
Abstract
Cattle domestication occurred at least twice independently and gave rise to the modern taurine and indicine cattle breeds. European cattle diversity is generally dominated by taurine cattle, although elevated levels of indicine ancestry have been recorded in several breeds from southern Europe. Here we use genome-wide high-density SNP genotyping data to investigate the taurine and indicine ancestry in southern European cattle, based on a dataset comprising 508 individuals from 23 cattle breeds of taurine, indicine and mixed ancestry, including three breeds from Central Italy known to exhibit the highest levels of indicine introgression among southern European breeds. Based on local genomic ancestry analyses, we reconstruct taurine and indicine ancestry genome-wide and along chromosomes. We scrutinise local genomic introgression signals and identify genomic regions that have introgressed from indicine into taurine cattle under positive selection, harbouring genes with functions related to body size and feed efficiency. These findings suggest that indicine-derived traits helped enhance Central Italian cattle through adaptive introgression. The identified genes could provide genomic targets for selection for improved cattle performance. Our findings elucidate the key role of adaptive introgression in shaping the phenotypic features of modern cattle, aided by cultural and livestock exchange among historic human societies.
Similar content being viewed by others


Introduction
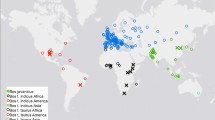
Livestock domestication is tightly intertwined with the evolution and welfare of human societies, contributing to the transition from a nomadic to sedentary lifestyle1,2,3,4. Understanding the origin, dispersal and evolution of globally important livestock species such as cattle and sheep has thus been the focus of numerous genetic investigations (e.g.5,6,7). European taurine cattle (Bos taurus) are believed to first have been domesticated from Bos primigenius primigenius in the Fertile Crescent ~10,000 years ago (YA)8,9. A second inferred domestication event occurred about 2,000 years later in the Indus valley, Central Asia, with Bos primigenius namadicus as ancestor of humped zebu cattle (Bos indicus)10,11. Following the expansion of agriculture, taurine cattle colonized Europe, Asia and Africa, and reached Southern Europe around 8,000 YA12. Similarly, indicine cattle were taken along by humans across South-East Asia, and later on into Africa13. The current geographical distribution of Eurasian cattle breed ancestry reflects these migration paths, with taurine cattle populating Europe and Northern Asia, and indicine Southern Asia, respectively14,15,16,17,18, and different degrees of admixture of taurine and indicine lineages giving rise to African taurine, African zebu, and their crosses known as Sanga cattle19,20.
Genome-wide analyses have provided evidence that several southern European breeds contain ancestry from both African taurine and indicine lineages15,21. One example are the geographically widespread Podolian cattle breeds22, whose name refers to their presumed origin in Moldova/Western Ukraine. Among Podolian cattle are some breeds from Central Italy which display phenotypes such as red coat in calves and white to light grey coat in adults, and a predisposition for draught and beef production, all typical traits of Podolian cattle breeds22. Such breeds – often referred to as Central Italian white cattle – also show the highest levels of indicine ancestry among southern European cattle12,15,21.
Indicine cattle are known to perform better than taurine at harsher conditions (e.g., feed- and heat-stress, resistance to parasites and diseases, drought23,24,25). To date, however, no assessment of the putative adaptive potential of the indicine-derived genomic component present in southern European cattle breeds has ever been undertaken.
Recent technological advances allow affordable use of high-density DNA arrays able to scan several thousand genome-wide markers for all of the major livestock species26,27. Such information has been used in several studies to successfully identify fine-scale gene-flow, selection signatures and association of allelic variants to quantitative traits in a variety of livestock species28,29,30. The acquisition of fitness-enhancing regions due to gene-flow – as the consequence of either natural or human-mediated events – is referred to as adaptive introgression31. Dense genome-wide information allowed the development of local ancestry inference (LAI) approaches able to assign ancestry along a chromosome and identify ancestry blocks32. Hence, adaptive introgression can be identified by LAI, by detecting those regions of fixed or nearly-fixed genome ancestry from a specific source population33. Currently, LAI has been mostly performed on high-density human datasets32,33,34,35, and medium-density DNA array data in canids and ovines36,37,38.
Here we survey the genomes of three autochthonous white cattle breeds from Central Italy, Chianina, Romagnola, and Marchigiana. We study the indicine-derived introgression in these Italian white cattle breeds using genome-wide HD SNP array, combining LAI and selection signature analysis to pinpoint such introgression at the chromosome level. We then infer the adaptive potential of the introgressed genomic regions, along with their putative geographical origin.
Materials and Methods
BovineHD Genotyping BeadChip (777k SNPs) genotypes from 501 individuals across 16 cattle breeds representing B. taurus and B. indicus were used (Table 1). Blood samples from 16 Chianina, 13 Marchigiana and 30 Romagnola individuals were provided by ANABIC ‘Associazione Nazionale Allevatori Bovini Italiani Carne’ (http://www.anabic.it/). Samples were collected by veterinarians between the years 2002–2011, complying with the EU Directive 86/609/EEC regulations which did not require the approval of animal welfare/ethics committee and with the agreement of breeders. The GenElute Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich) was used for total DNA extraction following the manufacturer’s instructions. Genotypes were produced at Laboratorio Genetica e Servizi (LGS) Cremona, Italy. BovineHD genotypes of the other 13 taurine and indicine breeds were kindly made available by Tad Sonstegard (previously unpublished data). BovineHD genotypes of seven additional breeds from Iberia, Italy, and Eastern Europe were available from Upadhyay et al.14,39 (Table 1).
SNP pruning of genotype data was performed using PLINK v1.740. Loci having a minor allele frequency <0.01 and call rate <0.9 were removed. Data were phased using SHAPEIT v2.r79041. SNPs located on sex chromosomes or with unknown map positions were removed. Linkage disequilibrium pruning was performed using the ‘–indep-pairwise’ function in PLINK, where SNPs with r2 > 0.25 were removed from sliding windows of 50 SNPs and a step size of five SNPs.
Summary stats and genetic structure
Observed heterozygosity values were calculated using custom scripts. Inbreeding levels for each population (FROH) were computed as the average of the individual proportion of Runs of Homozygosity (ROHs) to the total length of the genome; ROHs were computed using PLINK. Effective population size (Ne) was estimated using SNeP v1.1142 applying sample size correction for phased genotypes, and Sved & Feldman’s mutation rate modifier which performs well in inferring Ne for the most recent generations43.
A Neighbour-Net graph using Reynolds’ distances, computed with a custom script, was generated using SplitsTree v4.14.644. Maximum likelihood analysis of population structure was conducted using Admixture v1.3.045 for K values from 2 to 16, the latter corresponding to the total number of breeds sampled in our study. A principal component analysis (PCA) was performed to investigate the ordinal relationships between populations and individuals, using PLINK.
Analysis of local genomic ancestry
We used PCAdmix v1.046 to infer genomic local ancestry. For each chromosome and haploid individual, this software scans the target genome using a sliding-window approach, determining the relative ancestry proportions to the utilised reference populations. Window size depends on genome density; here default parameters were used (20 SNPs per window) as the optimal marker density suggested by the software authors matches the BovineHD SNPChip marker density (~1 SNP every 3 kb).
Local ancestry inference relies on reference populations representative of the putative ancestral populations that contributed to the current genomic composition in the target genome. Reference choice is key, along with sufficiently dense data along the chromosomes, to allow reliable ancestry inference37,46. In practical terms, reference choice is tricky, as recent demographic histories of the reference breeds can lead to confounding results46. An approach to partially overcome such issue requires to i) perform multiple analyses with different populations/breeds as representative of a same reference, ii) and then pool the results and identify consensus signals of ancestry, as implemented in the ‘Consistently Introgressed Windows of Interest’ (CIWI) framework37.
To identify reference-independent signals of introgression we applied the CIWI-framework using Chianina, Marchigiana, and Romagnola as target populations. Nine LAI analyses were performed for each target population using as reference breeds all the possible pairwise combinations of one taurine breed among Hereford, Fleckvieh, and Brown Swiss, and one indicine breed among Tharparkar, Gir, and Lohani. These reference breeds were chosen among those not showing mixed cluster components according to K = 2 from the Admixture analysis (Fig. 1). To reduce the possible bias due to productive trait selection, breeds selected for different productive purposes (milk, meat, and dual-purpose) were chosen.

To investigate the putative Eastern or Western origin of the main introgression signals, additional LAI tests focusing on chromosomes 7 and 18 (BTA7 and BTA18, respectively) were performed on genotype data of seven additional breeds, including two cattle breeds from the Iberian peninsula (Cardena and Lidia), two from Central and Southern Italy (Maremmana and Podolica), and three from Eastern Europe and Balkans region (Busha, Romanian grey and Boskarin; Table 1). These genotype data were merged with data used in the previous analysis and submitted to LAI using the same set of nine reference combinations as previously described. To further investigate the origin of BTA7 and BTA18 introgression, these regions were tested in Chianina using PCAdmix/CIWIs with the three indicine references (Tharparkar, Lohani, Gir), and one African taurine reference provided by 25 non-admixed N’Dama individuals (Fig. 1).
Selection signatures
Introgressed genomic portions are kept by selection if provide fitness enhancing variants. Hence, it is likely for a genomic region with adaptive significance to host a selective sweep. Here, we tested the presence of signatures of selection using the haplotype-based statistic nSL47 as implemented in the software Selscan v1.2.0a48. This approach uses the decay of haplotype homozygosity as a function of recombination distance to detect selection sweeps in genomic data47. Further, nSL is well suited to detect selection in loci at low-medium frequency, as in incomplete sweeps on standing variation47,49. Ancestral allele information was available from Rocha and colleagues50. Selection signature analysis was performed on Chianina BTA18 and BTA7. Results were first normalised, and then smoothed by means of cubic smoothed spline using R51.
Results
Genetic diversity and structure
After SNP pruning, 647,132 loci of the BovineHD SNP array were left for analysis. Observed heterozygosity ranged from 0.29 to 0.32 for European taurine breeds and the two Central African Sanga (Ankole-Watussi, Nganda), whereas the Central African N’Dama recorded 0.24. The three indicine breeds (Tharparkar, Lohani and Gir) recorded the lowest Ho values (0.19–0.22; Table 1).
Current Ne estimates from SNeP for European taurine breeds ranged between 98 and 152 (Table 1), with Brown Swiss showing the lowest (98), and Limousine and Piedmontese the highest values (138 and 144, respectively). African taurine recorded similar values (range 120–175). Among indicine cattle Gir recorded the highest Ne estimates (180), and Tharparkar the lowest (63; Table 1). Inbreeding values for European taurine ranged 0.17 (Piedmontese) to 0.34 (Hereford), similarly to the African breeds (0.17–0.31). The indicine population recorded higher inbreeding values on average (0.30–0.39), with highest values found in Tharparkar (Table 1).
In the Admixture analysis, estimates of CV decreased with increasing values of K from 1 to 16, with the main separation between B. taurus and B. indicus at K = 2 (Supplementary Fig. S1b). Admixture analysis at K = 2 split the dataset into three groups showing taurine, indicine and taurine x indicine ancestries (Fig. 1). These three groups overlapped with the European, Southeast Asian, and African origin of the breeds, respectively (Fig. 1, Supplementary Figs. S1a and S2). While most European breeds showed >97% taurine ancestry, approximately ~11–13% of indicine ancestry were recorded in three Central Italian breeds (Marchigiana, Romagnola, and Chianina). The three indicine breeds in our dataset grouped together at this clustering level. At K = 3 the African taurine component was identified; this cluster was found in African taurine N’Dama and comprised 30–47% of total ancestry of the two Sanga breeds Ankole-Watussi and Nganda (Fig. 1). Several African taurine N’Dama individuals show high levels of indicine ancestry, probably reflective of recent crossbreeding with indicine or indicine crossbreds. As found previously, levels of African taurine ancestry were identified in all of the southern European breeds: Fleckvieh (a Simmental-derived breed; ~5%), Limousine from central France (~8%), and Piedmontese from North-West Italy (~10%); the largest values were recorded in the three cattle breeds from Central Italy (Chianina, Marchigiana and Romagnola; range ~11–14%; Fig. 1).
In the PCA (Supplementary Fig. S6), the first principal component (PC) accounted for 18.5% of the variance and discriminated taurine and indicine cattle, mirroring the Admixture results obtained at K = 2. Similarly the second PC accounted for 4.4% of the variance and reflected Admixture results for K = 3, discriminating African taurines from other populations.
Local ancestry inference
We explored the relevant levels of indicine component in Chianina, Romagnola, and Marchigiana by means of local ancestry inference, comparing these three breeds with nine reference combinations and applied the CIWIs framework to aggregate PCAdmix results (Figs. 2 and 3). We identified eight regions of indicine-derived ancestry shared by the Central Italian white-cattle breeds within the top 5% of the genome-wide CIWI scores (Supplementary Table S1). Within these genomic regions, we identified 25 genes spread across seven chromosomes (1, 5, 7, 13, 15, 18, and 24; Table 2). More stringently, 24 of these genes were found within the top 1% of CIWI scores in at least one of the three breeds, but none of them shared by all the three breeds (Table 2).

Consistently Introgressed Windows of Interest (CIWIs) in three Central Italian white cattle breeds (Chianina, Marchigiana and Romagnola), identifying genomic regions of indicine-derived ancestry. Each grey horizontal bar corresponds to one cattle autosome (1–29), and local ancestry within each of the three breeds is shown in one row per breed along each chromosome. Indicine ancestry, when evidenced, is shown in blue, pink and green, respectively, for each breed. Grey indicates a lack of consistent evidence of indicine ancestry.

Local ancestry signals of indicine introgression into taurine cattle breeds, identified from chromosome painting, and identified Consistently Introgressed Windows of Interest (CIWIs) on BTA18. (A) Graphical representation of the chromosome painting results obtained using different combinations of reference populations of both taurine (red) and indicine (green) ancestry. (B) CIWI results for the same chromosome. See Table 1 for breed labels.
The strongest CIWI signal was recorded on BTA18 between 10.80 and 11.74 Mb (CW-18), with the highest values recorded in Chianina (Supplementary Fig. S5). Five genes: USP10, CRISPLD2, ZDHHC7, KIAA0513, and FAM92B (Table 2), were identified within the top 5% selection in all the three Central Italian cattle breeds. With the exclusion of USP10 all of these genes have previously been associated with residual feed intake (RFI) in indicine cattle52.
The second strongest CIWI signal was recorded in BTA7 between 47.75 and 49.38 Mb (spanning ~1.6 Mb; hereafter called CW-7). Out of the 18 genes included within this genomic region only three (H2AFY, NEUROG1, and CXCL14) were shared by all the Central Italian cattle breeds in the top 5% selection (Table 2). CXCL14 has been associated to body weight in cattle53. No direct phenotypic associations have been proposed for H2AFY in cattle, but splicing dysregulation of this gene has been associated with skeletal muscle regeneration disorders in murine models54. Further, the majority of the genes included in the CIWI – although below the top 5% threshold – have been directly associated to stature (CAMLG, DDX46, TXNDC15, CATSPER3, PITX155), body weight (SLC25A48, FBXL2153), and cartilage and body weight development/differentiation (LECT2, TGFBI56) in cattle.
Among the 24 genes also identified within the top 1% CIWI (Table 2), are CCDC14, ROPN1, and CPNE4 (BTA1), SBF2 (BTA15), and SERPINB10 (BTA24). The differential expression of CCDC14 and ROPN1 have been related to beef traits57,58, whereas CPNE4 and SBF2 have been associated with growth traits in horse and cattle, respectively59,60. SERPINB10 is involved in the amino acid metabolism and has been associated to feed efficiency traits in ruminants61.
Using seven additional breeds from Italy, the Balkan area, and Iberia (Table 1), we next performed additional CIWI analyses focused on the two main signals (CW-7 and CW-18) we identified using the BovineHD SNP panel (see above). Both genomic regions were identified as putatively introgressed in Maremmana and Podolica. Among the East European breeds, CW-7 was again identified in Busha and Romanian grey, whereas Boskarin did not show signal for either CW-7 or CW-18. Similarly, no signal was detected corresponding to CW-7 and CW-18 among the Iberian breeds. We then tested the hypothesis of a possible African ancestry for these two signals using the PCAdmix/CIWI framework with both African taurine and indicine breeds as reference and targeting Chianina. For both genomic regions, we recovered a signal of indicine (rather than African taurine) ancestry, overlapping with CW-18, and a partial overlap with the CW-7 (Supplementary Fig. S3).
Signatures of selection
Using nSL, we investigated the presence of signature of selection co-occurring in the genomic portions identified using the CIWIs approach to further test the adaptive significance of these putatively introgressed genomic regions. Indeed, one of the highest scoring selection sweeps perfectly overlapped CW-18 (Supplementary Fig. S4). Further, selection signals overlapped with CIWI signals identified in BTA1 and BTA24 (Supplementary Fig. S4). Conversely, we found no evidence of any marked selective sweep overlapping CIWI regions identified in BTA5, BTA7, BTA13, and BTA15 (Supplementary Fig. S4).
Discussion
We investigated patterns of local ancestry in several cattle breeds from Central Italy using high-density genome-wide polymorphism data. We identified signals of indicine ancestry at genes that in indicine breeds are associated with feeding efficiency. Further, we studied the occurrence of the same signals in several breeds from East and West Europe. Our results suggest adaptive introgression of indicine-derived alleles into Central Italian white cattle breeds, possibly as the result of several gene flow events.
Admixture between taurine and indicine cattle
Population structure analysis (Figs. 1 and 2) performed on genotype data identified >10% indicine derived ancestry in the white cattle breeds from Central Italy, and signals of 5–14% African taurine ancestry in several breeds from southern Europe - consistent with previous findings9,15,21. The three autochthonous Italian breeds remained in a joint Admixture cluster until K = 4, but at higher values of K the divergence of Romagnola from Chianina and Marchigiana is apparent (Supplementary Figs. S1a and S6). Among the three, Chianina is the only breed with no available accounts of introgressive crossbreeding. For Romagnola and Marchigiana, however, records from the 19th century describe the use of Chianina bulls to improve draught and body size traits of both breeds62. In addition, crosses between Marchigiana and Romagnola bulls were later performed to decrease the large Marchigiana body size which – after being crossed with Chianina – was too large and unsuited for grazing hilly pastures62. This documented admixture may explain the extensive shared ancestry of Chianina and Marchigiana at K = 13, and the smaller component of Romagnola ancestry in Marchigiana (Supplementary Fig. S1a). Our obtained clustering results therefore confirm a complex history of cross-breeding and enhancement along with strong selective breeding in these Italian cattle breeds.
In our dataset, genetic diversity of indicine breeds appears lower than that of European breeds (Table 1). This pattern contrasts with the documented reduced effective population size within the highly managed European breeds, and with previously published microsatellite and sequence data from other Asian indicine breeds10,63. The demographic patterns (e.g., bottlenecks) of our sampled indicine populations could in principle explain this discrepancy. However, although some indicine breeds were included in the BovineHD SNP array ascertainment panel, it is likely that our diversity results were impacted by ascertainment bias26. Hence, we caution that our analyses might underestimate the magnitude of indicine-derived alleles in southern European taurine breeds. Importantly, ascertainment bias impacts frequency-based statistics to a larger extent than multi-locus (e.g., Admixture) and haplotype-based (e.g., PCAdmix) statistics64,65,66,67. Regardless, our analysis suggest that southern European cattle show significant admixture levels with both indicine and African taurine cattle. Whether this is the result of one or several independent admixture event will require more extensive sampling of cattle breeds, and likely involve analysis of ancient DNA.
Signals of adaptive introgression
Chianina, Romagnola, and Marchigiana cattle were historically used mostly for draught, and coped well in rough terrain and poor quality pastures. Nowadays, these breeds are highly valued for their meat (e.g., Chianina and Marchigiana T-bones are used for the ‘Fiorentina steak’) and therefore subject to higher management standards to maximise production, but retained both hardiness and rusticity62. Our analyses identified multiple regions of putative indicine derived ancestry in Central Italian taurine cattle (Fig. 2), with the strongest signals recorded on BTA18 and BTA7 (Supplementary Fig. S5).
The genomic region CW-18 (~1 Mb) showed the highest CIWI signal genome-wide (Supplementary Fig. S5). Using a haplotype-based selection analysis (Supplementary Fig. S4), we identified four putative genes (KLHL36, USP10, KIAA0513, and FAM92B) which appear to have been the target of positive selection. KLHL36 belongs to the Kelch superfamily, which consists of a large number of structurally and functionally diverse proteins characterized by the presence of a Kelch-repeat domain68. Currently, no specific studies are available explaining the role of Kelch proteins on cattle physiology. However, studies on human data led to the identification of Kelch family members as regulators of skeletal muscle development and function68. USP10 has been suggested to have a role in regulating gluconeogenesis69, a metabolic pathway involved in the formation of glucose from non-carbohydrate precursors. In non-ruminant species this process is mostly activated during fasting, starvation, and more in general when no exogenous glucose is provided. Conversely, gluconeogenesis in ruminants occurs continuously as dietary carbohydrates are mostly metabolised in the rumen, and gluconeogenesis provides up to 90% of the glucose required70. To date, the involvement of USP10 in gluconeogenesis has only been recorded in pigs – a monogastric species. However, our findings suggest USP10 as a putative candidate for specific investigations on the feed efficiency of ruminants. KIAA0513 and FAM92B were among those genes identified in Chianina when the top 1% threshold was applied for CIWIs selection (Table 2). Both genes are part of a four gene cluster associated with RFI52. RFI is defined as the difference between actual feed intake and the feed intake required to meet maintenance requirements and growth, and reflects the ability of an animal to process food more efficiently and consequently thrive on poor fodder52,71. RFI seems to be a polygenic trait in both taurine and indicine cattle72. Importantly, indicine cattle are known for the good utilisation of low-quality fodder, higher growth rates, and lower weight losses during droughts13,73,74. Selection analysis confirmed that CW-18 harbours a selective sweep, hence we infer that the indicine-derived region CW-18 was favoured by positive selection, putatively allowing for more efficient consumption of poor quality fodder in Central Italian white cattle. Higher feed efficiency has likely been an advantageous trait in the past, especially during harsh environmental conditions with restricted food availability. However, feed efficiency in terms of improved ability to metabolize nutrients is still a highly desirable characteristic for cattle breeders as feed accounts for approximately 70–90% of the total cost in animal production, along with the inherent environmental cost of feed production. Hence, animals that use feed more efficiently positively affect both the costs and sustainability of beef production systems75,76. Furthermore, lower RFI in ruminants is also coupled with reduced generation of methane77, positively affecting greenhouse gas production, a critical parameter with regard to current climate change78,79.
We identified CW-7 (~1.6 Mb) as the second strongest signal of indicine-derived introgression. Signature of selection analysis did not show any evident selective sweep in CW-7 (Supplementary Fig. S4). However, signals for positive selection have previously been identified for the same genomic region in Pingzauer80. Within this region several genes are mapped related to body size, and muscle and bone development along with the growth-related CPNE4 and SBF2 genes we identified in BTA1 and BTA15, the former overlapping a signature of selection signal. It is possible that CW-7 alleles contribute to the increased body size in some cattle breeds, which is likely under positive selection for draught. Additionally, historical records dating back to Roman times mention the presence of thick-set and powerful cattle in Etruria and Latium and the use of white large cattle in ritual parades since Roman times (Columella, De Re Rustica, VI, 1–362,81). Our CW-7 results may thus mirror this anecdotal evidence. Overall, zebus tend to have longer legs and a more slender morphology than taurine cattle82,83. Hence, it is possible that the use of Chianina ancestors in public displays as ritual animals might have been one of the factors contributing to Chianina stature, as both large and tall animals were probably preferred and selected. Morphology has a relevant effect on the ability to adapt to different climates. In indicine cattle, high stature and relatively low body transversal diameters contribute to increase the body-surface/volume proportion, and together with dewlap, large ears and abundant and thin skin, to heat dissipation84. According to archaeological records at the time cattle reached southern Europe - and until 6,000–4,000 YA - the Mediterranean climate was hotter and drier85. Hence, it is possible that Chianina large but slender body type provided a combination of draught, elegance, and heat-resistance features which over time promoted and maintained its morphology.
Inference on the origin of the introgression
Using CW-18 and CW-7 as markers in several Iberian and Eastern European breeds we traced the origin of the indicine-derived components in the Central Italian white cattle breeds. Our results lend support to the hypothesis of multiple routes of introgression sourcing from the East. The strong signal we recorded for CW-18 was common to all the analysed Italian breeds (Chianina, Marchigiana, Romagnola, Maremmana, and Podolica), but was not present in any of the Iberian (Lidia and Cardena) or Eastern European breeds (Busha, Romanian Grey, and Boskarin). Conversely, CW-7 was recorded in all the Italian breeds but Romagnola, in two of the East European breeds, but not in the Iberian breeds. Further, local ancestry analysis assigned indicine rather than African taurine ancestry to both genomic regions (Supplementary Fig. S3). It is therefore possible that these two distinct patterns are reflective of at least two different introduction events from the Near East. Indeed, mitochondrial data suggest genetic proximity between central Italian and Podolian cattle22, and the CW-7 signal could be the consequence of gene-flow between indicine cattle and the cattle populations on the Podolian plateau, which subsequently spread towards southern Europe86. While the CW-18 signal identified in the Central Italian cattle breeds might result from a separate migration route (e.g., via a sea migration from the eastern Mediterranean region ~3,000 YA as suggested by mitochondrial analysis, or following the silk road route 2,000 years later87,88), single admixture scenarios coupled with founder effects could also explain the obtained results. Further investigations with more extensive sampling of local cattle populations will therefore be required to firmly assess these scenarios.
The eastern origin of indicine-derived ancestry in Central Italian white cattle breeds overlaps with the Podolian phenotypic features we can identify in such breeds: grey coat colour, long horns, and overall rusticity. Indeed, most of these traits are shared with the Central Italian white cattle breeds; moreover, recent findings based on mitochondrial analysis identified the genetic proximity of the Italian white cattle breeds with the Turkish grey, a Podolian cattle breed from Anatolia22. We tested the two migration hypotheses using genotype data of seven cattle populations. Although the haplotype-based analyses we utilised (e.g., PCAdmix) are fairly robust to small sample size (Table 1), further investigations using a larger number of individuals will be necessary to test and validate our results and provide a deeper level of detail on the underlying migration scenario. We predict that the availability of genome-wide SNPs from Turkish grey cattle would allow for greater detail comparing shared zebu and African cattle ancestry with Central Italian white cattle. Nevertheless, our Admixture results identified levels of African-derived ancestry component in the Italian breeds, in accordance with previous findings9,15,21.
Conclusions and outlook
Here we combine local ancestry analysis and the CIWI framework and provide the first evidence for adaptive introgression of alleles of indicine-derived ancestry into Central Italian white cattle breeds. Selective advantage of this introgression appears to result from improved feed efficiency and body size. We identified genes which might become target of ad-hoc physiological studies and targets for selection (and potentially gene editing), and eventually contribute to reduced production cost, as well as environmental burden in livestock farming (e.g., lower greenhouse gas production). Finally, for the first time, we used local ancestry information as a genomic fingerprint tool to discriminate among several migration scenarios and provided novel support for the hypothesis of multiple historical cattle migration events into the Italian peninsula.
Data availability
All the genomic data produced in this work are available from the Mendeley Data Repository: https://dx.doi.org/10.17632/znr5dy4x29.1.
References
Zeder, M. A. Domestication and early agriculture in the Mediterranean Basin: Origins, diffusion, and impact. Proc. Natl. Acad. Sci. 105, 11597–11604 (2008).
Clutton-Brock, J. The process of domestication. Mamm. Rev. 22, 79–85 (1992).
Aimé, C. et al. Human Genetic Data Reveal Contrasting Demographic Patterns between Sedentary and Nomadic Populations That Predate the Emergence of Farming. Mol. Biol. Evol. 30, 2629–2644 (2013).
Cavalli-Sforza, L. L., Luigi, L., Menozzi, P. & Piazza, A. The history and geography of human genes. (Princeton University Press, 1994).
Gibbs, R. A. et al. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 324, 528–32 (2009).
Chessa, B. et al. Revealing the history of sheep domestication using retrovirus integrations. Science 324, 532–6 (2009).
Alberto, F. J. et al. Convergent genomic signatures of domestication in sheep and goats. Nat. Commun. 9, 813 (2018).
Troy, C. D. et al. Genetic evidence for Near-Eastern origins of Europe cattle. Nature 410, 1088–1091 (2001).
Orozco-terWengel, P. et al. Revisiting demographic processes in cattle with genome-wide population genetic analysis. Front. Genet. 6, 1–15 (2015).
Chen, N. et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 9, 2337 (2018).
Chen, S. et al. Zebu cattle are an exclusive legacy of the South Asia neolithic. Mol. Biol. Evol. 27, 1–6 (2010).
Cymbron, T., Freeman, A. R., Malheiro, M. I., Vigne, J. D. & Bradley, D. G. Microsatellite diversity suggests different histories for Mediterranean and Northern European cattle populations. Proc. R. Soc. B Biol. Sci. 272, 1837–1843 (2005).
Lenstra, J. A., Felius, M. & Theunissen, B. Domestic cattle and buffaloes. in Ecology, Evolution and Behaviour of Wild Cattle (eds. Melleti, M. & Burton, J.) 30–38 (Cambridge University Press), https://doi.org/10.1017/CBO9781139568098.005 (2013).
Upadhyay, M. R. et al. Genetic origin, admixture and population history of aurochs (Bos primigenius) and primitive European cattle. Heredity (Edinb). 118, 169–176 (2017).
Decker, J. E. et al. Worldwide Patterns of Ancestry, Divergence, and Admixture in Domesticated Cattle. PLoS Genet. 10, e1004254 (2014).
Gautier, M., Laloë, D. & Moazami-Goudarzi, K. Insights into the Genetic History of French Cattle from Dense SNP Data on 47 Worldwide Breeds. PLoS One 5, e13038 (2010).
Pitt, D. et al. Demography and rapid local adaptation shape Creole cattle genome diversity in the tropics. Evol. Appl. https://doi.org/10.1111/eva.12641 (2018).
Sermyagin, A. A. et al. Whole-genome SNP analysis elucidates the genetic structure of Russian cattle and its relationship with Eurasian taurine breeds. Genet. Sel. Evol. 50, 37 (2018).
Kim, J. et al. The genome landscape of indigenous African cattle. Genome Biol. 18, 34 (2017).
Pitt, D. et al. Domestication of cattle: Two or three events? Evol. Appl. 12, 123–136 (2019).
McTavish, E. J., Decker, J. E., Schnabel, R. D., Taylor, J. F. & Hillis, D. M. New World cattle show ancestry from multiple independent domestication events. Proc. Natl. Acad. Sci. USA 110, E1398–406 (2013).
Di Lorenzo, P. et al. Mitochondrial DNA variants of Podolian cattle breeds testify for a dual maternal origin. PLoS One 13, e0192567 (2018).
Kumar, A. et al. Assessment of adaptability of zebu cattle (Bos indicus) breeds in two different climatic conditions: using cytogenetic techniques on genome integrity. Int. J. Biometeorol. 60, 873–882 (2016).
Mackinnon, M. J., Meyer, K. & Hetzel, D. J. S. Genetic variation and covariation for growth, parasite resistance and heat tolerance in tropical cattle. Livest. Prod. Sci. 27, 105–122 (1991).
Vajana, E. et al. Combining Landscape Genomics and Ecological Modelling to Investigate Local Adaptation of Indigenous Ugandan Cattle to East Coast Fever. Front. Genet. 9 (2018).
Matukumalli, L. K. et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS One 4, e5350 (2009).
Kijas, J. W., Hadfield, T., Naval Sanchez, M. & Cockett, N. Genome-wide association reveals the locus responsible for four-horned ruminant. Anim. Genet., https://doi.org/10.1111/age.12409 (2016).
Kijas, J. W. et al. Genome-Wide Analysis of the World’s Sheep Breeds Reveals High Levels of Historic Mixture and Strong Recent Selection. PLoS Biol. 10, e1001258 (2012).
Buzanskas, M. E. et al. Genome-wide association for growth traits in Canchim beef cattle. PLoS One 9, e94802 (2014).
Bomba, L. et al. Relative extended haplotype homozygosity signals across breeds reveal dairy and beef specific signatures of selection. Genet. Sel. Evol. 47, 25 (2015).
Hedrick, P. W. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol. Ecol. 22, 4606–4618 (2013).
Tang, H., Coram, M., Wang, P., Zhu, X. & Risch, N. Reconstructing genetic ancestry blocks in admixed individuals. Am. J. Hum. Genet. 79, 1–12 (2006).
Tang, H. et al. Recent Genetic Selection in the Ancestral Admixture of Puerto Ricans. Am. J. Hum. Genet. 81, 626–633 (2007).
Jarvis, J. P. et al. Patterns of Ancestry, Signatures of Natural Selection, and Genetic Association with Stature in Western African Pygmies. PLoS Genet. 8, e1002641 (2012).
Sanderson, J., Sudoyo, H., Karafet, T. M., Hammer, M. F. & Cox, M. P. Reconstructing Past Admixture Processes from Local Genomic Ancestry Using Wavelet Transformation. Genetics 200, 469–81 (2015).
VonHoldt, B. M. et al. A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res. 21, 1294–1305 (2011).
Barbato, M. et al. Genomic signatures of adaptive introgression from European mouflon into domestic sheep. Sci. Rep. 7, 7623 (2017).
Hu, X.-J. et al. The Genome Landscape of Tibetan Sheep Reveals Adaptive Introgression from Argali and the History of Early Human Settlements on the Qinghai–Tibetan Plateau. Mol. Biol. Evol. 36, 283–303 (2019).
Upadhyay, M. et al. Deciphering the patterns of genetic admixture and diversity in southern European cattle using Genome-wide SNPs. Evol. Appl. https://doi.org/10.1111/eva.12770 (2019).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–75 (2007).
Delaneau, O., Marchini, J. & Zagury, J.-F. A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2012).
Barbato, M., Orozco-terWengel, P., Tapio, M. & Bruford, M. W. SNeP: a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 6, 1–6 (2015).
Corbin, L. J., Liu, A. Y. H., Bishop, S. C. & Woolliams, J. A. Estimation of historical effective population size using linkage disequilibria with marker data. J. Anim. Breed. Genet. 129, 257–70 (2012).
Huson, D. H. & Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Brisbin, A. et al. PCAdmix: principal components-based assignment of ancestry along each chromosome in individuals with admixed ancestry from two or more populations. Hum. Biol. 84, 343–64 (2012).
Ferrer-Admetlla, A., Liang, M., Korneliussen, T. & Nielsen, R. On Detecting Incomplete Soft or Hard Selective Sweeps Using Haplotype Structure. Mol. Biol. Evol. 31, 1275–1291 (2014).
Szpiech, Z. & Hernandez, R. D. selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol. Biol. Evol. 31, 2824–7 (2014).
Vatsiou, A. I., Bazin, E. & Gaggiotti, O. E. Detection of selective sweeps in structured populations: A comparison of recent methods. Mol. Ecol. 25, 89–103 (2016).
Rocha, D., Billerey, C., Samson, F., Boichard, D. & Boussaha, M. Identification of the putative ancestral allele of bovine single-nucleotide polymorphisms. J. Anim. Breed. Genet. 131, 1–4 (2014).
R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing (2012).
Olivieri, B. F. et al. Genomic Regions Associated with Feed Efficiency Indicator Traits in an Experimental Nellore Cattle Population. PLoS One 11, e0164390 (2016).
Makina, S. O. et al. Genome-wide scan for selection signatures in six cattle breeds in South Africa. Genet. Sel. Evol. 47, 92 (2015).
Orengo, J. P., Ward, A. J. & Cooper, T. A. Alternative splicing dysregulation secondary to skeletal muscle regeneration. Ann. Neurol. 69, 681–690 (2011).
Randhawa, I. A. S., Khatkar, M. S., Thomson, P. C. & Raadsma, H. W. Composite Selection Signals for Complex Traits Exemplified Through Bovine Stature Using Multibreed Cohorts of European and African Bos taurus. G3: Genes|Genomes|Genetics 5, 1391–1401 (2015).
Sorbolini, S. et al. Detection of selection signatures in Piemontese and Marchigiana cattle, two breeds with similar production aptitudes but different selection histories. Genet. Sel. Evol. 47, 52 (2015).
Seong, J., Yoon, H. & Kong, H. S. Identification of microRNA and target gene associated with marbling score in Korean cattle (Hanwoo). Genes and Genomics 38, 529–538 (2016).
Nagai, L. & Augusto, E. Identification of genes involved in meat tenderness in Angus and Nellore using differential expression analysis. (UNIVERSIDADE ESTADUAL DE CAMPINAS, 2015).
Al Abri, M. A. et al. Genome-Wide Scans Reveal a Quantitative Trait Locus for Withers Height in Horses Near the ANKRD1 Gene. J. Equine Vet. Sci. 60, 67–73.e1 (2018).
Jahuey-Martínez, F. J. et al. Genomewide association analysis of growth traits in Charolais beef cattle1. J. Anim. Sci. 94, 4570–4582 (2016).
Cantalapiedra-Hijar, G. et al. Review: Biological determinants of between-animal variation in feed efficiency of growing beef cattle. Animal, https://doi.org/10.1017/S1751731118001489 (2018).
ARSIA. Risorse genetiche animali autoctone della Toscana. (Regione Toscana, 2002).
MacHugh, D. E., Shriver, M. D., Loftus, R. T., Cunningham, P. & Bradley, D. G. Microsatellite DNA variation and the evolution, domestication and phylogeography of taurine and zebu cattle (Bos taurus and Bos indicus). Genetics (1997).
Albrechtsen, A., Nielsen, F. C. & Nielsen, R. Ascertainment biases in SNP chips affect measures of population divergence. Mol. Biol. Evol. 27, 2534–2547 (2010).
McTavish, E. J. & Hillis, D. M. How do SNP ascertainment schemes and population demographics affect inferences about population history? BMC Genomics 16 (2015).
Malomane, D. K. et al. Efficiency of different strategies to mitigate ascertainment bias when using SNP panels in diversity studies. BMC Genomics 19, 22 (2018).
McVean, G. A genealogical interpretation of principal components analysis. PLoS Genet. 5, e1000686 (2009).
Gupta, V. A. & Beggs, A. H. Kelch proteins: Emerging roles in skeletal muscle development and diseases. Skelet. Muscle 4, 1–12 (2014).
Keller, J. et al. Effect of L-carnitine on the hepatic transcript profile in piglets as animal model. Nutr. Metab. 8, 1–10 (2011).
Young, J. W. Gluconeogenesis in Cattle: Significance and Methodology. J. Dairy Sci. 60, 1–15 (1977).
Koch, R. M., Swiger, L. A., Chambers, D. & Gregory, K. E. Efficiency of Feed Use in Beef Cattle. J. Anim. Sci. 22, 486–494 (1963).
Bolormaa, S. et al. Accuracy of prediction of genomic breeding values for residual feed intake and carcass and meat quality traits in Bos taurus, Bos indicus, and composite beef cattle1. J. Anim. Sci 91, 3088–3104 (2013).
Elzo, M. A. et al. Effect of breed composition on phenotypic residual feed intake and growth in Angus, Brahman, and Angus × Brahman crossbred cattle. J. Anim. Sci. 87, 3877–3886 (2009).
Frisch, J. E. & Vercoe, J. E. Food intake, eating rate, weight gains, metabolic rate and efficiency of feed utilization in Bos taurus and Bos indicus crossbred cattle. Anim. Prod. 25, 343–358 (1977).
Martello, L. S., da Luz e Silva, S., da Costa Gomes, R., da Silva Corte, R. R. P. & Leme, P. R. Infrared thermography as a tool to evaluate body surface temperature and its relationship with feed efficiency in Bos indicus cattle in tropical conditions. Int. J. Biometeorol. 60, 173–181 (2016).
Castro Bulle, F. C. P., Paulino, P. V., Sanches, A. C. & Sainz, R. D. Growth, carcass quality, and protein and energy metabolism in beef cattle with different growth potentials and residual feed intakes. J. Anim. Sci. 85, 928–936 (2007).
Hegarty, R. S., Goopy, J. P., Herd, R. M. & McCorkell, B. Cattle selected for lower residual feed intake have reduced daily methane production. J. Anim. Sci. 85, 1479–1486 (2007).
Hoffmann, I. Adaptation to climate change–exploring the potential of locally adapted breeds. Animal 7(Suppl 2), 346–362 (2013).
Bruford, M. W. et al. Prospects and challenges for the conservation of farm animal genomic resources, 2015–2025. Front. Genet. 6, 314 (2015).
Kukučková, V. et al. Genomic characterization of Pinzgau cattle: genetic conservation and breeding perspectives. Conserv. Genet. 1–18, https://doi.org/10.1007/s10592-017-0935-9 (2017).
Gargani, M. et al. Microsatellite genotyping of medieval cattle from central Italy suggests an old origin of Chianina and Romagnola cattle. Front. Genet. 6, 68 (2015).
Belavadi, V., Nataraja Karaba, N. & Gangadharappa, N. Agriculture under climate change: Threats, strategies and policies. (Allied Publishers Pvt. Ltd, 2017).
Geer, A. van der Animals in stone: Indian mammals sculptured through time. (Brill, 2008).
Seif, S. M., Johnson, H. D. & Lippincott, A. C. The effects of heat exposure (31 °C) on Zebu and Scottish Highland cattle. Int. J. Biometeorol. 23, 9–14 (1979).
Finné, M., Holmgren, K., Sundqvist, H. S., Weiberg, E. & Lindblom, M. Climate in the eastern Mediterranean, and adjacent regions, during the past 6000 years - A review. J. Archaeol. Sci. 38, 3153–3173 (2011).
Pieragostini, E., Scaloni, A., Rullo, R. & Di Luccia, A. Identical marker alleles in Podolic cattle (Bos taurus) and Indian zebu (Bos indicus). Comp. Biochem. Physiol. - B Biochem. Mol. Biol. 127, 1–9 (2000).
Pellecchia, M. et al. The mystery of Etruscan origins: novel clues from Bos taurus mitochondrial DNA. Proc. Biol. Sci. 274, 1175–9 (2007).
Flori, L. et al. A genomic map of climate adaptation in Mediterranean cattle breeds. Mol. Ecol., https://doi.org/10.1111/mec.15004 (2018).
Acknowledgements
Central Italian cattle samples were obtained from the project “PARAMOGGQUAL - Ricerca per l’individuazione di parametri oggettivi di qualità della carcassa e della carne rilevabili in filiera, dei principali fattori ambientali che la influenzano, e di indici genetici dei riproduttori per avviare la selezione dei bovini anche su parametri di qualità della carne” funded by the Italian Ministry of Agriculture, Food and Forestry Policies, MiPAAFT. This work was partially supported by the ‘Fondazione Romeo ed Enrica Invernizzi’, Milan, Italy.
Author information
Authors and Affiliations
Contributions
M.B. and P.A.-M. conceived the study. M.B. developed the analytical and informatics methods and drafted the manuscript. M.B., M.D. and E.-S.K. performed the analyses. M.B., F.H. and P.A.-M. contributed to the writing of the manuscript. M.B., F.H., M.U., L.C., R.N., R.C., T.S. and P.A.-M. contributed to data interpretation. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barbato, M., Hailer, F., Upadhyay, M. et al. Adaptive introgression from indicine cattle into white cattle breeds from Central Italy. Sci Rep 10, 1279 (2020). https://doi.org/10.1038/s41598-020-57880-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-57880-4
This article is cited by
-
Genetic legacy and adaptive signatures: investigating the history, diversity, and selection signatures in Rendena cattle resilient to eighteenth century rinderpest epidemics
Genetics Selection Evolution (2024)
-
Whole genome sequencing reveals signals of adaptive admixture in Creole cattle
Scientific Reports (2023)
-
Genetic improvement of economic traits in Murrah buffalo using significant SNPs from genome-wide association study
Tropical Animal Health and Production (2023)
-
Assessing Bos taurus introgression in the UOA Bos indicus assembly
Genetics Selection Evolution (2021)
-
On the origin and diversification of Podolian cattle breeds: testing scenarios of European colonization using genome-wide SNP data
Genetics Selection Evolution (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
