
Abstract
Mutations in whiB7 have been associated with both hypersusceptibility and resistance to various antibiotics in Mycobacterium tuberculosis (Mtb). Unlocking the secrets of antibiotic resistance in the bacterium, we examined mutations in the coding sequences of whiB7 of over 40,000 diverse Mtb isolates. Our results unveil the dominant c.191delG (Gly64delG) mutation, present in all members of the lineage L1.2.2 and its impact on WhiB7's conserved GVWGG-motif, causing conformational changes and deletion of the C-terminal AT-hook. Excitingly, we discovered six unique mutations associated with partial or total deletion of the AT-hook, specific to certain sublineages. Our findings suggest the selective pressures driving these mutations, underlining the potential of genomics to advance our understanding of Mtb's antibiotic resistance. As tuberculosis remains a global health threat, our study offers valuable insights into the diverse nature and functional consequences of whiB7 mutations, paving the way for the development of novel therapeutic interventions.
Similar content being viewed by others


Introduction
Tuberculosis (TB), is a severe bacterial disease that accounts for nearly 1.6 million human deaths and 10 million new cases worldwide annually1,2. The disease is caused by Mycobacterium tuberculosis (Mtb), consisting of nine major lineages (L1-9) with Lineages 1–4 (L1-4) distributed widely while the other lineages are more restricted to Africa3,4. Each lineage is further subdivided into several sublineages, which may have different epidemiological profiles.
The current treatment for TB involves a combination of several antibiotics; however, drug-resistant TB is a growing problem, with several hundred thousand cases reported annually5,6. One of the contributing factors to the drug resistance ability of Mtb is a transcription regulatory protein WhiB7. WhiB7 is one of the seven WhiB family of transcription regulators, specific to Actinobacteria. It interacts with the housekeeping sigma factor SigA, during transcription. WhiB7 contains a C-terminal motif called an AT-hook region (amino acid residues 80–91), which contains several positively charged amino acid residues, arginine and lysine, and binds to the AT-rich DNA regions upstream of transcription promoters of the regulated genes7,8, modulating their expression9. The SigA-WhiB7 interaction requires a triplet amino acid residue (EPW) motif adjacent to the conserved GVWGG motif, which forms a β-turn structure. The β-turn structure is located in a loop region and facilitate the protein–protein interaction10. WhiB7 is present across all non-pathogenic and pathogenic mycobacteria species including Mtb, M. smegmatis, and M. abscessus11. This protein has been linked to adaptive responses to antibiotic exposure and contributes to the intrinsic drug resistance of Mtb, playing part in multiple pathways including antibiotic export and chemical modifications of the antibiotics or their targets12. The protein has also been shown to be a redox-sensitive transcriptional regulator causing significant changes to thiol redox balance, which occurs shortly after antibiotic treatment9,13,14. The upregulation of whiB7 transcription has been shown to be in response to various antibiotics of various structural classes including aminoglycosides, macrolides as well as metabolic signals elicited by these different antibiotics9.
There have been studies which suggested that mutations in whiB7, either in the coding sequence or its promoter region, can increase drug resistance. For instance, the CRYPTIC consortium reported that, in some Mtb variants predominant in Southeast Asia, some mutations in whiB7 increased the minimum inhibitory concentration of ethionamide, suggesting that the mutations increase antibiotic-resistance to this drug2.
In contrast, there have been studies showing that disruption of whiB7 promotes the sensitivity of Mtb to some antibiotics. For example, Warit et al.13 found that a frameshift deletion mutation at the nucleotide position 191 of the gene caused the bacteria to become hypersusceptible to the macrolide clarithromycin. Others reported that the same mutation caused hypersusceptibility to clarithromycin15,16, and showed that the whiB7 LOF (loss of function) mutation could be found in the members of a specific sublineage of lineage 1. The L1.2.1 sublineage, described by Li et al.,15 as the mutant susceptible to clarithromycin, is equivalent to EAI2 defined by spoligotyping and has been recently renamed as L1.2.2,17,18 which will be used throughout this manuscript. It is predominant in Southeast Asia and accounts for 80% of TB cases in the Philippines19, and 10% in Thailand20, and has been subclassified into five sublineages including L1.2.2.2, equivalent to EAI2_NTB, mostly found in Thailand17,19,21. The other sublineages of L1.2.2 (L1.2.2.1, L1.2.2.3- L1.2.2.5) are equivalent to the spoligogroup EAI2_MNL. It should be noted that L1.2.1 is currently used to describe a newly discovered early branching sublineage of L1.2, isolated from patients in Europe, Papua, and Timor19. The finding that L1.2.2 is susceptible to clarithromycin opens an opportunity to explore the use of macrolides for the control of TB in the region. Establishing a connection between antibiotic susceptibility and lineage specificity is crucial because this prior information would help to direct the correct antibiotics in the treatment of Mtb of specific sublineages peculiar to different geographic locations.
Other studies which reported mutations in resistant genes associated with LOF and consequent susceptibility of Mtb variants that harbour the mutations include that of Walker et al.22 who characterised mutations within genes associated with resistance or susceptibility. Similarly, a recent study made an important contribution by identifying lineage-specific Mtb variants which have mutations on drug-resistant genes that confer hyper-susceptibility to varying antibiotics23.
Based on a large collection of whole genome sequences (WGS) of Mtb in the NCBI database, this study, aims to characterize the distribution of the mutations in whiB7 across all Mtb lineages, with a particular focus on those in the coding sequences. The results from this study may provide insights into the consequences of these mutations, for explaining hypersusceptibility or resistance to antibiotics.
Results
Mutations in whiB7
A multiple sequence alignment of 40,520 whiB7 coding sequences supplemented with its upstream and downstream sequences (200 bp on each side) was made to characterise a comprehensive set of whiB7 variants. The dataset comprised sequences from a diverse range of Mtb isolates from 8 lineages across all continents. A total of 1235 alleles were identified, 1007 (51.5%) of which were singleton, i.e., found only in one isolate. On the other hand, we found that there was one allele shared by 89.2% of the total sequences analysed (36,125 sequences), which coincidentally was also the allele of reference H37Rv strain. As such, we named this variant as the wt allele and used this allele as the reference allele for whiB7 mutation identification in this study. In our dataset, whiB7 of all L7 and L8 isolates had the wt allele. The proportion of the wt allele was lowest in L1 at 68.3% (3,317/4,853), followed by L5 at 85% (136/162). More than 90% of the sequences of the L2, L3, L4, and L6 isolates were of the wt alleles (Supplementary Table S1).
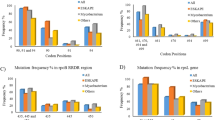
The number of mutations observed in whiB7 including the 200 bp of the flanking regions were 242. However, mutation analyses in this study focused on its coding sequence. One hundred and six mutations were identified in the coding sequence of whiB7, of which 90 were non-synonymous and 16 were synonymous. Seven nonsynonymous and one synonymous mutation were found within the AT-hook domain (Supplementary Table S2). Fifty-two of the 106 mutations were distributed across multiple isolates while 54 mutations were associated with singleton isolates. Forty of the 52 mutations, which occurred amongst multiple isolates, occurred specifically in a single sublineage (Fig. 1).

Distribution of mutations within the whiB7. Mutational landscape within the coding sequence of whiB7. A total of 106 mutations were identified, showcasing the diverse array of genetic alterations. About half of the mutations were singleton while the others occurred in more than one isolate. 40 of the latter were identified only in a single sublineage and referred to as sublineage-specific mutations. Our analysis was focused on mutations that occurred in 6 isolates or more.
Eight of the group-specific mutations were identified in a minimum of 6 isolates including c.70G > C, c.91G > A, c.125 T > C, c.173A > G, c.177G > T, c.191delG, c.200G > T, (Supplementary Table S2). The eighth mutation comprised a set of 4Mutations (c.245_246insTT, 242G > A, c.237C > A, and c.238A > C) occurring in some members of the L2.2.AA3.1. Examination of these mutations revealed that they were combined to form a single replacement of an eight-nucleotide sequence 237-CAAGCGTC-244 by a seven-nucleotide sequence 237-GCATCTT, which is referred to as the 4M mutation in this study. Two of the mutations, 191delG and 4M, caused frameshifts. Among other group-specific mutations were missense mutations c.70G > C, c.91G > A, c.125 T > C, c.173A > G, and c.200G > T which occurred among some members of sublineages L4.7, L1.1.1.11, L2.2.AA3.1, L1.1.2.217,24, and L5 respectively whereas c.177G > T was a silent mutation observed in L4.8.
The other 12 mutations including c.245_246insTT, 242G > A (parts of 4M) as well the long deletion 250_261GGACGTCCGCGC, occurred in more than one sublineage, thus indicating homoplasy. The sublineages which harboured these mutations were predominantly members of the L2 and L4 sublineages (Fig. 2).

Lineage distribution of the abundant whiB7 variants. The chord plot depicts some unique gene mutations on whiB7 and some with homoplasy. Multicoloured chords show the association of mutations at various nucleotide positions of the genes (depicted with prefix c. + nucleotide position) with the different Mtb sublineages (L1-L6). The thickness of the coloured chords corresponds to the number of mutated isolates. The chord plots were drawn using the CirclizeR package in R version 4.2.1. c.250_261del1 refers to c.250_261delGGACGTCCGCGC and c.254_261del2 refers to c.254_261delGTCCGCGC.
We confirmed 191delG13,15,16 in the whiB7 of 1098 isolates belonging to L1.2.2 (EAI2). There were only two isolates belonging to the L1.2.2.1 sublineage that did not harbour the mutation. The mutation accounted for 24.9% (1098/4395) of the whiB7 mutated population. The mutation disrupted the conserved GVWGG motif and caused a frameshift that changed the downstream amino acid sequences and the loss of the AT-hook sequence. This deletion has been reported to cause hypersusceptibility of mutants to clarithromycin13,15,16. We did not detect the mutation among L1.2.1 isolates, which are sisters of L1.2.2 isolates, reported in Europe, Papua, and Timor. This indicated that the mutation occurred after the separation of L1.2.1 and L1.2.2 but before the basal diversification of L1.2.2. Another group-specific mutation (c.173A > G) affecting 2.2% (26/1157) of the L1.1.2.2 isolates was also observed.
The 4M mutation occurred in 39 isolates among 1029 isolates belonging to L2.2.AA3.124. The frameshift caused by the mutation resulted in total amino acid residue changes from position 79 onwards (Fig. 3), including the sequence of the AT-hook region, and the delay of the translation termination to the amino acid position 167 instead of 92.

WhiB7 amino acid sequence alignment of the H37Rv sequence and various mutants with the affected AT-hook region. An alignment of some mutants highlighting the EPW conserved region, GVWGG motif, and the AT-hook region. The structure prediction was generated from Swiss-Model (https://swissmodel.expasy.org/) and subjected to quality assessment for accuracy and reliability of the predicted protein structure. The conserved EPW motif is highlighted with a red box. The β-turn GVWGG motif is shaded in grey with mutated residues shaded in blue. The ‘KRPRGRPRKDAV’ AT-hook region is highlighted with a blue box, with the core RGRP region in pink letters. (a) The wildtype sequence of WhiB7 in the H37Rv reference strain. (b) The mutation c.250_261delGGACGTCCGCGC, which occurred in 23 L4.1 isolates, 16 L2.2.AA1 isolates and 14 isolates in other sublineages. This mutation caused a deletion of 4 amino acid residues at the AT-hook region. (c) The c.192dupC mutation, which occurred in 13 L2.2.AA3.1 isolates resulting in a frameshift which led to the early termination of the protein at position 69. (d) The c.191delG mutation, which occurred in all L1.2.2 isolates, caused a frameshift and amino acid changes starting from position 64 and termination at position 77, resulting in the deletion of the entire AT-hook region. (e) The 4M mutation in 39 L2.2.AA3.1 isolates caused a frameshift and the elongation of the C terminal of the protein. (f) The other deletion at the AT-hook region, c.254_261delGTCCGCGC, which occurred in 3 L6 isolates and caused a frameshift and the elongation of the C terminal of the protein. (g) The protein structures of key variants showing the consistent presence of the EPW motif (red). The GVWGG motif (black) is disrupted in c.191delG and c.192dupC, resulting in complete deletions of the AT-hook region. The core region of the AT hook (RGRP, shown in pink) is absent in the c.250_261delGGACGTCCGCGC mutant.
The L2.2.AA3.1 isolates were primarily identified in China, India, and Vietnam, with the terminal clade exhibiting three distinct mutations as determined by phylogenetic analysis. Notably, 7.6% (78/1028) of the isolates bore one of the three mutations, i.e., 4M, c.192dupC or c.125 T > C, and were observed in four sister clades within the major terminal branch, with 48.1% (78/162) of these isolates originating from India (Fig. 4), with two 2 mutated isolates from Indonesia and two from Vietnam while the sources of the others were unknown. Most 4M and c.192dupC mutated isolates belonged to two terminal sister clades in the terminal branch while most c.125 T > C mutated isolates (24/26) formed another separate clade. All isolates of this clade were from India, as illustrated in Fig. 4. The c.192dupC mutation caused a frameshift and consequent amino acid residue changes starting from amino acid residue 65 as well as an early translation termination resulting in the truncation of the protein length from 92 to 68 amino acids and the loss of the AT-hook region.

A phylogenetic tree of L2.2.AA3.1, showing the isolates with mutations: 4M (orange), c.192dupC (green), and c125T > C (purple). (a) Phylogenetic tree of all 1026 L2.2.AA3.1 isolates rooted with an L2.1 isolate. (b) Phylogenetic tree of the terminal 187 isolates of L2.2.AA3.1 shows isolates with the three mutations. The numbers of the isolates with mutations 4M, c.192dupC, and c.125 T > C were 39, 13, and 26 respectively. The tree was generated with IQ-TREE v2.1.3 and visualized with FigTree v1.4.4 and GGtree package in R Studio v4.2.1. The list of all the isolates in the dataset is in Supplementary Table S3.
The c.192dupC and c.125 T > C mutations were observed to be independently occurring in L4.4.1.2 and L4.1.2.1. Additionally, isolates belonging to L2.2.M1.1 and L2.2.M4.5 harboured a subset of the 4M mutation, c.245_246insTT, and c.242G > A (Supplementary Table S2).
Mutation-induced changes to the AT-hook region of whiB7
Among relatively common mutations in whiB7, several lead to frameshifts and changes or loss of the amino acid sequences at the C-terminus, including the major mutation identified in sublineages L1.2.2 (c.191delG) and L2.2.AA3.1 (4M mutation and c.192dupC). The frameshifts generally occurred at positions before the AT-hook region of the gene, located at amino acid position 80 to 91 on its C-terminus25.
In addition, the mutations, c.250_261delGGACGTCCGCGC and c.254_261delGTCCGCGC directly affected the AT-hook region. The c.250_261delGGACGTCCGCGC mutation, occurred in 67 isolates belonging to 16 different sublineages, predominantly among L4.1 and L2.2.AA1, led to the loss of four amino acid residues from the AT-hook region, whereas the other mutation, 254_261delGTCCGCGC occurred only in some L6 isolates and caused the changes of the amino acid sequences, RGRP, from position 85 and elongation of the protein by 28 amino acids.
Discussion
Mtb is a global health threat with multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains posing a particular challenge for treatment26. Findings from many studies have established the associations between mutations in drug target genes in Mtb and drug susceptibility27. In this study, we identified mutations in the transcription regulator whiB7 in 40,520 isolates of Mtb through the analysis of their WGS.
We confirm the presence of 191delG in a common albeit geographically localized sublineage L1.2.2 (EAI2), which are known to be associated with clarithromycin resistance13,15,16. The consequent sensitivity to clarithromycin may provide a better opportunity to treat MDR-TB patients caused by the sublineage with the antibiotic. It is worth noting that the prescription rate of clarithromycin for respiratory infections in Thailand is generally low, in comparison to other antibiotics28, minimizing the unintentional exposure of Mtb to clarithromycin. Nevertheless, its use in multidrug regimens for treating MDR-TB patients caused by L1.2.2 remains to be investigated. In Thailand, L1 (the Indo-Oceanic Lineage) and L2 (the East-Asian lineage) are equally predominant while L1.2.2 accounted for about a quarter of L121. The rate of MDR cases was higher among L2 and the rate among L1.2.2 was less than 1% in northern Thailand20. Nevertheless, the burden and the MDR rate of L1.2.2 in the Philippines were much higher19. Globally the annual new cases of TB caused by the strains belonging to L1.2.2 was estimated to be around 600,00017, making exploring the use of clarithromycin worthwhile.
Evaluating the consequences of 191delG mutation revealed that it caused a dramatic change in the downstream amino acid sequence starting from the β-turn GVWGG motif, conserved among WhiB-like family of proteins10, resulting in the structural change and the complete loss of the AT-hook structure. The AT-hook structure is common among eukaryotic nuclear proteins but uncommon in bacteria. It is shown to enhance the binding of transcriptional regulatory complexes, comprising RNA polymerase-SigA holoenzyme, global regulators CarD and RbpA, and WhiB7, to the AT-rich DNA sequence motifs upstream of WhiB7-regulated promoters29. The role of these AT hook motifs, as well as the GVWGG-motif in conferring antibiotic resistance in vivo, has been established through targeted mutagenesis30. The study showed that different whiB7 mutants without the GVWGG-motif and AT hook region did not restore the wt resistance typically exhibited by intact WhiB7 thus revealing the importance of the AT-hook DNA binding domain as a requirement for optimal WhiB7 function. Therefore, these described consequences of 191delG suggest a molecular mechanism on how the mutation, contributing to the hypersusceptibility of L1.2.2 to clarithromycin naturally occurs.
The fact that almost all isolates of this genotype harbor the same mutation suggests that this mutation occurred in the early ancestor of L1.2.2. However, it is not clear whether the mutation has provided any selective advantages or contributed to the success or widespread of the sublineage, as increased sensitivity to an antibiotic by itself would not increase survival. However, WhiB7 is related to the expression of about 100 genes and transcriptional re-wiring of stress-response pathways can enhance tolerance to antibiotics31 or probably other environmental stress. Moreover, the occurrence of collateral sensitivity, where resistance to certain antibiotics may influence susceptibility to other antibiotics, has also been described31,32,33, suggesting that a reverse phenomenon may be possible.
The frameshift mutations with LOF effects, are not unique to L1.2.2 but also affect small proportions of other sublineages. Further observations have unveiled several mutations that interfere with the functionality of the AT-hook of WhiB7, either partially or completely. Our research has identified a few mutations in some L2.2.AA3.1 isolates that triggered frameshifts ahead of the AT-hook domain, such as the 4M mutation and c192dupC. Both mutations cause frameshifts and consequently the total loss of the AT-hook. c192dupC also disrupts the GVWGG motif. Unfortunately, the sensitivity to clarithromycin of the mutants was unknown.
Phylogenetic analysis revealed that the basal isolates of L2.2.AA3.1 were primarily identified in China24, whereas the mutants located in the terminal clade, which contained the 4M mutation, were predominantly found in India. This hints that the origin of the mutation occured after the sublineage was transferred from China.
There were also other mutations that affect the AT-hook directly. The c.250_261delGGACGTCCGCGC mutation caused the deletion of the core AT-hook sequence motif, RGRP, essential for tethering the protein to the DNA31. Interestingly the mutation was identified in 17 sublineages. The homoplasy suggested a positive selection of the RGRP loss in some situations. A homoplastic signal of selection pressure has been described for many drug resistance mutations34,35. c.254_261delGTCCGCGC, identified only in a few L6 isolates, also affects the RGRP motif and causes downstream frameshift, leaving only the first few amino acid residues of the AT-hook intact. Interestingly a recent study also revealed the presence of another mutation, R85C, affecting the RGRP motif of 25 MTB isolates in India36.
The mutations in whiB7 have various physiologic effects. It induces antibiotic resistance in Mtb against several antibiotics, including kanamycin and streptomycin, the second-line drugs for TB29. An earlier study by the Cryptic consortium2 used oligopeptide probes to link elevated minimum inhibitory concentration (MIC) of ethionamide to several substitutions within the AT-hook region. WhiB7 also plays an important role in cellular redox homeostasis. In Streptomyces, SigR, a sigma factor that is involved in the cellular response to oxidative stress, is controlled by WblC, an ortholog of Mtb WhiB7. In Mtb, the homologs of SigR, SigE and SigH, are also likely to be controlled by WhiB7, which aids the response to oxidative stress by activating genes that help the bacteria survive and restore their redox balance10.
whiB7 is regulated by VapC21, the toxin component of a toxin-antitoxin system VapBC2128. The VapBC toxin-antitoxin systems are differentially expressed in stress conditions, which Mtb may encounter during infection. Recently the SenX-RegX3 two-component system was also found to regulate the expression of whiB7 in response to phosphate starvation, acid stress, and hypoxia37. WhiB7 itself is autoregulated and is involved in regulating eis (enhanced intracellular survival, an N-acetyl transferase), tap (a multidrug transporter) and erm (ribosomal methyltransferase)12. The loss of transcription of erm by the loss of AT hook in the c191delG mutant is likely to contribute to the unusual sensitivity of L1.2.2. to clarithromycin38. It is possible that other mutations that cause the loss of AT hook region also lead to the sensitivity to clarithromycin.
While our study is limited by the lack of targeted gene knock-out experiments to confirm the causal relationship between LOF in the whiB7, and clarithromycin sensitivity, our results provide a starting point for further research into the role of this gene in TB drug resistance. A better understanding of the mechanism of WhiB7 and its AT hook region will be critical for the development of novel drugs targeting WhiB7. The availability of several natural mutations in WhiB7 will also provide an opportunity to study the roles of the transcription regulator in the physiology, survival, and pathogenesis of Mtb. Nevertheless, the scope of biochemical roles of WhiB7 is still unclear and the full implications of the mutations remain to be discovered.
Overall, the mutations in whiB7, which affect its AT-hook region, may cause both hypersusceptibility and resistance to different TB antibiotics. c.191delG causes the loss of both the β-turn GVWGG motif and the AT-hook region. There are other natural mutations that may affect only the AT-hook core motif (RGRP), the entire AT-hook region, or additionally the β-turn GVWGG motif. Further investigations are required to discern the exact effects of these mutations and their implications for Mtb antibiotic resistance.
Methods
Sample acquisition and sequencing
We downloaded 40,520 raw whole-genome sequencing (WGS) short read data from the NCBI Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra/) and the European Nucleotide Archive (ENA), which were mostly generated using the Illumina platform (https://www.ebi.ac.uk/ena). The samples were obtained from diverse geographic regions worldwide.
Data preprocessing and variant calling
We trimmed the raw sequencing reads to remove low-quality reads using Trimmomatic v0.3939 with the following parameters: sliding window trimming with a window size of 4 and a read quality threshold of 30. The trimmed reads were then aligned to the H37Rv reference genome (NC_000962.3) using BWA-MEM v0.7.1740. Picard's MarkDuplicates was used to identify and remove duplicate reads (https://github.com/broadinstitute/picard). Per-sample variant calling was performed using GATK HaplotypeCaller v3.841 with a haploid model, excluding bases with a quality score below 20. We used GATK GenotypeGVCFs to generate a single variant call format (VCF) file containing the variants in the whiB7 gene20.
SNV annotation and phylogenetic reconstruction
We annotated the SNVs using SnpEff v4.3t42 with the H37Rv reference genome. A VCF file was utilized to present genomic variants, which encompassed variant information from all samples and stored the variant data in a singular file. Multiple sequence alignment was performed using Aliview v1.17.143. We reconstructed the phylogenetic tree for the lineages using IQ-TREE v2.1.344 with ultrafast bootstrap supports from 1000 replications. The best-fit nucleotide substitution model was determined using ModelFinder v1.7.145. We used lineage 2.1 as an outgroup for rooting the tree of L2.2.AA3.1 isolates and tree visualization was performed using FigTree v1.4.4 and GGtree package in R Studio v4.2.1.
Protein structure homology modeling
We performed protein structure homology modeling of the WhiB7 alleles46,47. The amino acid sequence of the protein which contained the mutations were used to generate the model structure using ProMod348, a comparative modelling engine based on OpenStructure49. Structure prediction was done with the SWISS-MODEL platform (https://swissmodel.expasy.org/) which incorporates known structures and algorithms to predict the protein's structure based on its sequence and subsequently generating a model of the protein. The generated model was then subjected to quality assessment and validation using scores from QMEANDisCo50 and QMEAN Z51, ensuring the reliability of the predicted protein structure.
Mutation distribution among lineages
We constructed a chord plot to determine the association of the mutations and corresponding lineages using the Circlize package in RStudio v4.2.1.
Data availability
All data pertaining to the manuscript have been provided in the form of figures and tables. The sequence data are available in the NCBI Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra/) and the European Nucleotide Archive (ENA) (https://www.ebi.ac.uk/ena). Supporting information is available as Supplementary Tables S1-S3.
References
WHO. https://www.who.int/news-room/fact-sheets/detail/tuberculosis. (2022).
The CRyPTIC Consortium. Genome-wide association studies of global Mycobacterium tuberculosis resistance to 13 antimicrobials in 10,228 genomes identify new resistance mechanisms. PLoS Biol. 20, e3001755 (2022).
O’Neill, M. B. et al. Lineage specific histories of Mycobacterium tuberculosis dispersal in Africa and Eurasia. Mol. Ecol. 28, 3241–3256 (2019).
Freschi, L. et al. Population structure, biogeography and transmissibility of Mycobacterium tuberculosis. Nat. Commun. 12, 6099 (2021).
Lange, C. et al. anagement of patients with multidrug-resistant/extensively drug-resistant tuberculosis in Europe: A TBNET consensus statement. Eur. Respir. J. 44, 23–63 (2014M).
Dartois, V. A. & Rubin, E. J. Anti-tuberculosis treatment strategies and drug development: Challenges and priorities. Nat. Rev. Microbiol. 20, 685–701 (2022).
Burian, J. et al. The mycobacterial antibiotic resistance determinant WhiB7 acts as a transcriptional activator by binding the primary sigma factor SigA (RpoV). Nucleic Acids Res. 41, 10062–10076 (2013).
Wan, T. et al. Structural insights into the functional divergence of WhiB-like proteins in Mycobacterium tuberculosis. Mol. Cell 81, 2887-2900.e5 (2021).
Burian, J. et al. The mycobacterial transcriptional regulator whiB7 gene links redox homeostasis and intrinsic antibiotic resistance. J. Biol. Chem. 287, 299–310 (2012).
Bush, M. J. The actinobacterial WhiB-like (Wbl) family of transcription factors: The Actinobacterial WhiB-like (Wbl) family of transcription factors. Mol. Microbiol. 110, 663–676 (2018).
Cushman, J. et al. Increased whiB7 expression and antibiotic resistance in Mycobacterium chelonae carrying two prophages. BMC Microbiol. 21, 176 (2021).
Burian, J., Ramón-García, S., Howes, C. G. & Thompson, C. J. WhiB7, a transcriptional activator that coordinates physiology with intrinsic drug resistance in Mycobacterium tuberculosis. Expert Rev. Anti Infect. Ther. 10, 1037–1047 (2012).
Warit, S. et al. Genetic characterisation of a whiB7 mutant of a Mycobacterium tuberculosis clinical strain. J. Glob. Antimicrob. Resist. 3, 262–266 (2015).
Burian, J. & Thompson, C. J. Regulatory genes coordinating antibiotic-induced changes in promoter activity and early transcriptional termination of the mycobacterial intrinsic resistance gene whiB7: Regulatory genes that alter whiB7 transcription. Mol. Microbiol. 107, 402–415 (2018).
Li, S. et al. CRISPRi chemical genetics and comparative genomics identify genes mediating drug potency in Mycobacterium tuberculosis. Nat. Microbiol. 7, 766–779 (2022).
Shur, K. V. et al. The intrinsic antibiotic resistance to β-lactams, macrolides, and fluoroquinolones of mycobacteria is mediated by the whiB7 and tap genes. Russ. J. Genet. 53, 1006–1015 (2017).
Netikul, T. et al. Whole-genome single nucleotide variant phylogenetic analysis of Mycobacterium tuberculosis Lineage 1 in endemic regions of Asia and Africa. Sci. Rep. 12, 1565 (2022).
Napier, G. et al. Robust barcoding and identification of Mycobacterium tuberculosis lineages for epidemiological and clinical studies. Genome Med. 12, 114 (2020).
Phelan, J. E. et al. Mycobacterium tuberculosis whole genome sequencing provides insights into the Manila strain and drug-resistance mutations in the Philippines. Sci. Rep. 9, 9305 (2019).
Ajawatanawong, P. et al. A novel Ancestral Beijing sublineage of Mycobacterium tuberculosis suggests the transition site to Modern Beijing sublineages. Sci. Rep. 9, 13718 (2019).
Palittapongarnpim, P. et al. Evidence for host-bacterial co-evolution via genome sequence analysis of 480 Thai Mycobacterium tuberculosis lineage 1 isolates. Sci. Rep. 8, 11597 (2018).
Walker, T. M. et al. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: A retrospective cohort study. Lancet Infect. Dis. 15, 1193–1202 (2015).
Merker, M. et al. Phylogenetically informative mutations in genes implicated in antibiotic resistance in Mycobacterium tuberculosis complex. Genome Med. 12, 27 (2020).
Thawornwattana, Y. et al. Revised nomenclature and SNP barcode for Mycobacterium tuberculosis lineage 2. Microb. Genomics https://doi.org/10.1099/mgen.0.000697 (2021).
Lee, J.-H. et al. The WblC/WhiB7 transcription factor controls intrinsic resistance to translation-targeting antibiotics by altering ribosome composition. MBio 11, e00625-e1620 (2020).
Reeves, A. Z. et al. Aminoglycoside cross-resistance in mycobacterium tuberculosis due to mutations in the 5′ untranslated region of whiB7. Antimicrob. Agents Chemother. 57, 1857–1865 (2013).
Liu, Q. et al. Drug resistance gene mutations and treatment outcomes in MDR-TB: A prospective study in Eastern China. PLoS Negl. Trop. Dis. 15, e0009068 (2021).
Sharma, A. et al. VapC21 toxin contributes to drug-tolerance and interacts with non-cognate VapB32 antitoxin in Mycobacterium tuberculosis. Front. Microbiol. 11, 2037 (2020).
Lilic, M., Darst, S. A. & Campbell, E. A. Structural basis of transcriptional activation by the Mycobacterium tuberculosis intrinsic antibiotic-resistance transcription factor WhiB7. Mol. Cell 81, 2875-2886.e5 (2021).
Ramón-García, S. et al. WhiB7, an Fe-S-dependent transcription factor that activates species-specific repertoires of drug resistance determinants in actinobacteria. J. Biol. Chem. 288, 34514–34528 (2013).
Pál, C., Papp, B. & Lázár, V. Collateral sensitivity of antibiotic-resistant microbes. Trends Microbiol. 23, 401–407 (2015).
Baym, M., Stone, L. K. & Kishony, R. Multidrug evolutionary strategies to reverse antibiotic resistance. Science 351, aad3292 (2016).
Maeda, T., Kawada, M., Sakata, N., Kotani, H. & Furusawa, C. Laboratory evolution of Mycobacterium on agar plates for analysis of resistance acquisition and drug sensitivity profiles. Sci. Rep. 11, 15136 (2021).
Farhat, M. R. et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat. Genet. 45, 1183–1189 (2013).
Tantivitayakul, P. et al. Homoplastic single nucleotide polymorphisms contributed to phenotypic diversity in Mycobacterium tuberculosis. Sci. Rep. 10, 8024 (2020).
Rana, V. et al. Molecular epidemiology and polymorphism analysis in drug-resistant genes in M. tuberculosis clinical isolates from western and Northern India. Infect. Drug Resist. 15, 1717–1732 (2022).
Mahatha, A. C. et al. A systems approach to decipher a role of transcription factor RegX3 in the adaptation of Mycobacterium tuberculosis to hypoxic stress. Microbiology https://doi.org/10.1099/mic.0.001229 (2022).
Nash, K. A., Brown-Elliott, B. A. & Wallace, R. J. A novel gene, erm (41), confers inducible macrolide resistance to clinical isolates of Mycobacterium abscessus but is absent from Mycobacterium chelonae. Antimicrob. Agents Chemother. 53, 1367–1376 (2009).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at http://arxiv.org/abs/1303.3997 (2013).
Van der Auwera, G. A. et al. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinforma. https://doi.org/10.1002/0471250953.bi1110s43 (2013).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w 1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278 (2014).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Waterhouse, A. et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Guex, N., Peitsch, M. C. & Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 30, S162–S173 (2009).
Studer, G. et al. ProMod3—A versatile homology modelling toolbox. PLoS Comput. Biol. 17, e1008667 (2021).
Biasini, M. et al. OpenStructure : An integrated software framework for computational structural biology. Acta Crystallogr. D Biol. Crystallogr. 69, 701–709 (2013).
Studer, G. et al. QMEANDisCo—Distance constraints applied on model quality estimation. Bioinformatics 36, 1765–1771 (2020).
Benkert, P., Biasini, M. & Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343–350 (2011).
Acknowledgements
Olabisi Flora Davies-Bolorunduro is supported by the International Postdoctoral Fellowship at Mahidol University. The work is funded by a Multidisciplinary Research grant at Mahidol University, and the National Science and Technology Development Agency Emerging Infectious Diseases Genomics program. We thank members of the Pornchai Matangkasombut Center for Microbial Genomics (CenMiG) team, Thavin Bodharamik for useful discussions, and Thanakron Noppanamas for technical assistance with data analysis. We also appreciate Aye Nyein Phyu (Department of Epidemiology, Prince of Songkhla University) for her invaluable suggestions and some technical assistance with data analysis.
Author information
Authors and Affiliations
Contributions
O.F.D.-B. and P.P. conceived the study and wrote the manuscript, B.J. conducted sample acquisition, sequence analyses, S.N.P. analyses and phylogenetic reconstruction, W.R. annotated the SNVs, W.P. and O.F.D.-B. performed sequence data analysis, O.F.D.-B. performed protein structure modeling, visualization of lineage distribution of the abundant variants, P.A. provided critical input to finalize the draft and PP supervised the study. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Davies-Bolorunduro, O.F., Jaemsai, B., Ruangchai, W. et al. Analysis of whiB7 in Mycobacterium tuberculosis reveals novel AT-hook deletion mutations. Sci Rep 13, 13324 (2023). https://doi.org/10.1038/s41598-023-40152-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-40152-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
