
Abstract
Citrus canker is a bacterial disease caused by Xanthomonas citri subsp. citri (Xcc) that affects the citrus industry worldwide. Hrp pili subunits (HrpE), an essential component of Type III secretion system (T3SS) bacteria, play a crucial role in the pathogenesis of Xcc by transporting effector proteins into the host cell and causing canker symptoms. Therefore, development of antibodies that block HrpE can suppress disease progression. In this study, a specific scFv detecting HrpE was developed using phage display technique and characterized using sequencing, ELISA, Western blotting, and molecular docking. In addition, a plant expression vector of pCAMBIA-scFvH6 was constructed and agroinfiltrated into Nicotiana tabacum cv. Samson leaves. The hypersensitive response (HR) in the leaves of transformed and non-transformed plants was evaluated by inoculating leaves with Xcc. After three rounds of biopanning of the phage library, a specific human scFv antibody, named scFvH6, was identified that showed high binding activity against HrpE in ELISA and Western blotting. Molecular docking results showed that five intermolecular hydrogen bonds are involved in HrpE-scFvH6 interaction, confirming the specificity and high binding activity of scFvH6. Successful transient expression of pCAMBIA-scFvH6 in tobacco leaves was verified using immunoassay tests. The binding activity of plant-produced scFvH6 to detect HrpE in Western blotting and ELISA was similar to that of bacterial-produced scFvH6 antibody. Interestingly, tobacco plants expressing scFvH6 showed a remarkable reduction in HR induced by Xcc compared with control plants, so that incidence of necrotic lesions was significantly higher in non-transformed controls (≥ 1.5 lesions/cm2) than in the plants producing scFvH6 (≤ 0.5 lesions/cm2) after infiltration with Xcc inoculum. Our results revealed that the expression of scFvH6 in tobacco leaves can confer resistance to Xcc, indicating that this approach could be considered to provide resistance to citrus bacterial canker disease.
Similar content being viewed by others

Introduction
Citrus canker is one of the most economically important citrus diseases which is caused by Xanthomonas citri subsp. citri (Xcc)1. The bacterium causes destructive damage to several citrus cultivars by causing yellow chlorotic rings on leaves, stems, and fruits, inducing defoliation and fruit drop, and even reducing yield in severe cases1,2. Annually, millions of dollars have been spent to prevent and control citrus canker; however, the disease remains a major challenge with an undeniable economic impact on citrus production worldwide3. The bacteria use different strategies to successfully colonize host plants4. Type III secretion system (T3SS) is a key player in bacterial pathogenicity and acts through the injection of effector proteins into eukaryotic host cells4,5. T3SS is encoded by at least six operons of hrpA to hrpF, which are clustered in a 23-kb chromosomal region and play critical roles in the induction of hypersensitive response and bacterial pathogenicity5. T3SS translocates the bacterial effector protein into the cytosol of the host cell through an extracellular filamentous appendage, termed Hrp pilus, which acts as a channel between bacterial and plant cells6. Hrp pili have been characterized in T3SS-containing plant pathogens6,7. The structural component required for pilus formation in Xanthomonas is HrpE protein, which is a 9.7 kDa protein and shows high conservation among Xanthomonas strains, including Xcc, X. oryzae pv. oryzae, X. axonopodis pv. glycines, X. campestris pv. campestris, and X. campestris pv. vesicatoria6. In these bacteria, the assembly of HrpE into the pilus is essential for the secretion of different transcription activator-like (TAL) effectors, which play key roles in Xanthomonas-host interactions6. Therefore, HrpE exerts a critical role in bacterial pathogenicity due to transporting TAL effectors such as PthA into cytosol plants. The effector protein PthA is the most important factor in the production of canker symptoms in host plants8. So far, many methods have been attempted to control the disease using copper-based antimicrobials, uprooting and burning infected citrus trees, traditional breeding to generate canker-resistant citrus varieties, and transgenic expression of resistance genes and antimicrobial agents (AMPs)1,9. Citrus plants produced by transgenic overexpression and CRISPR genome editing approaches have shown promising results in conferring disease resistance10,11; however, only a few genetic engineering studies have shown enhanced canker resistance12. Recently, transgene-free canker-resistant citrus cultivars have been generated via Cas/gRNA ribonucleoprotein (RNP) complex editing of the coding region of canker susceptibility gene citrus sinensis lateral organ boundary 1 (CsLOB1), resulting in enabling canker resistance13.
Over the last three decades, the expression of recombinant proteins in plant cells against specific plant pathogens has received much attention as a new strategy to minimize losses and produce resistant plants14,15. Recombinant-antibody-mediated resistance in plants was first reported in the early 1990s16, and since then, different recombinant antibodies, e.g., fragment antigen-binding region (Fab), single chain variable fragments (scFvs), and single-domain antibody (sdAbs), have been expressed in plants against different plant pathogens17,18,19. Among the various forms of recombinant antibodies, scFv is the most applicable type, which retains antigen specificity and function of the full-size antibody20. Phage display is the most conducive technology for the development of recombinant scFv fragments. This technology screens libraries of random peptides to isolate fragments with high specificity and sensitivity and provides high-quality tests that are less time-consuming21. So far, this strategy has been used for plant expression of scFv antibodies against different targets17,22,23,24.
Some studies have demonstrated that the HrpE mutant of Xanthomonas was deficient in promoting disease symptoms in the host plants, indicating the critical role of HrpE in bacterial pathogenicity25. In addition, it was shown that HrpE can elicit plant immune response and hypersensitivity in non-host plants26. In support of these findings, HrpE could be a candidate for producing resistant plants through plant antibodies. In addition, because of HrpE conservation, anti-HrpE scFvs may recognize HrpE from different strains of Xanthomonas, resulting in broad-spectrum resistance.
We previously designed the specific primers from HrpE sequence submitted in GenBank under Acc. No. WP_011050291.1; and amplified HrpE gene (282 bp) from an Iranian A* isolate of Xcc (NIGEB-088). The PCR product was cloned into pTG-T19 vector, followed by subcloning into pET28a (+) bacterial expression vector bearing an N-terminal 6× His tag sequence. The recombinant protein of HrpE (rHrpE) was expressed in Escherichia coli strain Rosetta (DE3), and the integrity of the expressed protein was confirmed by Western blot analysis27.
In this study, we described an optimized protocol for generating highly sensitive and specific scFv detecting rHrpE using phage display technology. The binding activity of selected scFv to detect rHrpE was assessed by ELISA, Western blotting, and molecular docking. Furthermore, we promoted Nicotiana tabacum cv Samson transient expression of a specific anti-HrpE scFv antibody and examined the functional activity of plant-produced scFv against rHrpE and Xcc. An overview of the study design is shown in Fig. 1. This study provides insights into the development of Xcc-resistant plants by introducing anti-HrpE scFv antibody gene into citrus cultivars.

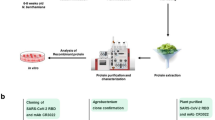
A schematic diagram of the experimental setup used in this study. (a) Representation of expression and purification of rHrpE, (b) selection of monoclonal scFv antibody from phage display library and characterization of selected scFv (scFvH6) against HrpE, (c) transient expression of scFvH6 antibody in tobacco plant model and characterization of plant-produced scFvH6 against HrpE at in vitro and in vivo levels. A. radiobacter: Agrobacterium radiobacter; ELISA: enzyme-linked immunosorbent assay; E. coli: Escherichia coli; IMAC: immobilized-metal affinity chromatography; IPTG: isopropyl ß-D-1-thiogalactopyranoside; rHrpE: recombinant HrpE; scFv: single-chain variable fragment; Xcc: Xanthomonas citri subsp. citri.
Results
Enrichment of phages with specific binding to rHrpE
To identify specific scFvs targeting HrpE, firstly, recombinant protein of HrpE was expressed in E. coli Rosetta strain. The results of SDS-PAGE and Western blotting showed the correct expression and purification of rHrpE at the expected size of 15 kDa with a concentration of about 700 µg/mL (Supplemental Fig. 1). To select antibodies against rHrpE, the human scFv library I was used. Antibodies were selected in microtiter plates against rHrpE in three rounds of biopanning. Based on the results, an increase in the phage recovery rate occurred during three rounds of selection, as far as an increase of 750-fold in the phage recovery rate of the final biopanning round was observed compared with the first round (Table 1). Monitoring the progression of biopanning rounds using polyclonal phage ELISA confirmed that the specificity of eluted phage displaying scFvs against rHrpE was significantly increased after three rounds of biopanning. There was no reaction between the phages obtained from biopanning rounds with BSA as a negative control (P < 0.0001) (Fig. 2a).

(a) Enrichment of Tomlinson I scFv library against rHrpE through three rounds of biopanning. (b) Phage-ELISA for screening of 94 individual phage clones obtained from the third round of panning against rHrpE. (c) The amino acid sequences of selected scFv antibody (scFvH6) with binding specificity to rHrpE (including depiction of CDRs, His-tag and c-Myc tag). (d) Western blot analysis to assay purification of scFH6 using anti-His tag antibody (1:10,000). BSA was considered as negative control. Data are represented as mean ± SD from three independent experiments. ****P < 0.0001, significantly different from control. BSA: bovine serum albumin; CDRs: complementarity-determining regions; M: Marker PageRuler Prestained Protein Ladder (Thermo Scientific, USA); rHrpE: recombinant HrpE.
Identification of a specific scFv antibody against recombinant HrpE
The output obtained after three rounds of biopanning was transformed into E. coli TG1 strain. The ability of 94 single clones to bind to rHrpE was determined by phage-ELISA. Based on the results, six enriched phage clones showed the highest signals for detecting rHrpE (Fig. 2b). The analysis of variations within the genes coding for scFv fragments using BstNI showed that all scFv genes had similar restriction endonuclease profiles (Supplemental Fig. 2), indicating the same sequence for all positive clones selected in phage-ELISA. This result was confirmed by DNA sequencing analysis of scFvs with high binding activity, revealing a distinct scFv sequence containing 6 complementarity-determining regions (CDRs): CDRHI-CDRHIII at positions 28–35, 53–60, and 99–107 and CDRLI-CDRLIII at positions 161–166, 184–187, and 223–231, respectively. This scFv was named scFvH6 and submitted to GenBank under Acc. No. OR513891. The complete gene sequence of scFvH6 is shown in Fig. 2c.
In vitro binding activity and cross-reactivity of scFvH6 antibody against HrpE
To characterize the efficiency of scFvH6 antibody against HrpE, we first expressed scFv in SHuffle strain of E. coli at the expected size (approximately 32 kDa) with yields about 420 µg/mL (Fig. 2d). Analysis of the specificity of scFvH6 by Western blotting exhibited a single band for rHrpE and native HrpE protein at the expected size of 15 kDa, whereas there was no reaction with BSA, demonstrating that scFvH6 specifically bound to HrpE (Fig. 3a).

Characterization of in vitro binding activity of scFvH6 against rHrpE and native HrpE in Xcc-infected samples using (a) Western blot analysis and (b) indirect ELISA. (c) Titration ELISA to detect the working concentration of scFvH6. BSA was used as negative control. Data are represented as mean ± SD from three independent experiments. ****P < 0.0001, significantly different from control. BSA: bovine serum albumin; M: Marker PageRule Prestained Protein Ladder (Thermo Scientific, USA); rHrpE: recombinant HrpE; Xcc: Xanthomonas citri subsp. citri.
To confirm the ability of scFvH6 to bind to native HrpE protein, indirect ELISA was carried out using Xcc-infected samples. The results showed that the highest absorption rate was observed for rHrpE protein (10 µg/mL), followed by samples one, five, and seven of Xcc-infected samples. The rate of adsorption of scFvH6 in all infected samples was more than twice those of the wells containing healthy samples. Absorbance of PthA was similar to BSA as the negative control, confirming non-cross-reactivity with other proteins (Fig. 3b).
To further characterize the binding activity of scFvH6 antibody to rHrpE, the ability of different concentrations of scFvH6 to detect rHrpE was checked by ELISA titration. The results demonstrated that scFvH6 detected rHrpE in a concentration-dependent manner, so that the lowest concentration detecting rHrpE was about 0.01 µg/mL (Fig. 3c).
In silico analysis of scFvH6 antibody affinity for HrpE
To investigate the structural basis of the interaction between scFvH6 and HrpE, the crystal structure of scFv was determined using I-TASSER server service. The Z-score predicted by ProSA indicated high-quality models (Supplemental Fig. 3). The results of docking outputs demonstrated a large binding interface between scFvH6 and HrpE, which was primarily comprising CDRH3, CDRL1, CDRL2, and CDRL3 of scFv fragment (Table 2). This interaction involved five hydrogen bonds and nine hydrophobic interactions. Consistently, most of the protein interferences were mediated by the light chain, so that 10 residues from scFvH6 light chain interacted with HrpE (Fig. 4).

3D structure presentation of the binding poses of HrpE (blue) and scFvH6 (green). Hydrogen bonds involved in scFvH6-HrpE interaction are shown in the 3D structure as pink lines, and total intermolecular analyses containing hydrogen bonds and Van der Waals forces of the interaction are also presented in 2D structure (A: antibody and B: antigen). 2D: two-dimensional; 3D: three-dimensional.
Expression of scFvH6 antibody fragments in plant leaves
To study the expression of scFvGH6 antibody in N. tabacum cv. Samson leaves, pCAMBIA-scFvH6 cassette was constructed and transformed into Agrobacterium radiobacter. The transformed agrobacteria were recognized by PCR using scFv-specific primers (supplemental Fig. 4) and infiltrated into tobacco leaves for transient expression. Temporal analysis of the expression and the integrity of scFvH6 in infiltrated leaves using Western blotting showed a detectable expression level of scFvH6 at the expected size (approximately 35 kDa) after 3 day post-inoculation (dpi), indicating the correct expression and folding of scFvH6 in plant cells (Fig. 5a). Therefore, this time point was selected to take plant material for total protein extraction.

Detection and characterization of expression of scFvH6 in leaves of Nicotiana tabacum cv. Samson. (a) Temporal analysis of the expression scFvH6 in transformed leaves during 1–3 days post inoculation (dpi) using Western blotting. (b) The binding activity of scFvH6 expressed in different transformed leaves to detect rHrpE using Western blotting. (c) The binding activity of plant-produced scFvH6 against rHrpE and native HrpE in Xcc-infected samples using Western blotting and (d) indirect ELISA. Data are represented as mean ± SD from three independent experiments. ****P < 0.0001, significantly different from control. B: bovine serum albumin; M: Marker PageRuler Prestained Protein Ladder (Thermo Scientific, USA); rH: recombinant HrpE; Xcc: Xanthomonas citri subsp. citri.
In vitro binding activity of plant-produced scFvH6 to HrpE
To determine functionality of the plant-produced scFvH6 antibody, the total proteins of the transformed plant were extracted and used as a primary antibody in immunoassay tests. Western blotting results showed that total proteins extracted from different agroinfiltrated leaves detected rHrpE at the expected size of 15 kDa (Fig. 5b). Additionally, the native form of HrpE was detected with plant-derived scFv antibody at the expected size in samples one, five, and seven of Xcc-infected plants (Fig. 5c).
In a parallel experiment, the results of indirect ELISA demonstrated that plant-produced scFvH6 detected rHrpE and Xcc-infected plants, whereas no reaction with healthy and negative control samples was found. Intestinally, samples one, five, and seven of Xcc-infected plants that were detected in Western blotting displayed the highest absorbance in ELISA among other infected samples, revealing differences in concentration of HrpE protein among the samples. These results showed that plant-produced scFvH6 is functional and strongly binds to HrpE (Fig. 5d). The binding activity of plant-derived scFvH6 antibody to HrpE was further compared with that of scFvH6 produced in bacterial cells. As expected, scFvH6 produced in both plant material and bacterial cells displayed similar binding properties (Supplemental Fig. 5).
Function of scFvH6 expressed in tobacco leaves to limit hypersensitive response activated by Xcc
To study the specificity of scFvH6, leaves of N. tabacum cv. Samson was initially inoculated with Xcc, PBS, and agrobacterium bearing pCAMBIA-scFvH6. Based on our results, necrotic lesions developed in leaves infiltrated by Xcc, indicating that Xcc induced HR in N. tabacum cv. Samson. Notably, the distinct necrotic lesions induced by Xcc inoculation first developed 2 dpi, whereas the controls, i.e., PBS and agrobacterium bearing pCAMBIA-scFvH6, were nearly symptomless (Supplemental Fig. 6a). Symptoms continued to develop through 3-day evaluation period. Notably, the size of the necrotic lesions developed by Xcc in tobacco leaves was approximately 1.5 cm2 after 3 dpi, whereas necrotic lesions were not considerable in treatment with pCAMBIA-scFvH6 or PBS (Supplemental Fig. 6b).
Subsequently, when the leaves were agroinfiltrated with pCAMBIA-scFvH6 and inoculated with Xcc 3 days later, HR was suppressed and necrotic lesions arising from Xcc were significantly limited compared with controls (Fig. 6a). Agroinfiltration of leaves with pCAMBIA (as vector control) and infiltration with PBS did not affect necrotic lesions arising from Xcc, so that necrotic area in these treatments was larger than 1.5 cm2 (Fig. 6b). These results showed that scFvH6 functionally blocked HrpE activity and interrupted the development of hypersensitive response in tobacco leaves against Xcc.

(a) Appearance of N. tabacum leaves infiltrated with agrobacterium bearing pCAMBIA-scFvH6w, agrobacterium bearing vector control (pCAMBIA1300), and PBS that after 3 days were inoculated with suspension of Xcc (108 CFU/mL). (b) Comparison of the size of the necrotic lesions developed by Xcc in different treatments (2 dpi). Data are represented as mean ± SD from six independent experiments. ****P < 0.0001, significantly different from control. PBS: phosphate-buffered saline; Xcc: Xanthomonas citri subsp. citri.
Discussion
The use of antibody fragments to confer resistance has been an effective approach against some plant diseases15,28,29. In these strategies, producing antibodies in plant cells can interfere in pathogenesis of pathogens and develop resistance to them28. Recently, the role of HrpE in the secretion of bacterial effector proteins has been characterized6. Accordingly, because of the crucial role of HrpE in Xcc pathogenesis, the present study was conducted to develop and characterize a specific phage display-derived scFv targeting HrpE. Moreover, transient expression of the anti-HrpE scFv antibody in N. tabacum cv. Samson leaves was assessed. Our results demonstrated that a unique scFv antibody, scFvH6, was selected after three rounds of biopanning, showing high binding activity against HrpE even at low concentrations (> 0.01 µg/mL). The high sensitivity and binding activity of specific scFvs to detect various antigens have been reported in different studies23,30. Additionally, the Western blotting result showed a single band at the expected size to detect rHrpE, indicating high specificity of scFvH6. Previous studies have demonstrated that scFv antibodies can be reliable tools with high specificity and sensitivity to detect different antigens17,22,23,31,32, which is in accordance with our results. Additionally, our previous work revealed that the use of specific scFvs against components of T3SS of Xcc can show high specificity for detecting Xcc infection in plants18,33. The results of molecular docking showed that four CDRs (CDRH3, CDRL1, CDRL2, and CDRL3) of scFvH6 were involved in the interaction with HrpE epitopes by forming five intermolecular hydrogen bonds. These interactions demonstrated a specific and strong binding of scFvH6 to HrpE and may explain the high binding activity of scFvH6 in immunoassay tests. Several studies have demonstrated that each of the six CDRs can be involved in antibody-antigen interactions; however, the involvement of CDR3 in antigen interactions has been mostly reported, which is in agreement with our results18,23,34.
Previous studies have demonstrated that transient gene expression in plants allows the study of in vivo protein function35,36. Agrobacterium infiltration is a widely used method for transient expression studies in plants and provides a cost-effective procedure for studying gene function36. It is well documented that the infiltration of plant leaves with agrobacterium bearing scFv genes can confer resistance to different pathogens18,24,32. In addition, we previously showed that transient expression of anti-PthA scFv antibody suppressed HR activated by Xcc in the plant model18. Different studies have discussed the critical role of HrpE in the pathogenesis of Xanthomonas. These studies have demonstrated that the HrpE mutant of Xanthomonas could not elicit HR in a non-host plant and pathogenicity in a host plant. Interestingly, this mutant failed to translocate effector proteins into host cells, resulting in a complete loss of Xanthomonas pathogenicity6,37. Supporting the role of HrpE in translocating effectors to plant cells, the transient expression of scFvH6 in non-host N. tabacum cv. Samson leaves was done to evaluate the inhibitory activity of this antibody on HR triggered by Xcc. Notably, tobacco is widely used for transient or stable gene expression, because it can be easily transformed by A. radiobacter35,38. Since Xcc can elicit HR at the site of infection in tobacco leaves, transient expression of scFvH6 in tobacco provides a short-term test to verify in vivo antibody function. Based on our results, agroinfiltration of N. tabacum cv. Samson leaves with pCAMBIA-scFvH6 construct led to the expression of scFvH6 in leaves at a detectable level after 3 dpi, which was relatively in agreement with other studies17,18,32. Notably, contrarily to these studies, non-detection of scFv protein in Western blotting has been a major challenge in other studies, which may be due to low-abundance antibody in plant cells17; however, several studies have demonstrated that plant-produced antibodies can confer resistance to specific pathogens even at low levels17,18,24,39. In our study, scFvH6 produced in plants could detect HrpE in immunoassay tests, supporting its correct expression and protein folding. Interestingly, both scFvH6 produced in bacteria and plant cells exhibited similar binding activities against rHrpE and cognate HrpE, which was in accordance with previous studies18,24,32.
Remarkably, the challenge of tobacco leaves expressing scFvH6 antibodies with Xcc significantly reduced the development of Xcc-induced necrotic lesions compared with the control plants non-expressing scFvH6, further confirming that the functional expression of anti-HrpE scFv antibody in plant cells and its inhibitory activity on T3SS of Xcc. These findings may explain the function of HrpE in the translocation of effector proteins from bacterial cells to plant cells6. The stability and functionality of scFv antibodies during transient expression in plants have been reported in other studies17,24,32.
Stable expression of recombinant antibodies in transgenic plants has been reported previously40. Transgenic plants producing specific scFvs confer resistance to different plant diseases19,28,39,41. Although there was no report about stable expression of scFvs to control Xcc, different genetic engineering approaches have been used to obtain Xcc-resistant plants10,12,13. The development of transgenic plants expressing antimicrobial peptides, resistance genes, and immune-related genes has been reported to increase canker resistance12. The use of CRISPR/Cas-mediated genome editing of the promoter or coding region of LOB1 has also introduced to confer citrus resistance to Xcc10,42. However, the use of transgenic plants may face concerns regarding food and environmental safety43. Recently, the use of the Cas/gRNA RNP complex to generate transgene-free crops has received much attention. This method presents an efficient transgene-free genome-editing strategy for conferring resistance to citrus Xcc13. The main limitation of our study was that the expression of scFvH6 was only estimated in the plant model, and we didn’t assess the scFv expression in host plants. However, it should be noted that agrobacterium infiltration is not used widely in citrus due to its low efficiency, therefore, there is a need for a more robust transient expression system in citrus leaves44,45. Additionally, the stable expression of scFvH6 in plants can help prove the ability of this antibody to control Xcc, which requires a time- and labor-intensive procedure and may not be provided by agrobacterium-mediated transformation46. Hence, our findings can provide preliminary insight into the development of more tolerant plants to Xcc in the future.
In conclusion, we successfully isolated a potent scFv antibody targeting HrpE of Xcc using the phage display technique, named scFvH6, which showed high binding activity in different in vitro assays and in silico analysis. Additionally, transient expression of scFvH6 in non-host tobacco leaves suppressed Xcc-triggering HR by inhibiting HrpE, probably because of the inability to transfer effector proteins to plant cells. The present study for the first time provides evidence for the transient expression of scFv targeting HrpE in a plant model; thus, further research is required to determine interventions of scFvs on HrpE activities in other Xanthomonas strains using stable expression in host plants.
Materials and methods
Plant materials and growth conditions
Leaf and stem samples from lime plants with blister-like lesions were collected from the southern regions of Iran, and their Xcc infection status was checked by PCR using hrpE-specific primers (Table 3, Supplemental Fig. 7). Additionally, N. tabacum cv Samson were cultivated in an insect-free greenhouse at 22 °C, 80% relative humidity, with a 16:8 h light: dark photoperiod.
Extraction of total proteins from plant samples
Extraction of plant proteins was conducted as described previously33. Briefly, plant leaves were ground to powder using liquid nitrogen, homogenized, and extracted in two volumes of extraction buffer (5 mM EDTA, 5 mM β-mercaptoethanol, 1× PBS, pH 7.5). Leaf extracts were vigorously vortexed and then centrifuged at 10,000×g for 15 min at 4 °C. The supernatants were filtered through 0.45 µm pore filters (Sartorius, Germany).
Production of HrpE recombinant protein
The HrpE gene of NIGEB-088 (Acc. No. GCF_002742455.1), an Iranian A* isolate of Xcc, was previously cloned into pET28a (+) with N-terminally 6× His tag27. To induce protein expression, E. coli Rosetta strain was transformed by pET28-HrpE plasmid and cultured in 2× YT broth (16 g/L tryptone, 10 g/L yeast extract, 5 g/L NaCl, pH 7.2) supplemented with 50 µg/mL kanamycin and 0.1% (w/v) glucose. Expression of protein was induced with 1 mM isopropyl ß-d-1-thiogalactopyranoside (IPTG) at OD600 = 0.4; and then the culture was incubated at 28 °C overnight. After that, the cells were harvested by centrifugation (10,000×g, 4 °C, 20 min) and lysed using sonication at 50% amplitude 10 s pulse settings for 3–5 min. Protein purification was performed using immobilized-metal affinity chromatography (IMAC) (Qiagen, Germany) under native conditions using buffers containing 20–250 mM imidazole gradient. Purified protein was separated on 12% SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane (Sigma-Aldrich, Germany). The membrane was blocked with 5% skimmed milk powder (Fluka, Germany) and then incubated with anti-His tag antibody (1:10,000) (Abcam, UK), followed by alkaline phosphatase (AP) conjugated anti-mouse antibody (1:7000) (Sigma-Aldrich, Germany). Protein signals were detected using the substrate 5-bromo-4-chloro-3-indolyl phosphate (BCIP) and nitro-blue tetrazolium (NBT) (Sigma-Aldrich, Germany). Protein concentration was estimated by measuring the sample absorbance at 280 nm.
Enrichment of phage display library against recombinant HpE
To select specific scFv antibodies, biopanning rounds were conducted on Tomlinson I scFv phage library (MRC, Cambridge, England, ReIn_0017), as described previously31. Briefly, an immunotube (Nunc Inc, Denmark) was coated with rHrpE (~ 100 µg/mL) and kept at 4 °C for 16 h. The coated immuno-tube was blocked with 3% (w/v) bovine serum albumin (BSA) (Fluka, Neu-Ulm, Germany), washed with 1× PBS, and incubated with phage suspension (~ 1013 cfu) to allow phage-protein binding at 37 °C for 2 h. After incubation, the immuno-tube was rinsed with PBST (1× PBS with 0.1% Tween-20) to remove unbound phages. The elution of antigen-bound phages was carried out using 100 mM trimethylamine, and the pH was neutralized by adding 1 M Tris–HCl (pH = 7.4). The eluted phages were amplified by reinfection of E. coli TG1 cells at OD600 = 0.4, and amplified phages were rescued from bacterial cells by superinfection with M13KO7 helper phage (about 109 pfu/mL). The amplified phages were used in the next round of biopanning. The recovery rate of eluted phages was determined after each round of biopanning23.
Specificity assessment of phage clones for detection of recombinant HpE
To assess the enrichment of antigen-specific clones, polyclonal phage-ELISA was conducted after each biopanning round as described previously31. Briefly, ELISA plates were coated with rHrpE (10 µg/mL), blocked with 3% skimmed milk powder, and washed with PBST. Eluted phages obtained from each round of biopanning were added to 96-well microtiter plates and incubated at 37 °C for 2 h. After washing with PBST, the plates were incubated with an anti-M13-HRP conjugate antibody (1:10,000) (Abcam, UK) at 37 °C for 2 h. The reactions were detected using 2,2-azino-di-3-ethylbenz-thiazoline sulfonate (ABST) (Ferramentas, Lithuania) substrate. After stopping the reaction by adding 100 μL 1 N H2SO4, the absorbance was measured at 405 nm using an ELISA plate reader (Tecan, Switzerland). The reactions were considered positive when the absorbance of the analyzed samples was more than twice the absorbance of BSA (3%).
Identification of specific recombinant phages for HrpE
To isolate monoclonal anti-HrpE binders, phages were further screened in a 96-well microtiter plate as described previously22. Briefly, E. coli TG1 cells were infected with phages obtained from the last round of biopanning. Bacterial cells bearing phage particles were plated on TYE medium agar supplemented with 1% (w/v) glucose and 100 µg/mL ampicillin. Individual colonies (94) were randomly picked and cultured in 96-well microtiter plates containing 2 × YT with 1% (w/v) glucose and 100 µg/mL ampicillin. Bacterial culture was superinfected with M13KO7 helper phage (about 109 pfu/mL) to rescue phage clones from bacterial cells and then incubated at 30 °C for 16 h. After incubation time, the culture supernatants containing the individual phage clones were separated by centrifugation (2000×g, 4 °C, 10 min) and used as primary antibodies to detect rHrpE in phage-ELISAs. Binding specificity of phages was assessed as described in polyclonal phage ELISA.
Characterization of scFvs antibodies
Endonuclease digestion and sequence analysis
The clones detecting rHrpE were amplified using pHEN primers designed by Oligo software version 7 (Table 3). The amplicons were digested with BstNI (Ferramentas, Lithuania) and separated using agarose gel electrophoresis (2% w/v) as described by Pansri et al.47. In addition, Sanger sequencing (Pishgam Biotech Co., Tehran, Iran) was done for positive clones using pHEN primers. The sequence alignment analysis was performed using IMGT/V-QUEST alignment tool (http://www.imgt.org).
Western blotting
To express scFv fragments, the phagemids were transformed into E. coli SHuffle strain. Protein expression was induced with 1 mM IPTG at OD600 = 0.4–0.6; and bacterial culture was incubated at 30 °C overnight. The specific scFv antibody was purified using IMAC under native conditions. Protein purification was analyzed using SDS-PAGE and Western blotting using anti-His tag antibody and AP-conjugated anti-mouse IgG antibody. Target proteins were visualized by adding BCIP/NBT substrate. Antibody concentration was determined by measuring absorbance at 280 nm.
To evaluate the ability of scFv antibody to detect antigen, rHrpE (10 μg/mL), total soluble proteins from healthy and Xcc-infected plant leaves (5 μL), and BSA as a negative control were separated on 12% SDS-PAGE; and transferred to PVDF membranes. The protein-transferred membranes were incubated with 1 µg/mL scFv as a primary antibody at 4 °C for 16 h. Target proteins were detected as described earlier.
Enzyme-linked immunosorbent assay (ELISA)
To assess specificity of the scFv antibody, indirect ELISA was used as previously described23. Briefly, 96-well microtiter plates were coated with healthy and Xcc-infected plant extracts, rHrpE (10 µg/mL) as a positive control, and BSA as a negative control; and kept at 4 °C for 16 h. Additionally, recombinant protein pthA of Xcc previously prepared by28 was used as an antigen for detection of cross-reactivity of selected scFvs. After blocking, wells were incubated with 1 µg/mL scFv antibody at 4 °C for 16 h, followed by an anti-c-myc tag antibody (1:5000) (Abcam, USA) at room temperature for 2 h. The bound scFv antibody was detected using goat-anti-mouse HRP secondary antibody (1:7000) (Abcam, UK), and colorimetric detection was performed by adding ABST substrate, as described earlier.
Additionally, to determine scFv antibody titer, a titration ELISA was performed23. To do this, a dilution series of the scFv antibody (0.00001, 0.0001, 0.001, 0.01, 0.1, 1, 10, 100 μg/mL) was prepared and used to detect rHrpE (10 μg/mL). scFvA2 antibody that exhibited very low absorption in phage-ELISA and BSA were considered as negative controls. The antibody-antigen reaction was detected as described earlier. The lowest concentration of scFv antibodies that detected rHrp was considered as the working concentration.
Homology modeling and molecular docking
To further characterize the binding activity of the selected scFv to HrpE, the interaction between these proteins was evaluated in silico. To do this, the tertiary structures (3D) of HrpE and scFvH6 were modeled using I-TASSER server service (https://zhanglab.ccmb.med.umich.edu/I-TASSER/). The predicted 3D structural models were refined by using ModRefiner server service (http://zhanglab.ccmb.med.umich.edu/ModRefiner/) to select structures closer to the native structure. The accuracy of the generated homology models was assessed using ProSA server service (https://prosa.services.came.sbg.ac.at/prosa.php). The mode of interaction between HrpE and scFvH6 was investigated using Haddock server service (https://haddock.science.uu.nl/services/HADDOCK2.2/). The selection of model was conducted based on the minimal local energy and the largest cluster size. The possible interactions between antibody and antigen were visualized using Pymol software version 1.5.0.1 (http://pymol.findmysoft.com). A detailed analysis of the visualization and 2D structural representations was accomplished using Ligplot plus software version (4.5.3).
ScFv-mediated inhibition of HrpE in vivo
scFv cloning into plant expression vector
The DNA sequence encoding anti-HrpE scFvH6 was obtained by digesting scFvH6 construct derived from scFv phage display library by NcoI/NotI restriction enzymes and ligated in pCAMBIA1300 plant expression vector under the control of 35S cauliflower mosaic virus (CaMV) promoter with Tobacco etch virus 5′ untranslated region (UTR), which previously prepared by Raeisi et al.18. The new vector, pCAMBIA-scFvH6, was transformed into A. radiobacter GV3101 strain (gentamicin, kanamycin, and rifampicin resistance) by heat shock method. The transformed agrobacteria were checked by PCR using scFv-specific primers (Table 3).
Transient expression of anti-HrpE scFv antibody in tobacco leaves
Transformed A. radiobacter was cultured in Luria broth (LB) medium supplemented with 10 mg/L rifampicin and 50 mg/L kanamycin at 28 °C with shaking at 150 rpm. Bacterial culture at OD600 = 1 were centrifuged (4300×g, 10 min, 4 °C); and bacterial cells were resuspended in agroinfiltration buffer (0.43% MS salts, 10 mM MES, 2% (w/v) sucrose, 200 mM acetosyringone, pH5.6) to an OD600 of 0.5 and incubated at room temperature for 3 h. Agroinfiltration of 6-week-old N. tabacum cv. samson plants was carried out by injecting a suspension of recombinant agrobacteria into the lower epidermis of intact leaves using a syringe without a needle. The suspension of Xcc (108 CFU/mL of NIGEB-088 solution in 1× PBS) and 1× PBS buffer were infiltrated into tobacco leaves as controls. After 3 days, agroinfiltrated leaf material was harvested and total soluble proteins were extracted using two volumes of extraction buffer. Western blotting was done to evaluate scFvH6 in leaf total proteins at different time points as previously described. All experiments were performed using six leaves of six independent biological replicates.
Characterization of plant-produced scFv antibody
To estimate functional ability of produced antibody in plant to detect Xcc, Western blot and indirect ELISA were done18. To do this, the total protein of the transgenic plant was extracted as previously described and used as a primary antibody to detect rHrpE (10 µg/mL) and native HrpE in protein extracts from Xcc-infected samples. Extracted proteins from healthy plants and BSA were considered as negative controls. The reactions were detected as described earlier.
scFvH6 transient expression in tobacco leaves and hypersensitive response evaluation against Xanthomonas citri subsp. citri (Xcc)
To evaluate the ability of plant-produced scFvH6 to reduce Xcc symptoms, recombinant agrobacteria bearing pCAMBIA-scFvH6 were infiltrated into the N. tabacum cv. Samson leaves as described18. Three days after infiltration, the agroinfiltrated areas were inoculated with a suspension of Xcc (108 CFU/mL). Plant responses were checked for 1–3 dpi. The ability of Xcc to elicit hypersensitive response (HR) on leaves was assessed by measuring the size of the necrotic lesions developed by Xcc.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8.0 (GraphPad Software, CA, USA). Data were checked for normality using Shapiro–Wilk’s test and statistically analyzed using one-way analysis of variance (ANOVA). Fisher's Least Significant Difference (LSD) test assessed the differences between means at P ≤ 0.05. The data were presented as mean ± standard deviation (SD) for at least three independent experiments.
Statement of compliance
Permission to collect lime plant samples was obtained from the collection curator of each region before sampling. This study complies with relevant institutional, national, and international guidelines and legislation.
Data availability
All data generated or analyzed that support the findings of this study are available from the corresponding author (H.R), upon reasonable request.
References
Ference, C. M. et al. Recent advances in the understanding of Xanthomonas citri ssp. citri pathogenesis and citrus canker disease management. Mol. Plant Pathol. 19, 1302–1318. https://doi.org/10.1111/mpp.12638 (2018).
Stover, E. et al. Incidence and severity of Asiatic citrus canker on diverse citrus and citrus-related germplasm in a Florida field planting. HortScience. https://doi.org/10.21273/HORTSCI.49.1.4 (2014).
Gabriel, D., Gottwald, T. R., Lopes, S. A. & Wulff, N. A. Bacterial pathogens of citrus: Citrus canker, citrus variegated chlorosis and Huanglongbing (eds Talon, M. et al.) 371–389 (Woodhead Publishing, 2020).
Zimaro, T. et al. The type III protein secretion system contributes to Xanthomonas citri subsp. citri biofilm formation. BMC Microbiol. 14, 96. https://doi.org/10.1186/1471-2180-14-96 (2014).
Meline, V. et al. Role of the acquisition of a type 3 secretion system in the emergence of novel pathogenic strains of Xanthomonas. Mol. Plant Pathol. 20, 33–50. https://doi.org/10.1111/mpp.12737 (2019).
Sheikh, T. M. M. et al. The type III accessory protein HrpE of Xanthomonas oryzae pv. oryzae surpasses the secretion role, and enhances plant resistance and photosynthesis. Microorganisms. https://doi.org/10.3390/microorganisms7110572 (2019).
Alvarez-Martinez, C. E. et al. Secrete or perish: The role of secretion systems in Xanthomonas biology. Comput. Struct. Biotechnol. J. 19, 279–302. https://doi.org/10.1016/j.csbj.2020.12.020 (2021).
Duan, S. et al. Functional characterization of the citrus canker susceptibility gene CsLOB1. Mol. Plant Pathol. 19, 1908–1916. https://doi.org/10.1111/mpp.12667 (2018).
Hao, G., Zhang, S. & Stover, E. Transgenic expression of antimicrobial peptide D2A21 confers resistance to diseases incited by Pseudomonas syringae pv. tabaci and Xanthomonas citri, but not Candidatus Liberibacter asiaticus. PLoS ONE. 12, e0186810. https://doi.org/10.1371/journal.pone.0186810 (2017).
Huang, X., Wang, Y. & Wang, N. Highly efficient generation of canker-resistant sweet orange enabled by an improved CRISPR/Cas9 system. Front. Plant Sci. https://doi.org/10.3389/fpls.2021.769907 (2022).
Borrelli, V. M. G., Brambilla, V., Rogowsky, P., Marocco, A. & Lanubile, A. The enhancement of plant disease resistance using CRISPR/Cas9 technology. Front. Plant Sci. https://doi.org/10.3389/fpls.2018.01245 (2018).
Sun, L., Ke, F., Nie, Z., Wang, P. & Xu, J. Citrus genetic engineering for disease resistance: Past, present and future. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20215256 (2019).
Su, H. et al. Generation of the transgene-free canker-resistant citrus sinensis using Cas12a/crRNA ribonucleoprotein in the T0 generation. Nat. Commun. 14, 3957. https://doi.org/10.1038/s41467-023-39714-9 (2023).
Schillberg, S., Raven, N., Spiegel, H., Rasche, S. & Buntru, M. Critical analysis of the commercial potential of plants for the production of recombinant proteins. Front. Plant Sci. 10, 720. https://doi.org/10.3389/fpls.2019.00720 (2019).
Satheeshkumar, P. K. Expression of single chain variable fragment (scFv) molecules in plants: A comprehensive update. Mol. Biotechnol. 62, 151–167. https://doi.org/10.1007/s12033-020-00241-3 (2020).
Tavladoraki, P. et al. Transgenic plants expressing a functional single-chain Fv antibody are specifically protected from virus attack. Nature. 366, 469–472. https://doi.org/10.1038/366469a0 (1993).
Yajima, W. et al. Expression of anti-sclerotinia scFv in transgenic Brassica napus enhances tolerance against stem rot. N. Biotechnol. 27, 816–821. https://doi.org/10.1016/j.nbt.2010.09.010 (2010).
Raeisi, H., Safarnejad, M. R., Alavi, S. M., Farrokhi, N. & Elahinia, S. A. Transient expression of an scFvG8 antibody in plants and characterization of its effects on the virulence factor pthA of Xanthomonas citri subsp. citri. Transgenic Res. 31, 269–283. https://doi.org/10.1007/s11248-022-00301-1 (2022).
Hemmer, C. et al. Nanobody-mediated resistance to Grapevine fanleaf virus in plants. Plant Biotechnol. J. 16, 660–671. https://doi.org/10.1111/pbi.12819 (2018).
Raeisi, H., Azimirad, M., Asadzadeh Aghdaei, H., Yadegar, A. & Zali, M. R. Rapid-format recombinant antibody-based methods for the diagnosis of Clostridioides difficile infection: Recent advances and perspectives. Front. Microbiol. 13, 1043214. https://doi.org/10.3389/fmicb.2022.1043214 (2022).
Alfaleh, M. A. et al. Phage display derived monoclonal antibodies: From bench to bedside. Front. Immunol. https://doi.org/10.3389/fimmu.2020.01986 (2020).
Raeisi, H. et al. Development and characterization of phage display-derived anti-toxin antibodies neutralizing TcdA and TcdB of Clostridioides difficile. Microbiol. Spectr. 11, e0531022. https://doi.org/10.1128/spectrum.05310-22 (2023).
Raeisi, H., Safarnejad, M. R. & Sadeghkhani, F. A new single-chain variable fragment (scFv) antibody provides sensitive and specific detection of citrus tristeza virus. J. Virol. Methods. 300, 114412. https://doi.org/10.1016/j.jviromet.2021.114412 (2022).
Safarnejad, M. R., Fischer, R. & Commandeur, U. Recombinant-antibody-mediated resistance against Tomato yellow leaf curl virus in Nicotiana benthamiana. Arch. Virol. 154, 457–467. https://doi.org/10.1007/s00705-009-0330-z (2009).
Weber, E. & Koebnik, R. Domain structure of HrpE, the Hrp pilus subunit of Xanthomonas campestris pv. vesicatoria. J. Bacteriol. 187, 6175–6186. https://doi.org/10.1128/jb.187.17.6175-6186.2005 (2005).
Gottig, N., Vranych, C. V., Sgro, G. G., Piazza, A. & Ottado, J. HrpE, the major component of the Xanthomonas type three protein secretion pilus, elicits plant immunity responses. Sci. Rep. 8, 9842. https://doi.org/10.1038/s41598-018-27869-1 (2018).
Raisi, H., Safarnezhad, M., Alavi, M., Ellahinia, A. & Farrokhi, N. Gene cloning and expression of Xanthomonas citri subsp. citri pilin. J. Agric. Biotech. 10, 35–48. https://doi.org/10.22103/jab.2018.2066 (2018).
Safarnejad, R. et al. Antibody-mediated resistance against plant pathogens. Biotechnol. Adv. 29, 961–971. https://doi.org/10.1016/j.biotechadv.2011.08.011 (2011).
Ko, K., Brodzik, R. & Steplewski, Z. Production of antibodies in plants: Approaches and perspectives. Curr. Top Microbiol. Immunol. 332, 55–78. https://doi.org/10.1007/978-3-540-70868-1_4 (2009).
Yang, H. et al. Screening of a ScFv antibody with high affinity for application in human IFN-γ immunoassay. Front. Microbiol. 9, 261. https://doi.org/10.3389/fmicb.2018.00261 (2018).
Raeisi, H., Safarnejad, M. R., Moeini, P., Safarpour, H. & Sokhansanj, Y. Isolation of single-chain variable fragment (scFv) antibodies for detection of Chickpea chlorotic dwarf virus (CpCDV) by phage display. Arch. Virol. 165, 2789–2798. https://doi.org/10.1007/s00705-020-04813-1 (2020).
Shahryari, F., Shams-bakhsh, M., Safarnejad, R., Safaie, N. & Kachoiee, S. Preparation of antibody against immunodominant membrane protein (IMP) of Candidatus Phytoplasma aurantifolia. Iran. J. Biotechnol. 11, 34–41. https://doi.org/10.5812/ijb.9305 (2013).
Raeisi, H., Safarnejad, M. R., Alavi, S. M., Elahinia, S. A. & Farrokhi, N. Applying the pthA effector protein of Xanthomonas citri subsp. citri for production of specific antibodies and its application for detection of infected plants. Plant Pathol. J. 102, 79–87. https://doi.org/10.1007/s42161-019-00385-5 (2020).
Ahmad, Z. A. et al. scFv antibody: Principles and clinical application. Clin. Dev. Immunol. 2012, 980250. https://doi.org/10.1155/2012/980250 (2012).
Garabagi, F., McLean, M. D. & Hall, J. C. Transient and stable expression of antibodies in Nicotiana species. Methods Mol. Biol. 907, 389–408. https://doi.org/10.1007/978-1-61779-974-7_23 (2012).
Tyurin, A. A., Suhorukova, A. V., Kabardaeva, K. V. & Goldenkova-Pavlova, I. V. Transient gene expression is an effective experimental tool for the research into the fine mechanisms of plant gene function: Advantages, limitations, and solutions. Plants https://doi.org/10.3390/plants9091187 (2020).
Tian, D. et al. The rice TAL effector-dependent resistance protein XA10 triggers cell death and calcium depletion in the endoplasmic reticulum. Plant Cell. 26, 497–515. https://doi.org/10.1105/tpc.113.119255 (2014).
Wu, B. et al. Harnessing a transient gene expression system in Nicotiana benthamiana to explore plant agrochemical transporters. Plants https://doi.org/10.3390/plants10030524 (2021).
Cervera, M. et al. Transgenic expression in citrus of single-chain antibody fragments specific to Citrus tristeza virus confers virus resistance. Transgenic Res. 19, 1001–1015. https://doi.org/10.1007/s11248-010-9378-5 (2010).
Diamos, A. G. et al. High level production of monoclonal antibodies using an optimized plant expression system. Front. Bioeng. Biotechnol. https://doi.org/10.3389/fbioe.2019.00472 (2020).
Song, X. et al. An antibody that confers plant disease resistance targets a membrane-bound glyoxal oxidase in Fusarium. New Phytol. https://doi.org/10.1111/nph.13806 (2015).
Peng, A. et al. Engineering canker-resistant plants through CRISPR/Cas9-targeted editing of the susceptibility gene CsLOB1 promoter in citrus. Plant Biotechnol. J. 15, 1509–1519. https://doi.org/10.1111/pbi.12733 (2017).
Tsatsakis, A. M. et al. Environmental impacts of genetically modified plants: A review. Environ. Res. 156, 818–833. https://doi.org/10.1016/j.envres.2017.03.011 (2017).
Acanda, Y., Welker, S., Orbović, V. & Levy, A. A simple and efficient agroinfiltration method for transient gene expression in Citrus. Plant Cell Rep. 40, 1171–1179. https://doi.org/10.1007/s00299-021-02700-w (2021).
Wu, H., Acanda, Y., Canton, M. & Zale, J. Efficient biolistic transformation of immature citrus rootstocks using phosphomannose-isomerase selection. Plants https://doi.org/10.3390/plants8100390 (2019).
Conti, G., Xoconostle-Cázares, B., Marcelino-Pérez, G., Hopp, H. E. & Reyes, C. A. citrus genetic transformation: An overview of the current strategies and insights on the new emerging technologies. Front. Plant Sci. https://doi.org/10.3389/fpls.2021.768197 (2021).
Pansri, P., Jaruseranee, N., Rangnoi, K., Kristensen, P. & Yamabhai, M. A compact phage display human scFv library for selection of antibodies to a wide variety of antigens. BMC Biotechnol. 9, 6. https://doi.org/10.1186/1472-6750-9-6 (2009).
Acknowledgements
The authors would like to acknowledge the members of the Iranian Research Institute of Plant Protection for providing research facilities.
Author information
Authors and Affiliations
Contributions
H.R. performed microbiological and molecular experiments; H.R., and M.R.S. designed and supervised the study; H.R., and M.R.S. carried out the data analysis; H.R., M.R.S., and M.O.A. reviewed and edited the manuscript; M.R.S., S.M.A., N.F., and S.A.E. critically revised the manuscript. All authors read the final version of the manuscript and approved the list of authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Raeisi, H., Safarnejad, M.R., Alavi, S.M. et al. Transient expression of anti-HrpE scFv antibody reduces the hypersensitive response in non-host plant against bacterial phytopathogen Xanthomonas citri subsp. citri. Sci Rep 14, 7121 (2024). https://doi.org/10.1038/s41598-024-57355-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-57355-w
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
