentous phage displaying antibody variable domains. nature 348, 552 (1990).
Marks, J. D. et al. By-passing immunization: human antibodies from V-gene libraries displayed on phage. Journal of molecular biology 222, 581–597 (1991).
Hoogenboom, H. R. et al. Antibody phage display technology and its applications. Immunotechnology 4, 1–20 (1998).
Clackson, T., Hoogenboom, H. R., Griffiths, A. D. & Winter, G. Making antibody fragments using phage display libraries. Nature 352, 624 (1991).
Parmley, S. F. & Smith, G. P. Antibody-selectable filamentous fd phage vectors: affinity purification of target genes. Gene 73, 305–318 (1988).
Clark, E. D. B. Refolding of recombinant proteins. Current Opinion in Biotechnology 9, 157–163 (1998).
Chen, R. Bacterial expression systems for recombinant protein production: E. coli and beyond. Biotechnology advances 30, 1102–1107 (2012).
Boder, E. T. & Wittrup, K. D. Yeast surface display for screening combinatorial polypeptide libraries. Nature biotechnology 15, 553 (1997).
Chao, G. et al. Isolating and engineering human antibodies using yeast surface display. Nat Protoc 1, 755–768, https://doi.org/10.1038/nprot.2006.94 (2006).
Gera, N., Hussain, M. & Rao, B. M. Protein selection using yeast surface display. Methods 60, 15–26, https://doi.org/10.1016/j.ymeth.2012.03.014 (2013).
Gu, X. et al. Novel strategy for selection of monoclonal antibodies against highly conserved antigens: phage library panning against ephrin-B2 displayed on yeast. PLoS One 7, e30680, https://doi.org/10.1371/journal.pone.0030680 (2012).
Morra, M. R. & Petty, I. T. Tissue specificity of geminivirus infection is genetically determined. The Plant Cell 12, 2259–2270 (2000).
Kabat, E. A., Te W, T., Foeller, C., Perry, H. M. & Gottesman, K. S. Sequences of proteins of immunological interest. (DIANE publishing, 1992).
Rouis, S. et al. Cloning and expression of functional single-chain Fv antibodies directed against NIa and coat proteins of potato virus Y. Journal of virological methods 137, 1–6 (2006).
Hairul Bahara, N. H. et al. Phage display antibodies for diagnostic applications. Biologicals 41, 209–216, https://doi.org/10.1016/j.biologicals.2013.04.001 (2013).
Notomi, T. et al. Loop-mediated isothermal amplification of DNA. 28, e63–e63 (2000).
Piepenburg, O., Williams, C. H., Stemple, D. L. & Armes, N. A. J. P. B. DNA detection using recombination proteins. 4 (2006).
Mackay, I. M., Arden, K. E. & Nitsche, A. J. N. a. r. Real-time PCR in virology. 30, 1292–1305 (2002).
Ahmad, Z. A. et al. scFv antibody: principles and clinical application. 2012 (2012).
Winter, G., Griffiths, A. D., Hawkins, R. E. & Hoogenboom, H. R. Making antibodies by phage display technology. Annual review of immunology 12, 433–455 (1994).
O’Brien, P. M. & Aitken, R. Antibody phage display: methods and protocols. Vol. 178 (Springer Science & Business Media, 2004).
Lee, C. M., Iorno, N., Sierro, F. & Christ, D. Selection of human antibody fragments by phage display. Nature protocols 2, 3001 (2007).
Eeckhout, D. et al. A technology platform for the fast production of monoclonal recombinant antibodies against plant proteins and peptides. Journal of Immunological Methods 294, 181–187, https://doi.org/10.1016/j.jim.2004.08.006 (2004).
Pohl, S. C., Schwarz, S., Frenzel, A. & Schirrmann, T. A cassette vector system for the rapid cloning and production of bispecific tetravalent antibodies. Antibodies 1, 19–38 (2012).
Dellaporta, S. L., Wood, J. & Hicks, J. B. A plant DNA minipreparation: version II. Plant molecular biology reporter 1, 19–21 (1983).
Clontech Laboratories, I. (Clontech Laboratories Mountain View, CA, 2009).
Vaughan, T. J. et al. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nature biotechnology 14, 309–314 (1996).
Sheets, M. D. et al. Efficient construction of a large nonimmune phage antibody library: the production of high-affinity human single-chain antibodies to protein antigens. Proceedings of the National Academy of Sciences 95, 6157–6162 (1998).
Knappik, A. et al. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides1. Journal of molecular biology 296, 57–86 (2000).
Kwon, M.-H. et al. A visible phagemid system for the estimation of Cre-mediated recombination efficiency. Journal of Immunological Methods 280, 165–173, https://doi.org/10.1016/s0022-1759(03)00261-8 (2003).
Rogers, S. O. & Bendich, A. J. Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant tissues. Plant molecular biology 5, 69–76 (1985).
Martin, A. C. Accessing the Kabat antibody sequence database by computer. Proteins: Structure, Function, and Bioinformatics 25, 130–133 (1996).
Nossal, N. G. & Heppel, L. A. The release of enzymes by osmotic shock from Escherichia coli in exponential phase. Journal of Biological Chemistry 241, 3055–3062 (1966).
Bornhorst, J. A. & Falke, J. J. In Methods in enzymology Vol. 326 245–254 (Elsevier, 2000).
Nallamsetty, S., Austin, B. P., Penrose, K. J. & Waugh, D. S. Gateway vectors for the production of combinatorially‐tagged His6‐MBP fusion proteins in the cytoplasm and periplasm of Escherichia coli. Protein Science 14, 2964–2971, https://doi.org/10.1110/ps.051718605 (2005).
Hartley, J. L., Temple, G. F. & Brasch, M. A. DNA cloning using in vitro site-specific recombination. Genome research 10, 1788–1795 (2000).
Li, H. et al. Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proceedings of the National Academy of Sciences of the United States of America (PNAS) 97, 773–778 (2000).
Zimmermann, S., Schillberg, S., Liao, Y.-C. & Fisher, R. Intracellular expression of TMV-specific single-chain Fv fragments leads to improved virus resistance in shape Nicotiana tabacum. Molecular Breeding 4, 369–379, https://doi.org/10.1023/A:1009638600492 (1998).
Acknowledgements
This research was supported by a grant (PJ01253403) from the Agricultural Biotechnology Development Program, Rural Development Administration of the Republic of Korea.
Author information
Authors and Affiliations
Contributions
S.-H.C., E.-J.K. and S.L. designed the experiment and concepts. S.-H.C., E.-J.K., E.-H.K. and M.-G.L. performed the experiments, analyzed the data and drafted the manuscript. S.C. and H.-S.B. assisted with the ELISA assay. H.-S.C. and Y.-G.L. prepared virus-infected plant samples and antigens, M.-G.L. and J.L. assisted for protein expression process. S.-H.C., E.-J.K., S.C., H.-S.B., E.-H.K. and M.-G.L. performed analysis and interpretation of data. All of the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cho, SH., Kil, EJ., Cho, S. et al. Development of novel detection system for sweet potato leaf curl virus using recombinant scFv. Sci Rep 10, 8039 (2020). https://doi.org/10.1038/s41598-020-64996-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-64996-0
This article is cited by
-
A Novel Specific Single-Chain Variable Fragment Diagnostic System for Viral Hemorrhagic Septicemia Virus
Marine Biotechnology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
