
Abstract
Homoplastic mutations are mutations independently occurring in different clades of an organism. The homoplastic changes may be a result of convergence evolution due to selective pressures. Reports on the analysis of homoplastic mutations in Mycobacterium tuberculosis have been limited. Here we characterized the distribution of homoplastic single nucleotide polymorphisms (SNPs) among genomes of 1,170 clinical M. tuberculosis isolates. They were present in all functional categories of genes, with pe/ppe gene family having the highest ratio of homoplastic SNPs compared to the total SNPs identified in the same functional category. Among the pe/ppe genes, the homoplastic SNPs were common in a relatively small number of homologous genes, including ppe18, the protein of which is a component of a promising candidate vaccine, M72/AS01E. The homoplastic SNPs in ppe18 were particularly common among M. tuberculosis Lineage 1 isolates, suggesting the need for caution in extrapolating the results of the vaccine trial to the population where L1 is endemic in Asia. As expected, homoplastic SNPs strongly associated with drug resistance. Most of these mutations are already well known. However, a number of novel mutations associated with streptomycin resistance were identified, which warrants further investigation. A SNP in the intergenic region upstream of Rv0079 (DATIN) was experimentally shown to increase transcriptional activity of the downstream gene, suggesting that intergenic homoplastic SNPs should have effects on the physiology of the bacterial cells. Our study highlights the potential of homoplastic mutations to produce phenotypic changes. Under selective pressure and during interaction with the host, homoplastic mutations may confer advantages to M. tuberculosis and deserve further characterization.
Similar content being viewed by others


Introduction
Mycobacterium tuberculosis (Mtb) is a successful human-adapted species that has undergone long-term coevolution with its human host1. Previous studies2,3 revealed the clonal expansion of Mtb, indicating a very low possibility of horizontal gene transfer between strains. Therefore, genetic variations in Mtb appear to be primarily derived from nucleotide substitutions, insertions, and deletions4,5. The global population of M. tuberculosis sensu stricto is grouped into five phylogenetic lineages, including L1 (Indo-Oceanic family); L2 (East Asian family); L3 (East-African Indian family); L4 (Euro-American); and L76,7.
In the past decade, several studies have demonstrated associations between Mtb lineages with demographic profiles, geographic distribution, transmission capacity, pathogenesis, cytokine induction, and drug resistance6,8,9,10.Once a strong association has been established, it may be explained through the identification of genotype-specific mutations, in coding or intergenic regions11,12,13. Some phenotypes, such as drug resistance, usually occur in several phylogenetic lineages. Mutations that promote drug resistance can be identified by correlating the resistance phenotypes with convergent mutations or homoplastic single nucleotide polymorphisms (SNPs). These mutations in the Mtb isolates would be expected to enhance survival in the presence of anti-TB drugs14,15,16,17,18. In addition, associations have been found between homoplastic mutations with increased Mtb transmission rate19 and TB disease phenotypes, such as tuberculous meningitis20. The studies primarily examined homoplastic SNPs in coding regions and did not examine the distribution of the homoplastic SNPs in relation to the genotypes. In most cases, when identified, intergenic homoplastic SNPs have not been experimentally evaluated.
We previously sequenced 1,170 Mtb isolates and characterized the sublineage-specific SNPs. Our analysis demonstrated correlations of Mtb genotypes with ages, ethnicity, HIV infection, and drug resistance21. All isolates were obtained from the same geographic area and health service system, and likely to be share some selective pressures. Here, we characterized the distribution of homoplastic SNPs in both intergenic and coding regions of the Mtb genomes that were identified in five isolates or more. This analysis provided some clues on the functions of the genes and their relevance to Mtb survival in the host environment. Furthermore, the functional effect of a homoplastic mutation in a regulatory region was experimentally examined in this study.
Results
Distribution of homoplastic SNPs in M. tuberculosis genome
The homoplastic SNPs were identified in 1,170 clinical isolates, with 480, 521, 11 and 158 isolates belonging to L1 to L4, respectively. 1,229 homoplastic SNPs were identified in 5 isolates or more. 1,121 SNPs (91%) were in the coding sequences of 589 annotated genes and 108 SNPs in intergenic regions (Table 1 and Supplementary Table S1).
As we used the genomes of the H37Rv strain, belonging to L4.9, as the reference for identifying SNPs, the numbers of total identified SNPs would depend on the lineages of the studied isolates, which might also correlate with the numbers of homoplastic SNPs. Indeed, L1, the most distant lineage from L4, harbored the most numbers of total SNPs as well as homoplastic SNPs, with the average of 207 homoplastic SNPs per isolate compared to the average of 74 for L4 and 180 for L2. However, the average ratio of homoplastic SNPs to total SNPs of Mtb L2 isolates was 0.13, significantly higher than other Mtb lineages (One way ANOVA, Kruskal Wallis test, p < 0.001) (Fig. 1a), which were 0.099 for L1, 0.11 for L3 and 0.089 for L4.

(a) The plots between the ratios of the numbers of homoplastic SNPs per total SNPs and the total SNPs of the 1,170 Mtb isolates. The ratios of homoplastic SNPs per total SNPs were markedly different between lineages so that the major groups from left to right are L4 (0.089 ± 0.011), L2 (0.130 ± 0.013), L3 (0.110 ± 0.006), and L1 (0.099 ± 0.015), respectively. The small leftmost group represents L4.8 which is most closely related to H37Rv, a member of L4.9. (b) The ratio of the number of homoplastic SNPs to total SNPs of each Mtb gene categorized into 8 functional categories: i) virulence and detoxification (VF, 226 genes), ii) lipid metabolism (LM, 238 genes), iii) information pathway (IP, 232 genes), iv) cell wall and cell process (CW, 751 genes), v) pe/ppe family protein (PE_PPE, 168 genes), vi) intermediary metabolism and respiration (IMR, 898 genes), vii) regulatory protein (RP, 193 genes), viii) conserved hypothetical protein (CHP, 1,163 genes). Each dot represents a gene that harbored at least a SNP. Statistical differences were evaluated with the nonparametric Kruskal-Wallis test. Asterisks (***) indicate the statistical significance (p < 0.001).
We further compared the average ratios of homoplastic SNPs per total SNPs in 8 different functional categories of the genes, as shown in Fig. 1b. The pe/ppe genes had a significantly higher average ratio than those of the others (One way ANOVA, Kruskal Wallis test, p < 0.001). This is consistent with the high density of homoplastic SNPs (number of SNPs per kb) of pe/ppe genes, which was significantly higher than that of information pathway, intermediary metabolism, and lipid metabolism with p-value <0.01 (Table 1). Next, we compared the number of homoplastic SNPs in the genes of each functional group in 4 different Mtb lineages. The average ratio of homoplastic SNP to total SNPs of pe/ppe family of L4 was significantly lower than those of L1, L2 and L3 (One way ANOVA, Kruskal Wallis test, p < 0.001) (Supplementary Figure S1).
More than one-third of the identified homoplastic SNPs occurred in the pe/ppe gene family, with 96% in the coding regions as shown in Table 1. However, among the pe/ppe gene family, the distribution of SNPs in coding sequences was uneven with 90 (54%) pe/ppe genes harboring no homoplastic SNPs in the studied population. In contrast the homoplastic nonsynonymous SNPs arose to a density of more than 0.5% of nucleotides in 5 ppe genes, namely ppe18, ppe19, ppe57, ppe59 and ppe60, as shown in Supplementary Table S2, indicating a high level of genetic variation. It should be noted that ppe18, ppe19 and ppe60 were highly homologous while ppe57, ppe58 and ppe59 were a separate homologous group22. ppe57, ppe59, and ppe60 were previously reported to be highly polymorphic23. Compared to the other gene categories, the percentage of pe/ppe genes containing the homoplastic SNPs in the regulatory region was also high (6% of total pe/ppe genes) (Table 1).
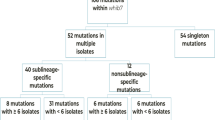
Homoplastic SNPs were not distributed evenly in all 4 Mtb lineages, as shown in Supplementary Table S3. 40% of the homoplastic SNPs were identified only in a single lineage. These SNPs could be identified only when the bacterial samples were classified into sublineages. 10% of the homoplastic SNPs were distributed in all 4 Mtb lineages (Supplementary Table S3).We identified some SNPs in all studied isolates except the ones belonging to the same clades as the H37Rv strain or L4.9. The clades could be L4.8 and L4.9 or L4.5, L4.8 and L4.9 or L4.3, L4.4, L4.5, L4.8 and L4.9. The SNPs, which were absent in these monophyletic groups, were not considered as homoplastic SNPs. Approximately 30% of the homoplastic SNPs occurred in more than 100 clinical M. tuberculosis isolates (Supplementary Figure S2).
In addition to the functional classification, the genes of M. tuberculosis were also classified based on whether they were essential, contributed to virulence, or exhibited antigenicity. We compared the density of nsSNPs per kb in each category (Supplementary Table S4). The homoplastic nsSNPs were more common in non-essential than essential genes. Nevertheless, the frequencies of nsSNPs identified in virulence and antigenic protein genes were not statistically different from non-virulence and non-antigenic genes, respectively. 57 (25%) of the 226 homoplastic nonsynonymous SNPs in antigenic protein genes caused missense mutations in T cell epitopes as shown in Supplementary Table S5, with 53 (93%) of them being in pe/ppe family and ESX family proteins.
PPE proteins share a relatively conserved N-terminal domain with a proline-proline-glutamic acid motif. Interestingly, the N-terminal regions of PPE18, PPE57, and PPE60 proteins interact with the TLR2 receptor present in macrophages24,25,26 and modify the host responses by inducing IFN-γ production25,27,28. These genes are candidates for TB vaccine development. To explore more on the possible cause of genetic variations among the 5 ppe genes, we calculated dN/dS values of the whole genes, their T cell epitope regions and non-T cell epitope regions as shown in Table 2. The dN/dS of T cell epitopes of the five ppe genes were generally less than one, and usually less than the non-T cell epitopes, suggesting purifying selection, even though the density of homoplastic SNPs was high. The only exception is the dN/dS of the T cell epitopes of ppe57, of L2 isolates which was more than one, suggesting positive selection (Table 2).
PPE18 is a subunit of a promising TB vaccine candidate, M72/AS01E, which has been evaluated in a phase 2b clinical study29. Of the 65 nucleotide variants identified in PPE18 from the 1,170 L1-L4 M. tuberculosis isolates shown in Supplementary Table S6, 37 SNPs were found in more than 5 isolates and 42 SNPs (65%) being non-synonymous. 11 of the 22 homoplastic nsSNPs (50%) resulted in changes in the T cell epitopes (Fig. 2a). Eight homoplastic nsSNPs were located in the relatively conserved N-terminal domain, which is involved in the interaction with TLR2 of host cells. L1 isolates harbored 20 of the 22 homoplastic nsSNPs while the L2 isolates harbored 11.Three previously reported mutations G704T, G860A, and T926C30,31 were commonly present. G860A, a mutation affecting a T cell epitope, was actually present in all L1 isolates and all Beijing strains (L2.2), but not the Proto-Beijing sublineage (L2.1) as shown in Supplementary Figure S4. G704T (Fig. 3) and T926C were found in 86% and 53% of L1 isolates, respectively but not L2. The presence of G860A in all L1 and the Beijing strains were also confirmed in a set of the complete genomes of 6 L1 and 35 L2 isolates deposited in Genbank (Supplementary Table S7). G704T were present in all 6 complete L1 genomes and T926C were present in 5 L1 genomes while both were not found in any complete genomes of L2. C88A, also affecting a T cell epitope, was present almost exclusively to a sublineage of the Beijing strains, L2.2.1 Asia Ancestral 3, with 76% (112/146) harboring the SNP (Fig. 3). Four L4 isolates also contained the SNP. In the complete genome set, it was present in one of the two isolates that belonged to the Asia Ancestral 3 sublineage and 23% (26/111) of the L4 isolates. Novel mutations identified in the T cell epitopes included G96C and T365C. The G96C substitution was found in 78 L1 isolates (17%), affected the same T cell epitope as C88A and was predicted to impair the function of PPE18, as shown in Supplement Table S1. The presence of two double mutations, G163A-T164C and A787C-G788A, resulted in amino acid changes, Val55Thr and Ser263His respectively.

A diagram of M. tuberculosis PPE18 (a) and PPE60 (b) proteins. PPE18 is composed of 391 amino acids, with amino acids 1–180 classified as the N-terminal domain responsible for TLR-2 binding. The PPE60 protein is 393 amino acids in length, with amino acid residues 6–163 being the N-terminal domain, while amino acids 309–389 contained the C-terminal domain. T cell epitope regions were mapped as solid lines. The locations of homoplastic nsSNPs were shown. Asterisks indicate nsSNPs affecting the T cell epitopes and circles represent nsSNPs outside the T cell epitope regions.

Position of homoplastic C1339436A SNP (C88A) and G1340052T SNP (G704T) of ppe18 in phylogenetic tree of 1,170 M. tuberculosis isolates. The phylogeny was reconstructed using Bayesian Interference (BI) methods, which included 4 major lineages (L1: Indo-Oceanic family, L2: East Asian family, L3: East-African Indian family, L4: Euro-American) and 38 sublineages belonging to L1, L2 and L4. Red terminal lines correspond to Mtb isolates carrying homoplastic C88A, and green terminal lines represent the isolates carrying homoplastic G704T of ppe18. The C88A mutation mostly presents in 76% of L2.2.1 Asia Ancestral 3 isolates, 1 isolate of L2.2.1 Asia Ancestral 4 and 4 isolates of L4.4. While, the G704T SNP was found in 86% of L1 isolates and 3 isolates of L4.5.2. Color background shading represents all isolates of that sublineage carrying the homoplastic SNPs.
PPE60 is a component of the ID87/GLA-SE vaccine candidate32, which has comparable efficacy to the BCG vaccine in mice32. 30 homoplastic SNPs (18 nsSNPs, 11 sSNPs, and 1 premature stop codon mutation shown in Supplementary Table S6) were found with 3 nsSNPs (16%) resulting in changes to amino acid residue sequences in T cell epitopes and 1 nsSNP leading to a premature stop codon (Fig. 2b).
Associations between homoplastic SNPs and the demographic and clinical characteristics of the patients
The demographic and clinical characteristics of TB patients were previously reported21. The associations of all 1,229 homoplastic variants and various TB phenotypes including anti-TB resistance, treatment outcomes, AFB smear positivity and HIV status were evaluated (Supplementary Table S8). We found significant associations after Bonferroni correction between homoplastic SNPs with anti-TB resistance and treatment outcomes but not with HIV status and AFB smear positivity.
There were 11 homoplastic SNPs associated with the drug resistance phenotypes. Eight of them are already well known. However, associations between streptomycin resistance to mutations in aglA (T922C, p = 2.15E-13), pe_pgrs7 (G2353A, p = 4.20E-13), and sseA (G826A, p = 1.01E-12) have never been described and required further investigations. There were several SNPs associated with poor outcomes of treatment, either failure or death within a year after the start of treatment, which deserved further investigations.
Functional characterization of a homoplastic SNP in the 5′ regulatory region of DATIN (Rv0079)
108 SNP loci were found in the 5′ upstream region of 87 Mtb genes. Promoters were predicted using the Neural Network Promoter Prediction program (BDGP) and Bacterial promoter prediction program BPROM. Homoplastic SNPs were predicted in 22 promoter regions with five having higher promoter scores greater than 0.8 (Supplementary Table S1). Three of the five promoters were predicted to regulate conserved hypothetical proteins with unknown functions. Only one, (−47 A/T), located 47 bp upstream the start codon of a pathogenesis-related DATIN (Rv0079) gene. Rv0079 encodes a conserved hypothetical protein and is the only DosR regulon-encoded gene that harbors a homoplastic SNP upstream of the coding sequence. While DATIN (dormancy associated translation inhibitor) is important for the latency state of M. tuberculosis, the homoplastic SNP was identified in only 10 isolates, although these were distributed among all 4 lineages, as shown in Supplementary Table S1. This indicated that the mutations at this particular position likely occurred independently several times.
There is no reliable computational method to predict the effect of the −47 A/T mutation in Rv0079. To evaluate the effect of the mutation on expression of Rv0079, we constructed a promoter fusion with a downstream gfp gene. The 150 bp-long DNA segment upstream of the start codon of DATIN displayed promoter activity as expected in M. smegmatis and M. tuberculosis H37Ra. Plasmids containing T at the −47 position exhibited significantly higher GFP fluorescence levels than plasmids containing A at the same position, p-value <0.001 (Fig. 4). Remarkably, GFP fluorescence was 10 times greater in M. tuberculosis H37Ra transformed with the promoter-gfp reporter plasmid containing T at −47 compared to the reporter with A at this position (mean RFU = 23,031 and 2,300 units, respectively). Thus the homoplastic A/T SNP in Rv0079 regulatory region enhanced promoter activity of a downstream gene. It is quite interesting to prove whether this homoplastic SNP would have effect on level of DATIN activity in clinical isolates.

Effect of the homoplastic −47 A/T SNP on the expression of a downstream gfp gene. The promoter activity was expressed in relative fluorescence levels per unit of optical density (RFU; fluorescence level/OD) of GFP. The activity of promoter with T nucleotide at −47 was compared to the promoter with A nucleotide in M. smegmatis (a) and M. tuberculosis H37Ra (b). Mycobacterial cells carrying the pFPV2 plasmid served as a positive control for GFP expression, whereas cells without the plasmid served as the negative control. The presented data were averaged RFU from three independent experiments. Asterisks represent significant difference; p-value <0.001.
Identification of the transcriptional start site (TSS) in Rv0079 (DATIN)
To establish the exact location of the −47 A/T mutation in the Rv0079 promoter, 5' RACE was performed to determine the TSS. This analysis revealed the TSS is at the A nucleotide 37 bp upstream of the DATIN start codon (Fig. 5). Therefore, the homoplastic −47 A/T is 10 bases upstream of the TSS, inside the −10 promoter of Rv0079. The potential −35 and −10 promoter sequences are predicted to be TGTCCG-15 bp-GACAAG starting at the position −35 bases and −14 bases upstream the TSS, respectively.

Characterization of the regulatory sequence upstream of Rv0079 (DATIN). GTG is the start codon of Rv0079. The transcriptional start site (TSS) was identified at nucleotide 37 upstream the start codon (arrow), with the RACE chromatogram shown below. The predicted −10 and −35 promoter sequences were indicated (boxed). The double-underlined letters represent the putative ribosomal binding site (RBS) of Rv0079.
Discussion
In this work, we explored the distribution of homoplastic SNPs in a cohort of M. tuberculosis. The 1,170 M. tuberculosis isolates examined in this study were from a single province in Thailand, and were likely to be exposed to similar environments. The identified homoplastic SNPs should provide some clues on the mechanisms of survival or pathogenesis of the bacteria.
Our analysis revealed the presence of homoplastic SNPs in all categories of genes. They were less common in the essential genes, as expected. Approximately 36% of identified homoplastic SNPs were in the pe/ppe family, although more than half of the pe/ppe genes did not harbor any homoplastic SNPs. This suggested the diverse functions of the protein family with the more polymorphic proteins probably playing important roles in interaction with the host cells. The pe/ppe family consists of 99 pe and 69 ppe genes, accounting for 7% of total M. tuberculosis coding potential22. Many proteins in this family are cell surface-associated proteins or secreted proteins and involved in the pathogenesis of M. tuberculosis. The pe/ppe proteins also act as potent antigens that induce both host humoral and cell-mediated immune responses22,33. There are evidences that at least some pe/ppe genes are under selective pressure23. Our analysis was consistent with this prediction and also indicated that selective pressures were variable among different polymorphic pe/ppe proteins. Both pe and ppe genes share similar N-terminal sequences, allowing homologous recombination within and between pe/ppe genes, which may contribute to the high mutation rates observed in this gene family34. Recombination of pe/ppe genes may be responsible for producing a large source of antigenic variation in M. tuberculosis35 with the potential to modulate the host immune response.
The homology between members of pe and ppe gene families makes identification of SNPs in the gene family less reliable than the other groups of genes. Nucleotide variants in pe/ppe genes are therefore generally excluded from phylogenetic analysis. In order to increase the reliability of the identification of SNPs in pe/ppe genes, we used two SNPs identification methods and analyzed only the SNPs identified by both methods. We also analyzed the presence of common homoplastic SNPs in the complete genomes of M. tuberculosis deposited in Genbank, the results of which were consistent with the results from the set of 1,170 clinical isolates.
Three of the most polymorphic PPE proteins, PPE18, PPE57, and PPE60, have been shown to interact with TLR2 receptors. Many polymorphisms have been identified in TLR236 with at least two mutations, R677W37 and R753Q38,39, have been known to impact tuberculosis and other mycobacterial infections. These residues are within the extracellular domain of TLR2 and may be involved in PPE-TLR2 interactions. The mutations in the ppe genes may provide a molecular mechanism for human-Mtb co-evolution. However, the structural consequences of the SNPs in ppe18, ppe 57 and ppe 60 remained to be determined.
There were differential associations between the pe/ppe polymorphisms and lineages. The ppe18 was most polymorphic among L1 isolates while ppe57 was more polymorphic among L2 isolates (Table 2 and Supplementary Table S6). The selective pressure acting on pe/ppe genes appeared to depend on sublineages, as indicated by dN/dS. For example, ppe18 was highly polymorphic among L1 isolates, yet this gene was under purifying selective pressure in all lineages. For most lineages, the selective pressure on ppe57 was purifying selection; however, the protein was under positive selective pressure in L2 which appeared to drive them to be more polymorphic.
PPE18 is a component of the M72/AS01E vaccine candidate, a fusion protein derived from two Mtb antigens, pepA and PPE18. Administration of the M72/AS01E vaccine to HIV-negative adults with latent TB infections had a 54% efficacy in preventing the development of active TB disease29. However, sequences of ppe18 among 225 clinical isolates collected from Arkansas and Turkey displayed substantial variability, especially among isolates belonging to principal genetic group 1, which are mostly L1 and L2 strains30. The deviation of ppe18 sequences from the H37Rv (L4) strain used to produce the vaccine candidate was reported to be high among three isolates belonging to the EAI family (L1)31. In contrast, Mortier et al.40 demonstrated high diversity in only one of the four EAI isolates. Here we showed variations in ppe18 from all 4 major Mtb lineages, particularly L1. The numbers of nonsynonymous mutations (both homoplastic and non-homoplastic SNPs) in the isolates belonging to L1-L4 were 35, 16, 7 and 16 loci, respectively (Supplementary Table S6). Three ppe18 mutations were common among L1 isolates with G860A affecting a T cell epitope and also common in the Beijing strains. Furthermore, homoplastic nsSNPs were found to affect promiscuous epitopes that can bind to multiple HLA alleles41. Examination of binding between PPE18 epitopes and various DRB1 of MHC class II indicates that the M72/AS01E vaccine may not be recognized well in people from many countries, including Thailand, Vietnam, Indonesia, and the Philippines41. These countries are all endemic to L1 strains with highly variable ppe18 sequences. The co-evolution between ppe18 and host cell antigen recognition indicates that precaution is needed when extrapolating data from the clinical trials in Africa to Southeast Asia. Vaccine epitope diversity could induce variable host immune responses and, therefore, may affect the novel TB vaccine efficacy42,43.
Ruesen et al.20 reported the homoplastic mutations in Rv0218 and the absence of Rv3433c and nanK to be highly correlated with tuberculous meningitis. However, the SNPs in these genes were not found in this study, which included patients with pulmonary tuberculosis.The same study also identified homoplastic mutations in six pe/ppe proteins associated with pulmonary tuberculosis disease. Here we confirmed the presence of three homoplastic nsSNPs: A270T in pe/pgrs18, L111W in pe/pgrs19, and D236G in pe/pgrs2620.
The homoplastic SNPs can be intragenic or intergenic. Methods to predict the effect of intragenic SNPs are fairly well-developed while predicting the effects of intergenic SNPs is more difficult due to the considerable variability in the sequences of regulatory elements44.
In this study, we experimentally evaluated the effect of the homoplastic SNP upstream of Rv0079 (DATIN) as it is the only DosR regulated gene that harbors the intergenic SNP. The genetic variations SNP substantially increased the expression of the downstream gfp reporter gene. The effect on promoter activity is presumably similar in native bacterial cells. The Rv0079 (DATIN) gene is a member of dormancy survival regulon (DosR regulon)45,46 and the DATIN protein interacts with the 30S ribosome and inhibits protein synthesis in M. tuberculosis47. In addition, DATIN could interact with the TLR2 receptor and stimulate the secretion of pro-inflammatory cytokines (IFN-ℽ, TNF-α, IL-1β, and IL-8) by mononuclear cells48. These cytokines play significant roles in granuloma formation. Therefore, the SNP identified in the intergenic region upstream of the Rv0079 (DATIN) gene may alter the cellular processes for entry into a dormant period. The association of a homoplastic substitution in DATIN regulatory region and adaptability of clinical Mtb to enter dormancy warrants further investigation. Although the prevalence of the SNP upstream DATIN was not high, this work demonstrated that screening for homoplastic SNPs could be a method for identifying function-affecting SNP in regulatory regions. To our knowledge, this is the first report demonstrating the functional effect of an intergenic homoplastic SNP.
In conclusion, this study evaluated homoplastic SNPs, which were not reported in most WGS studies of Mtb. The possible functional consequences of SNPs were examined and the information obtained may be useful for understanding the bacterial-host interaction, and developing new technology against TB43 such as identifying new drug targets as well as understanding the effects of vaccines.
Materials and Methods
Bacterial strains
1,170 Mtb isolates, analyzed by WGS, were obtained from pulmonary TB patients in Chiangrai province, Northern Thailand between 2003–2010. The isolates were previously classified into four major lineages, with 480, 521, 11 and 158 isolates belonging to L1–4, respectively. The three major Mtb lineages were further divided into 38 sub-lineages49.
Genome sequencing data and Variant calling
Genome sequencing data of all Mycobacterial isolates obtained from our previous study49 were further analyzed, using two different variant callers, GATK50 and SAMtools51. The intersecting SNPs were used for further analysis. Criteria for single nucleotide variant filtering was used to meet high-confidence variants, according to the study of Faksri et al.52; mapping quality >50, base alignment quality >20, >10 reads covering each site. Insertions and deletions were excluded from this study. The SNP calling workflow was shown in Supplementary Figure S3.
Genomic sequence data availability
Whole genome sequence data used in this study were deposited at European Nucleotide Archive (ENA) of EMBL-EBI under the accession numbers ERP006738.
Homoplastic SNPs identification
Phylogenetic trees were previously constructed based on 70,937 SNPs by Maximum Likelihood (ML) and Bayesian Inference (BI) methods21. Both phylogenetic methods resulted in the same sublineage classifications, although the structure of the tree inside each sublineage might be slightly different. All identified SNPs were mapped in the phylogenetic trees. SNPs that were found only in a single sublineage were excluded from further analysis. The SNPs were considered as homoplastic if they occurred in two or more sublineages and did not form a monophyletic clade. The reference H37Rv strain belongs to L4, the SNPs present in all L1, L2 and L3 isolates but not in any of L4 isolates were considered to be specific to L4 and not considered as homoplastic SNPs. The homoplastic SNPs found in less than 5 isolates (approximately 0.5% of Mtb isolates) and SNPs present in mobile genetic elements and prophages were excluded from further analysis.
Prediction of the functional consequences of homoplastic SNPs on protein functions and T cell epitopes
The effects of nonsynonymous mutations on protein functions were determined with the online tool, PredictSNP 1.053, using three algorithms (SIFT, Polyphen-1, and SNAP). The programs reported the possible effects as N: neutral effect (no effect) and A: deleterious effect on protein function. Analysis of the mutations altering amino acid sequences in T cell epitopes was performed using the information catalogue in the Immune Epitopes Database (www.iedb.org). This database contains a large number of experimentally-proved T cell epitopes in M. tuberculosis54. The dN/dS ratio was calculated from all SNP types identified in our study and analyzed with the MEGA7 program55. The functional gene categories were extracted from Tuberculist56
Identification of SNPs in the predicted promoter regions
Homoplastic SNPs residing within 200 bp upstream of the start codons from annotated genes were identified as intergenic SNPs. The promoter regions were predicted from the 5' non-coding sequences using the Neural Network Promoter Prediction computer program57 and Bacterial promoter prediction program BPROM58.
SNP identification in complete genomes of M. tuberculosis deposited in Genbank
Complete genomes of Mtb were obtained from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/genome/browse#!/prokaryotes/166/). The number of genomes were 6 for L1, 35 for L2, 2 for L3 and 111 for L4. The sublineage of each isolate was identified using previously published criteria21. Homologous genes to the ones in the H37Rv strains that contained homoplastic SNPs and subsequently the SNPs were identified.
Experimental characterization the effect of the homoplastic SNP upstream of the DATIN
The effect of an intergenic homoplastic mutation on transcriptional activity was performed using a green fluorescence protein (GFP) reporter assay. Briefly, 150 bp-long DNA fragments upstream of the start codon of DATIN (Rv0079) were amplified, using DATIN-F and -R primers (Supplementary Table S9), from both M. tuberculosis H37Rv and a clinical Mtb strain belonging to sublineage 1.1.1.2 which carried the homoplastic SNP in Rv0079 intergenic region. The PCR products were purified, cloned into a promoter-less GFP plasmid59 and transformed into E. coli strain XL1-Gold (Stratagene, USA). A clone harboring the DNA segment was selected and the recombinant plasmid was sequenced to confirm the correct sequence. The plasmid was purified and transformed into M. smegmatis mc2155 and M. tuberculosis H37Ra ATCC 25177 by electroporation. Mycobacterial cells carrying the recombinant plasmid were grown on Middlebrook 7H11 agar plates (Difco, USA) containing 30 µg/ml of kanamycin. GFP expression was measured using a Synergy H1 hybrid Multi-mode microplate reader (Biotek, USA) in the bottom-reading mode. The excitation wavelength was 485 nm and the emission wavelength was 535 nm, with 7H9-OADC as blank. The fluorescence of samples was measured in triplicate wells and repeated three times on different occasions and compared to the wild type strains.
5′ Rapid Amplification of cDNA Ends (RACE) assay
Total RNA of the M. tuberculosis H37Ra strain was extracted and converted to cDNA. Homopolymeric A-tails were added to the cDNA using the terminal deoxynucleotidyl transferase (TdT) enzyme (Promega, USA). Nested PCR amplification was carried out using gene-specific antisense primers and oligo d(T) anchor primer (Supplementary Table S9). The product of the 5′ RACE reaction was subsequently cloned into pGEM-T Easy vector (Promega, USA) and transformed into E. coli XL-1Blue. Finally, DNA sequencing (Bioneer, Korea) using T7 and SP6 universal primers was performed for identification of the 5′ end of the cDNA.
Statistically analysis
Statistical analyses were performed using SPSS software (version 21.0, SPSS Inc, Chicago, IL). Statistical difference between two groups or among 8 functional groups were evaluated using the nonparametric Mann-Whitney U test and nonparametric Kruskal-Wallis test, respectively. The associations between the homoplastic SNPs and anti-TB resistance, treatment outcomes, AFB smear positivity and HIV status were carried out by Chi-square test. The associations are considered significant at the alpha level of 0.05 with Bonferroni-correction for multiple testing by dividing by the number of homoplastic SNPs (1,229 SNP loci) and 7 tested phenotypes, resulting in the p-value threshold of 5.80 × 10−6.
References
Brites, D. & Gagneux, S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol Rev 264, 6–24 (2015).
Hershberg, R. et al. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol 6, e311 (2008).
Smith, N. H., Hewinson, R. G., Kremer, K., Brosch, R. & Gordon, S. V. Myths and misconceptions: the origin and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol 7, 537–544 (2009).
Namouchi, A., Didelot, X., Schock, U., Gicquel, B. & Rocha, E. P. After the bottleneck: Genome-wide diversification of the Mycobacterium tuberculosis complex by mutation, recombination, and natural selection. Genome Res 22, 721–734 (2012).
Supply, P. et al. Linkage disequilibrium between minisatellite loci supports clonal evolution of Mycobacterium tuberculosis in a high tuberculosis incidence area. Mol Microbiol 47, 529–538 (2003).
Gagneux, S. et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci USA 103, 2869–2873 (2006).
Gagneux, S. & Small, P. M. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis 7, 328–337 (2007).
Romagnoli, A. et al. Clinical isolates of the modern Mycobacterium tuberculosis lineage 4 evade host defense in human macrophages through eluding IL-1beta-induced autophagy. Cell Death Dis 9, 624 (2018).
Sarkar, R., Lenders, L., Wilkinson, K. A., Wilkinson, R. J. & Nicol, M. P. Modern lineages of Mycobacterium tuberculosis exhibit lineage-specific patterns of growth and cytokine induction in human monocyte-derived macrophages. PLoS One 7, e43170 (2012).
Yuen, C. M., Kurbatova, E. V., Click, E. S., Cavanaugh, J. S. & Cegielski, J. P. Association between Mycobacterium tuberculosis complex phylogenetic lineage and acquired drug resistance. PLoS One 8, e83006 (2013).
Gonzalo-Asensio, J. et al. Evolutionary history of tuberculosis shaped by conserved mutations in the PhoPR virulence regulator. Proc Natl Acad Sci USA 111, 11491–11496 (2014).
Mikheecheva, N. E., Zaychikova, M. V., Melerzanov, A. V. & Danilenko, V. N. A Nonsynonymous SNP Catalog of Mycobacterium tuberculosis Virulence Genes and Its Use for Detecting New Potentially Virulent Sublineages. Genome Biol Evol 9, 887–899 (2017).
Rose, G. et al. Mapping of genotype-phenotype diversity among clinical isolates of mycobacterium tuberculosis by sequence-based transcriptional profiling. Genome Biol Evol 5, 1849–1862 (2013).
Doolittle, R. F. Convergent evolution: the need to be explicit. Trends Biochem Sci 19, 15–18 (1994).
Farhat, M. R. et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat Genet 45, 1183–1189 (2013).
Grandjean, L. et al. Convergent evolution and topologically disruptive polymorphisms among multidrug-resistant tuberculosis in Peru. PLoS One 12, e0189838 (2017).
Hazbon, M. H. et al. Convergent evolutionary analysis identifies significant mutations in drug resistance targets of Mycobacterium tuberculosis. Antimicrob Agents Chemother 52, 3369–3376 (2008).
Mortimer, T. D., Weber, A. M. & Pepperell, C. S. Signatures of Selection at Drug Resistance Loci in Mycobacterium tuberculosis. mSystems 3 (2018).
Nebenzahl-Guimaraes, H. et al. Transmissible Mycobacterium tuberculosis Strains Share Genetic Markers and Immune Phenotypes. Am J Respir Crit Care Med 195, 1519–1527 (2017).
Ruesen, C. et al. Large-scale genomic analysis shows association between homoplastic genetic variation in Mycobacterium tuberculosis genes and meningeal or pulmonary tuberculosis. BMC Genomics 19, 122 (2018).
Ajawatanawong, P. et al. A novel Ancestral Beijing sublineage of Mycobacterium tuberculosis suggests the transition site to Modern Beijing sublineages. Sci Rep 9, 13718 (2019).
Fishbein, S., van Wyk, N., Warren, R. M. & Sampson, S. L. Phylogeny to function: PE/PPE protein evolution and impact on Mycobacterium tuberculosis pathogenicity. Mol Microbiol 96, 901–916 (2015).
Phelan, J. E. et al. Recombination in pe/ppe genes contributes to genetic variation in Mycobacterium tuberculosis lineages. BMC Genomics 17, 151 (2016).
Nair, S. et al. The PPE18 of Mycobacterium tuberculosis interacts with TLR2 and activates IL-10 induction in macrophage. J Immunol 183, 6269–6281 (2009).
Su, H. et al. Mycobacterium tuberculosis PPE60 antigen drives Th1/Th17 responses via Toll-like receptor 2-dependent maturation of dendritic cells. J Biol Chem 293, 10287–10302 (2018).
Xu, Y. et al. PPE57 induces activation of macrophages and drives Th1-type immune responses through TLR2. J Mol Med (Berl) 93, 645–662 (2015).
Chen, J. et al. Novel recombinant RD2- and RD11-encoded Mycobacterium tuberculosis antigens are potential candidates for diagnosis of tuberculosis infections in BCG-vaccinated individuals. Microbes Infect 11, 876–885 (2009).
Lindestam Arlehamn, C. S. et al. Memory T cells in latent Mycobacterium tuberculosis infection are directed against three antigenic islands and largely contained in a CXCR3+CCR6+ Th1 subset. PLoS Pathog 9, e1003130 (2013).
Van Der Meeren, O. et al. Phase 2b Controlled Trial of M72/AS01E Vaccine to Prevent Tuberculosis. N Engl J Med 379, 1621–1634 (2018).
Hebert, A. M. et al. DNA polymorphisms in the pepA and ppe18 genes among clinical strains of Mycobacterium tuberculosis: implications for vaccine efficacy. Infect Immun 75, 5798–5805 (2007).
Homolka, S., Ubben, T. & Niemann, S. High Sequence Variability of the ppe18 Gene of Clinical Mycobacterium tuberculosis Complex Strains Potentially Impacts Effectivity of Vaccine Candidate M72/AS01E. PLoS One 11, e0152200 (2016).
Windish, H. P. et al. Protection of mice from Mycobacterium tuberculosis by ID87/GLA-SE, a novel tuberculosis subunit vaccine candidate. Vaccine 29, 7842–7848 (2011).
Brennan, M. J. The Enigmatic PE/PPE Multigene Family of Mycobacteria and Tuberculosis Vaccination. Infect Immun 85 (2017).
Gey van Pittius, N. C. et al. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol Biol 6, 95 (2006).
Akhter, Y., Ehebauer, M. T., Mukhopadhyay, S. & Hasnain, S. E. The PE/PPE multigene family codes for virulence factors and is a possible source of mycobacterial antigenic variation: perhaps more? Biochimie 94, 110–116 (2012).
Mukherjee, S., Huda, S. & Sinha Babu, S. P. Toll-like receptor polymorphism in host immune response to infectious diseases: A review. Scand J Immunol, e12771 (2019).
Ben-Ali, M., Barbouche, M. R., Bousnina, S., Chabbou, A. & Dellagi, K. Toll-like receptor 2 Arg677Trp polymorphism is associated with susceptibility to tuberculosis in Tunisian patients. Clin Diagn Lab Immunol 11, 625–626 (2004).
Hu, L., Tao, H., Tao, X., Tang, X. & Xu, C. TLR2 Arg753Gln Gene Polymorphism Associated with Tuberculosis Susceptibility: An Updated Meta-Analysis. Biomed Res Int 2019, 2628101 (2019).
Pattabiraman, G., Panchal, R. & Medvedev, A. E. The R753Q polymorphism in Toll-like receptor 2 (TLR2) attenuates innate immune responses to mycobacteria and impairs MyD88 adapter recruitment to TLR2. J Biol Chem 292, 10685–10695 (2017).
Mortier, M. C., Jongert, E., Mettens, P. & Ruelle, J. L. Sequence conservation analysis and in silico human leukocyte antigen-peptide binding predictions for the Mtb72F and M72 tuberculosis candidate vaccine antigens. BMC Immunol 16, 63 (2015).
McNamara, L. A., He, Y. & Yang, Z. Using epitope predictions to evaluate efficacy and population coverage of the Mtb72f vaccine for tuberculosis. BMC Immunol 11, 18 (2010).
Gong, W., Liang, Y. & Wu, X. The current status, challenges, and future developments of new tuberculosis vaccines. Hum Vaccin Immunother 14, 1697–1716 (2018).
Danilenk, V. N. Developing a technological platform to createinnovative TB drugs active against multidrug-resistant strains. RussAcad Sci 89, 144–150 (2019).
Newton-Foot, M. & Gey van Pittius, N. C. The complex architecture of mycobacterial promoters. Tuberculosis (Edinb) 93, 60–74 (2013).
Park, H. D. et al. Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol Microbiol 48, 833–843 (2003).
Voskuil, M. I. et al. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med 198, 705–713 (2003).
Kumar, A. et al. Mycobacterium tuberculosis DosR regulon gene Rv0079 encodes a putative, ‘dormancy associated translation inhibitor (DATIN)’. PLoS One 7, e38709 (2012).
Kumar, A. et al. Dormancy Associated Translation Inhibitor (DATIN/Rv0079) of Mycobacterium tuberculosis interacts with TLR2 and induces proinflammatory cytokine expression. Cytokine 64, 258–264 (2013).
Palittapongarnpim, P. et al. Evidence for Host-Bacterial Co-evolution via Genome Sequence Analysis of 480 Thai Mycobacterium tuberculosis Lineage 1 Isolates. Sci Rep 8, 11597 (2018).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Faksri, K. et al. Comparative whole-genome sequence analysis of Mycobacterium tuberculosis isolated from tuberculous meningitis and pulmonary tuberculosis patients. Sci Rep 8, 4910 (2018).
Bendl, J. et al. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol 10, e1003440 (2014).
Vita, R. et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res 47, D339–D343 (2019).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 33, 1870–1874 (2016).
Lew, J. M., Kapopoulou, A., Jones, L. M. & Cole, S. T. TubercuList–10 years after. Tuberculosis (Edinb) 91, 1–7 (2011).
Reese, M. G. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput Chem 26, 51–56 (2001).
Solovyev V, S. A. Automatic Annotation of Microbial Genomes and Metagenomic Sequences In: Metagenomics and its Applications in Agriculture. Nova Science Publishers. Biomedicine and Environmental Studies, 61–78 (2011).
Tantivitayakul, P., Panapruksachat, S., Billamas, P. & Palittapongarnpim, P. Variable number of tandem repeat sequences act as regulatory elements in Mycobacterium tuberculosis. Tuberculosis (Edinb) 90, 311–318 (2010).
Acknowledgements
We thank Dr. Pakorn Aiewsakun for his valuable comments. This work was funded mainly under the Science and Technology Research Partnership for Sustainable Development (SATREPS) project (Grant no.JP17jm0110010) by AMED and the Japan International Cooperation Agency (JICA) and Department of Medical Sciences, Ministry of Public Health, Thailand. PT and PP were supported by Mahidol University Research Center grant.
Author information
Authors and Affiliations
Contributions
P.T., P.P. analyzed, interpreted and draft the manuscript. T.J., W.R., N.S., A.D., W.V. analyzed the sequences. P.T. carried out experiments. S.M., K.T. guided the analysis. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tantivitayakul, P., Ruangchai, W., Juthayothin, T. et al. Homoplastic single nucleotide polymorphisms contributed to phenotypic diversity in Mycobacterium tuberculosis. Sci Rep 10, 8024 (2020). https://doi.org/10.1038/s41598-020-64895-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-64895-4
This article is cited by
-
Analysis of whiB7 in Mycobacterium tuberculosis reveals novel AT-hook deletion mutations
Scientific Reports (2023)
-
Insight into pathogenomics and phylogeography of hypervirulent and highly-lethal Mycobacterium tuberculosis strain cluster
BMC Infectious Diseases (2023)
-
Comparative genomics of drug-resistant strains of Mycobacterium tuberculosis in Ecuador
BMC Genomics (2022)
-
A genome epidemiological study of mycobacterium tuberculosis in subpopulations with high and low incidence rate in Guangxi, South China
BMC Infectious Diseases (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
