
Abstract
Autosomal dominant cerebral cavernous malformations (CCM) are leaky vascular lesions that can cause epileptic seizures and stroke-like symptoms. Germline mutations in either CCM1, CCM2 or CCM3 are found in the majority of patients with multiple CCMs or a positive family history. Recently, the first copy number neutral inversion in CCM2 has been identified by whole genome sequencing in an apparently mutation-negative CCM family. We here asked the question whether further structural genomic rearrangements can be detected within NGS gene panel data of unsolved CCM cases. Hybrid capture NGS data of eight index patients without a pathogenic single nucleotide, indel or copy number variant were analyzed using two bioinformatics pipelines. In a 58-year-old male with multiple CCMs in his brain and spinal cord, we identified a 294 kb insertion within the coding sequence of CCM2. Fine mapping of the breakpoints, molecular cytogenetic studies, and multiplex ligation-dependent probe amplification verified that the structural variation was an inverted unbalanced insertion that originated from 1p12-p11.2. As this rearrangement disrupts exon 6 of CCM2 on 7p13, it was classified as pathogenic. Our study demonstrates that efforts to detect structural variations in known disease genes increase the diagnostic sensitivity of genetic analyses for well-defined Mendelian disorders.
Similar content being viewed by others

Introduction
Cerebral cavernous malformations (CCM; MIM: 116860, 603284, 603285) are irregular clusters of enlarged and thin-walled vessels that can present as sporadic or autosomal dominant cerebrovascular disease. Aside from epileptic seizures and headaches, CCM patients may present with stroke-like symptoms due to chronic or acute bleeding events1. Pathogenic germline variants have been identified in CCM1 (also known as KRIT1)2,3, CCM24,5, and CCM3 (PDCD10)6. The mutational spectrum primarily includes nonsense, frameshift, splice, and copy number variants (CNVs). 282 unique CCM1, 84 CCM2 and 75 CCM3 variants are classified as disease-causing in the Human Gene Mutation Database (HGMD Professional 2019.3)7. Depending on the inclusion criteria for genetic analyses, mutation detection rates of 87 to 98% have been reported for familial CCM cases and up to 60% for sporadic ones8,9,10,11. The mutation detection rate in the latter group could even be higher if patients with associated developmental venous anomalies or a history of radiotherapy to the brain were excluded1,12. Although pathogenic variants in a yet unknown CCM4 candidate gene have been discussed for unresolved cases13, we recently were able to identify the first copy number neutral inversion in CCM2 in an apparently mutation-negative CCM family by whole genome sequencing (WGS)14.
Disease-causing structural variants (SVs) may explain a part of the “missing heritability” in rare diseases15. SVs are defined as structural and quantitative chromosomal rearrangements that compromise cytogenetically visible and submicroscopic variants16,17. They contribute to phenotypic variation but can also cause human disease17. In fact, rare SVs are even more likely to be deleterious than rare single nucleotide variants (SNVs)18,19. Deletions, duplications, insertions, translocations, and inversions may directly disrupt the organization of a disease gene or affect its transcriptional regulation by positional effects16,17. Using a multi-platform WGS approach, Chaisson and colleagues have demonstrated in 2019 that more than 27,000 SVs (≥50 bp) can be found per human genome20. In contrast, an individual human genome harbors approximately 4,000,000 to 5,000,000 SNVs and up to 800,000 indels, which are defined as insertions or deletions with a length of up to 49 bp18. However, the identification of SVs remains much more challenging than SNV or indel calling in a diagnostic setting. While deletions and duplications of exonic sequences can be reliably detected with qPCR, multiplex ligation-dependent probe amplification or NGS-based CNV detection algorithms, copy number neutral SVs or deletions and duplications in non-coding regions of known disease-associated genes may escape targeted genetic approaches.
In this study, we have analyzed the hybrid capture NGS gene panel data of eight genetically unresolved CCM index patients for the presence of SVs in CCM1, CCM2 or CCM3. An interchromosomal insertion which led to an interruption of exon 6 of the CCM2 gene was identified in a sporadic CCM patient.
Results
Clinical findings
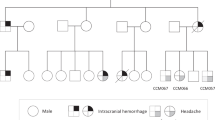
The male index patient III:1 (pedigree 1, Fig. 1a) was referred to genetic counselling at the age of 58 years because of multiple symptomatic CCMs. Magnetic resonance imaging (MRI) documented at least nine cavernous lesions with surrounding hemosiderin deposits in his brain and spinal cord (Fig. 1b). One brainstem cavernoma and three CCMs in his thoracic, cervical, and lumbar spine had already been resected because of acute bleeding events at the age of 44, 50, and 58, respectively (Fig. 1c, Supplementary Video S1). The index patient also reported an atrial septal defect and a traumatic L1 vertebral body fracture at the age of 55 years which had been treated with balloon kyphoplasty. On physical examination, he presented a partial loss of vibration and fine touch sensation below the level of T10 and impaired sensation of temperature on the contralateral side. In addition, he had an ataxic gait with right foot drop. The index patient’s father (II:2) died of gastric cancer when he was 72 years old. Cavernous lesions or neurological symptoms that would be suggestive for CCM had not been documented for him. The mother of the index patient (II:3) passed away at the age of 90 years with no signs of CCM either. Both children (IV:1 and IV:2) are healthy, and there were no other relatives who had CCMs or reported epileptic seizures, stroke-like symptoms or chronic headaches.

Multiple cerebral and spinal cavernomas in a sporadic CCM patient. (a) Pedigree of the CCM index case from pedigree 1 (III:1, arrow). (b) Repetitive magnetic resonance imaging (MRI) of the index patient’s brain and spinal cord showing progression of CCM disease with two cavernous malformations in the pons (I, II), intramedullary lesions in the cervical (III, IV) and thoracic spinal cord (V) as well as a CCM in the cauda equina (VI). MRI images were acquired between 2005 and 2019. White arrowheads indicate CCMs. (c) Intraoperative photograph of the cauda equina cavernous malformation.
Identification of an interchromosomal insertion
Gene panel sequencing of CCM1, CCM2, and CCM3 for index patient III:1 (Fig. 1) and routine bioinformatics analyses identified no pathogenic SNV, indel or CNV. However, a high number of split reads in CCM2 was noticed in NGS gene panel data of this proband. These deviant reads could be grouped into two clusters based on their mapping. Reads of the first group mapped with one part to the 5' half of exon 6 of CCM2, which is located on 7p13, and with the other part to 1p11.2 or 1q21.1. Due to known sequence homologies, aberrant mapping on chromosome 1 could not be further specified. Split reads of the second group partially mapped to the 3' half of CCM2 exon 6 but also to 1p12. Notably, detection of deviant reads required the SeqNext module of the Sequence Pilot tool for mapping and alignment. Using the mapping and alignment information of the MiSeq Reporter Software resulted in significantly reduced read depth at positions [hg19] chr7:45,108,098-45,108,101. However, split reads were filtered out by SeqNext and no variant was called since there were no reads that covered a heterozygous deletion of these four nucleotides (Fig. 2a).

Identification of an unbalanced interchromosomal insertion in CCM2. (a) Read alignment of the hybrid capture NGS data of III:1. Shown is a part of exon 6 of the CCM2 gene. The coverage plot indicated significantly reduced read depths at positions [hg19] chr7:45,108,098-45,108,101 but no reads were found that covered a deletion of 4 bp. (b) Schematic depiction of the identified interchromosomal insertion. Material from 1p12-p11.2 was found as an inverted insertion in exon 6 of the CCM2 gene on 7p13. (c) Verification of the chromosomal rearrangement [46,XY.ish ins(7;1)(p13;1p11)(RP11-425C16+,RP11-111G2 +)] by fluorescence in situ hybridization. There is a known crosshybridization of the applied probe RP11–425C16 in 1q21.1, which does not need to be considered further taking into account all other data presented here. (d) MLPA analysis of the centromere region of chromosome 1 indicated three copies of the NOTCH2 gene (1p12) which is part of the inserted fragment.
Based on the NGS data of index patient III:1, we suspected an interchromosomal insertion from 1p12-p11.2 into the coding region of CCM2 on 7p13 (Fig. 2b). This rearrangement was finally made visible by fluorescence in situ hybridization (FISH) on metaphase chromosomes from cultured blood lymphocytes of the index patient (Fig. 2c). The inserted fragment covers a genomic region of 294 kb that contains the complete NOTCH2 (MIM: 600275), ADAM30 (604779), and NBPF7 (613997) genes as well as parts of REG4 (609846). MLPA analysis with a centromere kit further indicated the presence of three copies of NOTCH2 which is the only gene in the inserted fragment with an associated phenotype (Fig. 2d). Loss-of-function mutations and pathogenic missense variants in this gene have been reported in patients with Alagille (MIM: 610205) and Hajdu-Cheney syndrome (102500). However, the index patient did not meet the clinical diagnostic criteria of either syndrome.
Fine mapping of the breakpoints
We next amplified the breakpoints of the interchromosomal insertion for its precise molecular characterization. Sanger sequencing allowed us to fine map the breakpoints to [hg19] chr1:120,347,265, chr1:120,641,440, and chr7:45,108,098–45,108,101, respectively. It also verified that the fragment of 1p12-p11.2 was inserted in an inverted orientation on 7p13 and flanked by a small insertion and a deletion-insertion variant in which the four base pairs with reduced sequencing depths were replaced by six nucleotides of unknown origin (Fig. 3). While the breakpoint on 1p11.2 locates within a long interspersed nuclear element (LINE), no repeat elements were found in close proximity to the breakpoints on 1p12 or 7p13. As the heterozygous SV interrupts exon 6 of the CCM2 gene, it was classified as pathogenic for CCM. In summary, the results of our molecular, cytogenetic, and molecular cytogenetic studies demonstrated that index patient III:1 is a heterozygous carrier of a submicroscopic, unbalanced, interchromosomal, and inverted insertion.

Fine mapping of the breakpoints by Sanger sequencing. Scheme of the normal and inverted insertion alleles (middle panel). PCR and sequencing primers (chr1: chr1-F1 + R1 and chr1-F2 + R2; chr7: CCM2-F + R) are depicted as blue, red, and green arrows, respectively. PCR products and chromatograms of the normal allele on chr1 (chr1-F1/R1 and chr1-F2/R2) are depicted in the upper panel. PCR products and chromatograms of the inverted insertion on chr7 (CCM2-F/chr1-F2 and chr1-R1/CCM2-R) are depicted in the lower panel. Fine mapping also revealed an additional small insertion and an indel variant at the breakpoints. C = healthy control; P = index patient; – = negative control; Ex = exon.
Screening for SVs in mutation-negative CCM patients
We finally wanted to know if SVs can also be identified in the hybrid capture NGS data of seven additional CCM patients without any pathogenic SNV, indel or CNV in CCM1, CCM2 or CCM3 that had been analyzed between 2017 and 2019 (Supplementary Table S1). Therefore, we chose the Agilent SureCall tool, which allowed us to search for translocation events based on the presence of split reads in their NGS data. The index patient III:1 from family 1 was used as a positive control. The tool precisely re-identified his SV but did not detect any further chromosomal rearrangements in the other CCM cases. Furthermore, no split reads were found by visual inspection of their NGS data.
Discussion
In this study, we have identified the first interchromosomal insertion, sometimes also referred to as interchromosomal insertional translocation (IT), in a CCM patient. Notably, current genetic analysis cannot clarify the molecular basis of CCM disease in 2 to 13% of familial cases and up to 40% of sporadic CCM patients1. Besides phenocopies, somatic mosaicism and pathogenic variants in an unknown CCM4 gene or non-coding regions of CCM1, CCM2 or CCM3 have been discussed as possible explanations for these patients21,22,23. However, the identification of an interchromosomal insertion in one of eight unsolved CCM cases and the copy number neutral inversion in CCM2, that we have reported recently14, demonstrate that SVs also need to be considered. It is remarkable that both rearrangements identified so far have been detected in CCM2 which is the largest of the three CCM genes. However, future studies will have to show whether CCM2 is more susceptible to the occurrence of SVs or whether further genomic rearrangements can be detected in CCM1 and CCM3 with more sensitive techniques.
ITs and other SVs have a higher prevalence than previously thought20,24,25. FISH confirmation and parental follow-up studies of CNVs that had been identified by clinical array comparative genomic hybridization (aCGH) analysis revealed that ITs can be found in approximately 1 of 500 patients referred to aCGH analysis24. This observation suggests that their prevalence might also be underestimated in patients with CCM or other inherited disorders for whom aCGH is not part of standard genetic analyses. Sophisticated bioinformatics algorithms have been developed to identify SVs from short-read NGS data based on inconsistent paired-end mapping, the presence of split reads or changes in read depth26,27. While CNVs can be efficiently detected with NGS-based approaches, inversions and ITs that do not cause gain or loss in the number of CCM1, CCM2 or CCM3 alleles might be regularly missed with targeted gene panel sequencing. Furthermore, the analytical sensitivity of the available bioinformatics tools is still incomplete. Notably, only one of the two tools that were used for mapping and alignment in our current study allowed us to detect aberrant reads in the NGS data analysis for the index patient from pedigree 1.
The search for chromosomal rearrangements is becoming more and more important in genetic research projects that try to solve undiagnosed cases with rare diseases. While the detection of ITs within the coding region of a gene seems to be relatively straightforward with special bioinformatics tools and filter criteria, insertions into intronic, promotor or cis-regulatory regions that can also impair gene regulation16 can hardly ever be detected with PCR- or hybrid capture-based target enrichment strategies and short-read sequencing. Recently, short-read WGS and a new bioinformatics pipeline revealed disease-causing SVs in 16 out of 477 patients of the Undiagnosed Diseases Network (UDN)28. However, third-generation sequencing technologies will probably further improve the diagnostic sensitivity for the identification of chromosomal rearrangements as the probability of spanning the whole SV and the mappability in repetitive regions are higher for long reads29,30. In a cohort of 1,324 undiagnosed probands with rare diseases from the NIHR BioResource research study, three cases with a complex pathogenic SV were identified by short-read WGS and a duplication-inversion-duplication event of unknown clinical significance was resolved in another proband by long-read sequencing31. Whole genome long-read sequencing also identified a heterozygous ~2.2 kb deletion in PRKAR1A which is a known disease gene for autosomal dominant Carney complex in a patient with multiple tumours for whom initial targeted PRKAR1A sequencing and short-read WGS had not revealed any pathogenic variant32. Several other examples illustrate that long-read sequencing enables effective SV calling and could help to solve undiagnosed cases33. Therefore, long-read WGS is a promising next step for the yet unsolved CCM cases of our cohort. These have already been checked for CNVs in CCM1, CCM2, and CCM3 but third-generation sequencing technologies would allow the detection of CNVs and copy number neutral SVs in a genome-wide approach. Nevertheless, the combination of multiple sequencing technologies might be still necessary to achieve sufficient insertion and inversion detection sensitivities20.
The identification of a pathogenic SNV, indel, CNV or SV is essential for genetic counselling of CCM families and always raises the question whether it is an inherited or a de novo variant. As both parents of the index patient from pedigree 1 died without CCMs, epileptic seizures or stroke-like symptoms, one might speculate that his SV is a de novo mutation. In line with this hypothesis, a recent analysis of the Genome Aggregation Database (gnomAD) Consortium suggested a de novo mutation rate of 0.35 SVs per generation (95% confidence interval: 0.18–0.52)25. Given that a significant number of SVs might have been missed with short-read WGS, this projection likely underestimates the real number of de novo SVs per genome. However, we can also not exclude a cryptic inherited case in pedigree 1 since the penetrance of CCM is incomplete and up to 45% of CCM2 mutation carriers remain asymptomatic9. Indeed, Nowakowska and colleagues demonstrated that a significant number of apparently de novo, interstitial CNVs that had been found in patients with multiple congenital anomalies or mental retardation were actually the result of an unbalanced transmission of a derivative chromosome from one parent with a balanced insertional translocation34. Furthermore, the transmission from an obviously unaffected parent that carries the same unbalanced rearrangement as its affected child has also been reported24. Unfortunately, DNA samples of the index patient’s parents were not available and there were no other affected family members to address the origin of the chromosomal rearrangement or to demonstrate co-segregation of the SV and CCM disease.
In conclusion, our study adds the first interchromosomal insertion to the CCM mutation spectrum and suggests that hard-to-detect SVs might account for more CCM cases than previously thought. Together with other literature reports of copy number neutral chromosomal rearrangements in known disease genes14,35,36, our results support the hypothesis that efforts to detect SVs might be more promising than the search for novel candidate genes for well-defined Mendelian disorders.
Methods
Study population and ethical considerations
Genomic DNA was isolated from peripheral blood lymphocytes of all study participants with written informed consent using the NucleoSpin Blood L Kit according to the manufacturer’s instructions (Macherey-Nagel, Düren, Germany). A Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, USA) was used to measure DNA concentrations before NGS target enrichment. DNA purity was determined on a NanoPhotometer instrument (Implen, München, Germany). All procedures performed in this study involving human participants were in accordance with the 1964 Helsinki declaration and its later amendments.The study protocol was approved by the local ethics committee of the University Medicine Greifswald (BB 047/14) and written informed consent was obtained from index patient III:1 to publish the medical information and images that are presented here.
Target enrichment, next-generation sequencing, and bioinformatics analyses
All exons (±20 bp) of CCM1 (Locus Reference Genomic sequence: LRG_650t1), CCM2 (LRG_664t2), and CCM3 (LRG_651t1) were defined as target regions for NGS gene panel analysis. A Nextera Rapid Capture Custom Enrichment Kit (Panel ID: 113402; Illumina, San Diego, USA) or an Agilent SureSelect custom library (Panel ID: 3152261, Agilent Technologies, Santa Clara, USA) and a SureSelect Reagent Kit (Agilent Technologies) were used for target enrichment and library preparation according to the manufacturers’ instructions, respectively. Pre- and post-capture libraries were analyzed on a 2100 Bioanalyzer instrument (Agilent Technologies). Indexed libraries were pooled and sequenced on a MiSeq instrument (Illumina) as 2 × 150 bp paired-read runs. The MiSeq Reporter Software (Illumina) was used for demultiplexing and FASTQ file generation. Mapping and alignment were independently performed for each sample with the MiSeq Reporter Software and the SeqNext module of the Sequence Pilot software (JSI medical systems, Ettenheim, Germany). The SeqNext module was also used in a read depth-based approach to identify CNVs in CCM1, CCM2, and CCM3 as described previously37. BAM files that had been generated with the MiSeq Reporter Software were further analyzed for translocation events with the SureCall 4.1.1.5 software (Agilent Technologies), and aligned reads were grouped by the mate chromosome in the Integrative Genomics Viewer38.
Sanger sequencing and multiplex ligation-dependent probe amplification
Specific primer pairs were designed to amplify the suspected breakpoints of a novel interchromosomal insertion identified in CCM2. Primer sequences are available upon request. PCR products were purified with the ExoSAP-IT Cleanup Reagent (Thermo Fisher Scientific). Sanger sequencing was performed on a SeqStudio Genetic Analyzer (Thermo Fisher Scientific) following established protocols. The SALSA MLPA P181-B1 Centromere mix 1 (MRC-Holland, Amsterdam, The Netherlands) was used according to the manufacturer’s instructions to determine copy number variations of a part of the NOTCH2 gene (MIM: 600275) which is located on chromosome 1p12 (hg19: 120,454,176–120,612,317).
Chromosome analysis and molecular cytogenetic studies
Karyotyping was performed following standard procedures on metaphases obtained from PHA stimulated blood lymphocytes. Fluorescence in situ hybridization (FISH) was used to confirm the interchromosomal insertion using standard protocols. The following BAC clones were used: RP11–425C16 in 1p12 (hg19: 120,176,963–120,358,983) and RP11–111G20 in 7p12.3~13 (hg19: 45,283,437–45,448,789).
Ethical approval
All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional and/or national research committee (University Medicine Greifswald; BB 047/14) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Data availability
All relevant data generated or analysed during this study are included in this published article and its supplementary information files.
References
Spiegler, S., Rath, M., Paperlein, C. & Felbor, U. Cerebral cavernous malformations: an update on prevalence, molecular genetic analyses, and genetic counselling. Mol. Syndromol. 9, 60–69, https://doi.org/10.1159/000486292 (2018).
Laberge-le Couteulx, S. et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat. Genet. 23, 189–193, https://doi.org/10.1038/13815 (1999).
Sahoo, T. et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum. Mol. Genet. 8, 2325–2333, https://doi.org/10.1093/hmg/8.12.2325 (1999).
Liquori, C. L. et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am. J. Hum. Genet. 73, 1459–1464, https://doi.org/10.1086/380314 (2003).
Denier, C. et al. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 74, 326–337, https://doi.org/10.1086/381718 (2004).
Bergametti, F. et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 76, 42–51, https://doi.org/10.1086/426952 (2005).
Stenson, P. D. et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 136, 665–677, https://doi.org/10.1007/s00439-017-1779-6 (2017).
Spiegler, S. et al. High mutation detection rates in cerebral cavernous malformation upon stringent inclusion criteria: one-third of probands are minors. Mol. Genet. Genomic Med. 2, 176–185, https://doi.org/10.1002/mgg3.60 (2014).
Denier, C. et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann. Neurol. 60, 550–556, https://doi.org/10.1002/ana.20947 (2006).
Stahl, S. et al. Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: in-frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex. Hum. Mutat. 29, 709–717, https://doi.org/10.1002/humu.20712 (2008).
Cigoli, M. S. et al. PDCD10 gene mutations in multiple cerebral cavernous malformations. PLoS One 9, e110438, https://doi.org/10.1371/journal.pone.0110438 (2014).
Akers, A. et al. Synopsis of guidelines for the clinical management of cerebral cavernous malformations: consensus recommendations based on systematic literature review by the Angioma Alliance Scientific Advisory Board Clinical Experts Panel. Neurosurgery 80, 665–680, https://doi.org/10.1093/neuros/nyx091 (2017).
Liquori, C. L. et al. Low frequency of PDCD10 mutations in a panel of CCM3 probands: potential for a fourth CCM locus. Hum. Mutat. 27, 118, https://doi.org/10.1002/humu.9389 (2006).
Spiegler, S. et al. First large genomic inversion in familial cerebral cavernous malformation identified by whole genome sequencing. Neurogenetics 19, 55–59, https://doi.org/10.1007/s10048-017-0531-7 (2018).
Maroilley, T. & Tarailo-Graovac, M. Uncovering missing heritability in rare diseases. Genes (Basel) 10, https://doi.org/10.3390/genes10040275 (2019).
Spielmann, M., Lupianez, D. G. & Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 19, 453–467, https://doi.org/10.1038/s41576-018-0007-0 (2018).
Feuk, L., Marshall, C. R., Wintle, R. F. & Scherer, S. W. Structural variants: changing the landscape of chromosomes and design of disease studies. Hum. Mol. Genet. 15(Spec No 1), R57–66, https://doi.org/10.1093/hmg/ddl057 (2006).
Eichler, E. E. Genetic variation, comparative genomics, and the diagnosis of disease. N. Engl. J. Med. 381, 64–74, https://doi.org/10.1056/NEJMra1809315 (2019).
Chiang, C. et al. The impact of structural variation on human gene expression. Nat. Genet. 49, 692–699, https://doi.org/10.1038/ng.3834 (2017).
Chaisson, M. J. P. et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 10, 1784, https://doi.org/10.1038/s41467-018-08148-z (2019).
Batra, S., Lin, D., Recinos, P. F., Zhang, J. & Rigamonti, D. Cavernous malformations: natural history, diagnosis and treatment. Nat. Rev. Neurol. 5, 659–670, https://doi.org/10.1038/nrneurol.2009.177 (2009).
Riant, F. et al. Deep intronic KRIT1 mutation in a family with clinically silent multiple cerebral cavernous malformations. Clin. Genet. 86, 585–588, https://doi.org/10.1111/cge.12322 (2014).
McDonald, D. A. et al. Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: evidence for a common biochemical pathway for CCM pathogenesis. Hum. Mol. Genet. 23, 4357–4370, https://doi.org/10.1093/hmg/ddu153 (2014).
Kang, S. H. et al. Insertional translocation detected using FISH confirmation of array-comparative genomic hybridization (aCGH) results. Am. J. Med. Genet. A 152A, 1111–1126, https://doi.org/10.1002/ajmg.a.33278 (2010).
Collins, R. L. et al. An open resource of structural variation for medical and population genetics. bioRxiv, 578674, https://doi.org/10.1101/578674 (2019).
Tattini, L., D’Aurizio, R. & Magi, A. Detection of genomic structural variants from next-generation sequencing data. Front Bioeng. Biotechnol. 3, 92, https://doi.org/10.3389/fbioe.2015.00092 (2015).
Alkan, C., Coe, B. P. & Eichler, E. E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 12, 363–376, https://doi.org/10.1038/nrg2958 (2011).
Holt, J. M. et al. Identification of pathogenic structural variants in rare disease patients through genome sequencing. bioRxiv, 627661, https://doi.org/10.1101/627661 (2019).
De Coster, W. & Van Broeckhoven, C. Newest methods for detecting structural variations. Trends Biotechnol. 37, 973–982, https://doi.org/10.1016/j.tibtech.2019.02.003 (2019).
Mantere, T., Kersten, S. & Hoischen, A. Long-read sequencing emerging in medical genetics. Front Genet 10, 426, https://doi.org/10.3389/fgene.2019.00426 (2019).
Sanchis-Juan, A. et al. Complex structural variants in Mendelian disorders: identification and breakpoint resolution using short- and long-read genome sequencing. Genome Med. 10, 95, https://doi.org/10.1186/s13073-018-0606-6 (2018).
Merker, J. D. et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet. Med. 20, 159–163, https://doi.org/10.1038/gim.2017.86 (2018).
Mitsuhashi, S. & Matsumoto, N. Long-read sequencing for rare human genetic diseases. J. Hum. Genet. 65, 11–19, https://doi.org/10.1038/s10038-019-0671-8 (2020).
Nowakowska, B. A. et al. Parental insertional balanced translocations are an important cause of apparently de novo CNVs in patients with developmental anomalies. Eur. J. Hum. Genet. 20, 166–170, https://doi.org/10.1038/ejhg.2011.157 (2012).
Brigida, I. et al. A novel genomic inversion in Wiskott-Aldrich-associated autoinflammation. J. Allergy Clin. Immunol. 138, 619–622 e617, https://doi.org/10.1016/j.jaci.2016.03.007 (2016).
Vuillaume, M. L. et al. Whole genome sequencing identifies a de novo 2.1 Mb balanced paracentric inversion disrupting FOXP1 and leading to severe intellectual disability. Clin. Chim. Acta 485, 218–223, https://doi.org/10.1016/j.cca.2018.06.048 (2018).
Much, C. D. et al. Novel pathogenic variants in a cassette exon of CCM2 in patients with cerebral cavernous malformations. Front. Neurol. 10, 1219, https://doi.org/10.3389/fneur.2019.01219 (2019).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26, https://doi.org/10.1038/nbt.1754 (2011).
Acknowledgements
This study was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, RA 2876/2–1). We would like to thank the patients for their participation in the study. Dr. W. Schröder is thanked for fruitful discussions.
Author information
Authors and Affiliations
Contributions
R.A.P., U.F., and M.R. designed the study. R.A.P., K.S., S.Sp., E.G. and M.R. analyzed the NGS data. P.D. and A.S. took care for the patient and participated in interpretation of the genetic, clinical and magnetic resonance imaging data. A.W., T.L., and C.A.H. performed and analyzed the cytogenetic analyses. R.A.P., U.F., and M.R. drafted the manuscript and all authors participated in final draft revisions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pilz, R.A., Schwefel, K., Weise, A. et al. First interchromosomal insertion in a patient with cerebral and spinal cavernous malformations. Sci Rep 10, 6306 (2020). https://doi.org/10.1038/s41598-020-63337-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-63337-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
