
Abstract
Growing evidence indicates that purinergic signalling is involved in the pathogenesis of chronic obstructive pulmonary disease (COPD) and in the vascular remodelling that occurs in other disorders; however, its role in initial vascular changes of COPD is not entirely known. We hypothesised that expression of genes regulating extracellular ATP and adenosine levels would be altered in the lung and systemic arteries of COPD patients. Quantitative real-time PCR was performed to analyse the relative expression of 17 genes associated with purinergic signalling and inflammation in lungs and intercostal arteries of never smokers (NS) (n = 16), non-obstructed smokers (NOS) (n = 17) and COPD patients (n = 21). Gene expression of ATP-degrading enzymes was decreased in both tissues of NOS and COPD patients compared to NS. NT5E expression (gene transcribing for an AMP hydrolyzing ectonucleotidase) was increased in both tissues in NOS compared to the other groups. P1 and P2 receptors did not show changes in expression. Expression of genes associated with inflammation (interleukin-13) was upregulated only in lung tissues of COPD. These findings suggest that the expression of different extracellular ATP-degrading enzymes is altered in smokers (NOS and COPD patients), promoting inflammation. However, the high NT5E expression found only in NOS could compensate this inflammatory environment.
Similar content being viewed by others


Introduction
Chronic obstructive pulmonary disease (COPD) is a highly prevalent chronic respiratory disease and a major cause of mortality and morbidity worldwide1. Smoking has long been recognised as the main risk factor2, but not all smokers develop the disease. The factors that determine the development of COPD in susceptible smokers are complex and may involve genetic and epigenetic factors, altered immune regulation, and abnormal repair mechanisms3,4.
COPD is associated with vascular alterations in both pulmonary and systemic arteries5. In advanced stages of the disease, the risk of developing pulmonary hypertension increases due to the loss of pulmonary vessels caused by emphysema, pulmonary vascular remodelling, and chronic hypoxaemia6. However, in the pulmonary arteries of patients with moderate COPD, an association between endothelial dysfunction6, structural changes7, and the presence of an inflammatory infiltrate in the adventitial layer8 has been also described.
Recently, morphometric studies carried out on the systemic and pulmonary arteries of patients with mild-moderate COPD have demonstrated a significant correlation between the initial changes that occur in the walls of both systemic and pulmonary arteries. These systemic and pulmonary changes are characterised by a thickening of the intima and a decrease in the vascular lumen, which are observed to a greater degree in COPD patients than in smokers without airflow obstruction9. To date, the mediators involved in these processes are not well known.
Numerous studies have indicated that purinergic signalling plays a major role in the development of COPD10,11,12 and in the vascular remodelling that occurs in other disorders like idiopathic pulmonary fibrosis13. Purinergic signalling involves purines (ATP and its hydrolysis products) and pyrimidines (mainly UTP) that act as extracellular ligands for the widely-expressed purinergic receptors, P2 (activated by nucleoside tri-/diphosphates among others) and P1 (activated by adenosine and others)14. It is well known that ATP and adenosine levels are increased in the lungs of COPD patients15,16, which could be associated with COPD development. Extracellular levels of ATP and adenosine are controlled by membrane proteins called ectonucleotidases, which include four families: the ectonucleoside triphosphate diphosphohydrolase (ENTPDase) family, the ectonucleotide pyrophosphatase/phosphodiesterase (ENPP) family, the alkaline phosphatase (AP) family, and the 5′-nucleotidase family, of which only one member (NT5E/CD73) is expressed on the membrane17.
The present study explored the expression of the genes involved in purinergic signalling and inflammation in the lung and vascular systemic tissues of never smokers (NS), non-obstructed smokers (NOS) and stable COPD patients. We performed quantitative real-time PCR on peripheral lung tissue and intercostal arteries (representative systemic vascular tissue) to examine the expression of 17 genes of interest.
Results
Gene expression studies in the lungs of NOS and COPD patients compared to NS
Gene expression of ATP-degrading enzymes and DPP4
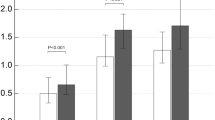
The gene expression of ATP-degrading enzymes and DPP4, which binds adenosine deaminase (ADA), is shown in Fig. 1A. All the ATP-degrading enzymes studied (ENTPD1, ENTPD2 and ENTPD3) showed a similar expression pattern in both NOS and COPD patients, with decreases in ENTPD1 and ENTPD2 expression and no changes in ENTPD3 expression. ENPP1 expression was upregulated, while the gene expression of the adenosine-producing enzyme NT5E/CD73 was up to one-fold higher in NOS, but unchanged in COPD patients. ADA and DPP4 expression was downregulated in both NOS and COPD patients.

Gene expression analyses in the lung. Changes in the mRNA expression of genes for: NOS vs. NS and COPD vs. NS. Results are expressed as fold change (log2) relative to NS. Bar plots represent median ± log2 of RQmax and log2 of RQmin. (A) The expression of ENTPD1, ENTPD2, ADA and DPP4 was downregulated in NOS and COPD patients. ENPP1 expression was upregulated in NOS and COPD patients while NT5E expression was upregulated in NOS and unchanged in COPD. No changes were found in ENTPD3 expression. (B) P2RX2, P2RX7 and P2RY2 expression was downregulated in COPD patients and NOS as well as the expression of ADORA1 in COPD patients. ADORA2A and ADORA2B expression levels were similar to NS in both groups. ADORA3 expression in NOS and COPD patients along with ADORA2A expression in COPD patients was upregulated. No changes were found in ADORA1 expression in NOS. (C) IL-13 expression was upregulated whereas CCL18 expression was downregulated in NOS and COPD patients. No expression of IL-4 was found. *Significantly different from NS (p < 0.05).
Gene expression of P1 and P2 receptors
The gene expression of the purinergic P1 and P2 receptors is shown in Fig. 1B. In the lungs of NOS and COPD patients, gene expression of all the ATP receptors studied (P2RX2, P2RX7 and P2RY2) was downregulated. In the case of adenosine receptors, ADORA1 expression was downregulated only in COPD patients, while ADORA3 expression was upregulated in both groups. ADORA2A and ADORA2B expression in NOS and COPD patients was similar to that in NS.
Expression of the genes associated with inflammation
Among the three genes encoding inflammatory molecules (IL-13, IL-4 and CCL18) that were analysed in pulmonary tissue (Fig. 1C), only IL-13 expression showed significant changes, as expected, with increases observed in both NOS and COPD patients. IL-4 expression was not amplified probably due to the lack of tissue expression. CCL18 expression was decreased in NOS and COPD patients compared to NS.
Gene expression studies in the intercostal arteries of NOS and COPD patients compared to NS
Gene expression of ATP-degrading enzymes and DPP4
Similar patterns of gene expression of the ATP-degrading enzymes were found in the intercostal arteries compared to pulmonary tissue (Fig. 2A). ENTPD1, ENTPD2 and ENTPD3 expression was downregulated in NOS, while ENTPD2 and ENTPD3 expression was also reduced in COPD patients. As observed in the lung samples, ENPP1 expression was upregulated in the intercostal arteries of both NOS and COPD patients. NT5E expression in the intercostal arteries was upregulated in NOS, as also seen in the lung tissue, but was reduced by up to one-fold in the intercostal arteries of COPD patients, which differed to that observed in the lungs. ADA and DPP4 expression was upregulated in the intercostal arteries of both groups, which was opposite to that observed in the lungs.

Gene expression studies in the intercostal arteries. Changes in the mRNA expression of genes for: NOS vs. NS and COPD vs. NS. Results are expressed as fold change (log2) relative to NS. Bar plots represent median ± log2 of RQmax and log2 of RQmin. (A) ENTPD2 and ENTPD3 expression was downregulated in NOS and COPD patients, as well as ENTPD1 in NOS and NT5E in COPD patients. ENPP1, ADA and DPP4 expression levels were increased in both groups. ENTPD1 expression in COPD patients and NT5E expression in NOS were also upregulated. (B) P2RX2, ADORA2A and ADORA2B expression was downregulated in NOS. P2RX7 expression was also downregulated in both groups. P2RY2, ADORA1 and ADORA3 expression in NOS and COPD patients and P2RX2 and ADORA2B expression in COPD patients were also upregulated. No changes were found in ADORA2A expression in COPD patients. (C) No expression of IL-13 or IL-4 was found. CCL18 expression was upregulated in NOS and downregulated in COPD patients. *Significantly different from NS (p < 0.05).
Gene expression of P1 and P2 receptors
The expression patterns of the P1 and P2 receptors differed between the intercostal arteries and pulmonary tissue (Fig. 2B). In the intercostal arteries, P2RX2 expression was upregulated in COPD patients and downregulated in NOS (P2RX2 expression was downregulated in the lung tissues of both groups). P2RX7 expression also differed between the tissues in both groups. Unlike in lung tissue, P2RY2 expression was upregulated in the intercostal arteries of both groups. Whereas ADORA1 expression was downregulated in the lung samples of COPD patients, it was upregulated by up to one-fold in the intercostal arteries of both groups, this being significant for the COPD patients when compared to NS. There were no major differences in the expression levels of ADORA2A, ADORA2B and ADORA3 between the tissues.
Expression of the genes associated with inflammation
IL-13 and IL-4 expression was not amplified in the intercostal arteries, probably due to a lack of expression in these tissues (Fig. 2C). In NOS, CCL18 expression differed between the intercostal arteries and lung tissue, being upregulated by up to one-fold in the intercostal arteries (Fig. 2C).
Figure 3 summarises the main and most interesting results in the format of a heat map. To facilitate the understanding of our findings regarding the enzymes associated with purinergic signalling and their relationship with inflammation, we have highlighted the genes that promote or reduce inflammation.

Schematic of the genes associated with purinergic signalling that were analysed. Heat map of the genes over- or underexpressed (log2 of RQ) in NOS and COPD patients compared to NS. Genes that promote or reduce inflammation are highlighted (red for promotion and green for reduction).
No significant differences were found in the expression of the purinergic pathway genes in lung or intercostal artery after adjusting for sex and diabetes. Furthermore, to analyse potential susceptibility according to gender, independently of tobacco consumption, we compared the gene expression specifically in the non-smoker group and found no significant differences (p > 0.05) in the expression of purinergic pathway genes.
Comparison of gene expression levels in the lung and intercostal arteries between COPD patients and NOS
Changes in the gene expression levels in pulmonary tissue and intercostal arteries in COPD patients compared to NOS are shown in Table 1. All the ATP-degrading enzymes studied were downregulated in the lung, but upregulated or unaffected in the intercostal arteries of COPD patients compared to NOS. NT5E expression was downregulated in both tissues, while ADA expression was upregulated in the lung and downregulated in the intercostal arteries.
In the lung, P2RX2, P2RX7, ADORA1 and ADORA3 expression levels were downregulated, while P2RY2, ADORA2A and ADORA2B expression was upregulated. In the intercostal arteries, all the genes for the P1 and P2 receptors, except ADORA3, were upregulated. Regarding the genes associated with inflammation, IL-13 expression was upregulated in pulmonary tissue, but not expressed in the intercostal arteries. IL-4 expression was not observed in the lung or intercostal arteries. CCL18 expression was downregulated in both tissues.
Discussion
The focus of our study was to characterise the expression patterns of the genes associated with purinergic signalling in the early phases of COPD. We also compared gene expression between pulmonary tissue and systemic arteries from the same patients to identify the mechanisms that initiate and perpetuate this disease and detect any potential molecules involved with the systemic vascular changes observed in COPD. This is the first study to explore the expression of the genes associated with purinergic signalling in the lung and systemic arteries of COPD patients using TaqMan low-density arrays.
Our results suggest that the downregulation of ATP-degrading and adenosine-producing enzymes in the lung and intercostal arteries could produce a pro-inflammatory state in COPD patients compared to NOS. Although ATP-degrading enzymes are also downregulated in both tissues in NOS, adenosine-generating enzymes are upregulated (Figs 1A and 2A), suggesting a compensatory mechanism that exists only in NOS. We found an imbalance between the enzymes regulating extracellular ATP and adenosine levels in the early stages of COPD that favoured ATP accumulation (acting as a pro-inflammatory molecule through the P2 receptors) and lowered adenosine levels (acting as anti-inflammatory molecules through the P1 receptors), possibly causing the overall pulmonary inflammation18. This imbalance could be overcome in NOS by an enhanced expression of NT5E, which encodes the main enzyme (CD73) regulating extracellular adenosine levels. Figure 4 summarizes all this information and represents the possible molecular mechanism in which changes in the expression of purinergic signalling genes favour the accumulation of extracellular ATP that may promote inflammation in COPD patients. By contrast, NOS balanced the levels of extracellular pro-and anti-inflammatory molecules through the increase of NT5E/CD73 expression.

Graphic image representation of the possible mechanisms of lung inflammation in COPD involving purinergic signaling pathway. See text for details and further information (second paragraph of the discussion section). Pro-inflammatory molecules: ATP and derivates (dark and light blue and pink circles). Anti-inflammatory molecules: adenosine (yellow square). The figure was created using Servier Medical Art according to a Creative Commons Attribution 3.0 Unported License guidelines 3.0 (https://creativecommons.org/licenses/by/3.0/). Simplification and colour changes were made to the original cartoons.
Previous studies have shown that tobacco induces an accumulation of extracellular ATP in the human respiratory tract, leading to high ATP concentrations in COPD patients, even after smoking cessation19. By activating P2 receptors, ATP induces macrophages and neutrophils to secrete pro-inflammatory molecules and the mediators of tissue degradation, thus contributing to the chronic inflammation characteristic of COPD15. Extracellular adenosine levels are also increased in the lungs of patients with severe COPD. In accordance with this, previous studies have shown that the enzymatic activity of NT5E/CD73 is increased in the lung tissue of patients with severe COPD compared to smokers with mild obstruction20. In addition, COPD patients with acute exacerbations have decreased ADA enzymatic activity (inactivates adenosine), thus favouring its extracellular accumulation21. Adenosine has immunomodulatory functions; thus, its role may be important in COPD. In fact, adenosine receptors have been proposed as possible therapeutic targets in the treatment of COPD22. However, all the studies to date have focused on the advanced stages of the disease (GOLD stages III and IV) to assess the roles of ATP and adenosine in the pathophysiology of COPD. Given the dual role of adenosine depending on the situation, i.e., acting as an anti-inflammatory molecule in processes associated with acute lung diseases23,24 or as a pro-inflammatory agent with tissue remodeling functions in chronic lung diseases25, it is important to distinguish between the early and late stages of COPD when determining the role of adenosine in this disease. Furthermore, some adenosine receptors also act as anti- or pro-inflammatory molecules depending on the stages (acute or chronic) of lung injury during which they are activated26.
As seen in Fig. 3, NOS expressed more genes favouring a non-inflammatory state than COPD patients in both pulmonary and systemic tissues when compared to NS. The downregulated ectonucleotidases play a larger role in inflammation secondary to tobacco in the lung than in the arterial tissue of the same patients. This observation is in line with the previous studies of our group demonstrating an underexpression of ENTPDase1/CD39 in the lungs of COPD patients27. NT5E/CD73, which is expressed less in the lungs of COPD patients than NOS, is markedly decreased in systemic arteries. NT5E/CD73 has been reported to play an important anti-inflammatory role that is associated with anti-fibrotic activity and a reduced production of pro-inflammatory cytokines in the aortic artery, most likely by activating adenosine A2a receptors28. This would be interesting to investigate further in future studies assessing the role of CD73 in cardiovascular risk.
Classically, the inflammation leading to COPD has been described as a type I inflammation predominantly involving neutrophils. In line with this, the inflammatory parameters analysed in our patients, such as C-reactive protein (CRP) and leukocyte blood counts, were higher in COPD patients than NOS, even though these differences were not significant (Table 2). However, new evidence has emerged that type 2 or eosinophilic inflammation also plays a role in some COPD patients29,30. For this reason, we decided to analyse both IL-13 and IL-4 gene expression as they are involved in eosinophilic inflammation. Our results on IL-13 expression in lung tissue provide a plausible explanation for why NOS show less inflammation than patients with moderate COPD (Fig. 1C). We analysed IL-4 and IL-13 expression to determine the type of inflammatory response elicited in our COPD patients. IL-13 expression was significantly elevated in the pulmonary tissues of NOS and COPD patients, suggesting a Th2-derived inflammatory response (Fig. 1C). IL-13 was not expressed in the intercostal arteries of NOS or COPD patients, suggesting that this type of inflammation is not relevant in the initial systemic vascular changes in smokers and patients with moderate COPD. As for IL-4, we could not assess any amplification of this gene in the lung or intercostal arteries (Figs 1C and 2C). We were not able to determine if this was because there was no expression or whether this was due to methodological issues. CCL18, a chemokine involved in vascular changes31, showed reduced expression in the lungs and systemic arteries of COPD patients. Other studies have shown a similar gene expression pattern of CCL18 in COPD patients and smokers31.
There were differences in purinergic signalling between patients with moderate COPD and NOS. However, other studies have found some similarities in the expression profiles of other genes between patients with moderate COPD and NOS, which have not been observed in patients with severe COPD32. For this reason, we believe that this study should be complemented with future studies investigating pulmonary levels of ATP and adenosine and the genetic expression of purinergic signalling enzymes in patients with severe COPD (GOLD stages III and IV) too.
This study had several strengths and limitations. Performing a genetic analysis on patients in the early stages of COPD enabled us to check for disease-initiating mechanisms that are more difficult to detect at later stages of the disease. Nevertheless, it has to be pointed out that this is an exploratory study to generate new hypotheses about the pathophysiological changes that occur in the first steps of COPD. Almost none of the p-values of the results comparing gene expression levels between the groups were statistically significant, probably because of the exploratory nature of the study and/or the small number of patients included. The study population had primary treatable lung cancer and, therefore, lung cancer could have been a possible introduced bias. However, we assumed that any bias introduced would have been the same for all the subjects. Moreover, this was the only way of obtaining fresh tissue samples from the patients along with clinical and functional data.
In summary, this preliminary study suggests that the expression patterns of different extracellular ATP-degrading enzymes are altered in a manner that promotes inflammation in both NOS and patients with early COPD, with a compensatory mechanism possibly occurring only in NOS. ENTPD1, ENTPD2 and NT5E might be relevant in the pathophysiology of COPD. Future studies are needed to confirm this hypothesis.
Materials and Methods
Subjects
Fifty-four patients who underwent a lobectomy or pneumonectomy of a solitary pulmonary nodule at Bellvitge University Hospital were included in this study. Demographic, clinical and pre-operative pulmonary function assessment (spirometry, lung volumes and carbon monoxide diffusing capacity) data were collected for all the subjects before surgery. None of the patients presented severe systemic comorbidities, atelectasis or obstructive pneumonitis. Moreover, they had not received chemotherapy or radiotherapy prior to surgery. According to their previous smoking history and the results of the pulmonary function tests, subjects were classified as follows: 16 never smokers (NS), 17 non-obstructed smokers (NOS) and 21 stable COPD patients. COPD was diagnosed based on current GOLD guidelines33. In the COPD group, most of the patients had early stages of disease (14 GOLD I and five GOLD II). All the participants signed an informed consent in accordance with the principles outlined in the Declaration of Helsinki. The study was approved by the local ethics committee (CEIC, ref. PR330/15). General characteristics and lung function measurements of the three groups are summarised in Table 2.
Sample collection and processing
The samples used in this study were obtained and processed as previously described9. They have been used in previous studies published by our research group9,27,31.
RNA processing
Total RNA was isolated and purified as previously described27,31.
TaqMan low-density arrays
Gene expression analysis was performed by real-time PCR using Custom TaqMan low-density arrays (TLDAs; Applied Biosystems, Foster City, CA, USA). The 17 genes analysed in this study are listed in Table 3, classified by their functional groups. One µg of total RNA was retrotranscribed into cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Before performing PCR, cDNA was pre-amplified using Custom TaqMan PreAmp Pools, following the manufacturer’s protocol (Applied Biosystems). PCR reactions were prepared with the TaqMan Gene Expression Master Mix, following the manufacturer’s protocol (Applied Biosystems), and samples were run in triplicate on an ABI Prism 7900HT Real-Time PCR System (Applied Biosystems). Data were collected using SDS Software version 2.4 (Applied Biosystems) and used for subsequent analysis.
Gene expression analysis
Data analysis was performed using the Relative Quantification application module on the Thermo Fisher Cloud online software (https://www.thermofisher.com/es/es/home/cloud.html). Relative quantification (RQ) was based on the comparative cycle threshold (Ct) method using GAPDH (Hs99999905_m1) as an endogenous control. For differential expression analysis, a limma-modified t-test34 was used to calculate the ΔΔCt value [ΔΔCt = mean ΔCt value (target samples) - mean ΔCt value (control samples)]. RQ was calculated from these ΔΔCt values (RQ = 2−ΔΔCt) and used for fold change calculations. Results are expressed as fold changes in logarithms to base 2 (log2) of the RQ values.
Heat maps were created with the plotly and ggplot2 R packages version 3.5 (R Foundation for Statistical Computing, Vienna, Austria; https://www.R-project.org/).
Statistical analysis
Continuous variables were compared by Student’s t-test and expressed as mean ± standard deviation (SD). Qualitative variables were compared with the chi-square test. Comparisons between the groups were evaluated by one-way analysis of variance (ANOVA) and an overall p-value was calculated. Adjusted analyses were performed using unbalanced demographic variables (gender and diabetes). Statistical analysis was performed using IBM SPSS version 19.0 (IBM Corp., Armonk, NY, USA). A p-value less than 0.05 indicated statistical significance.
References
Barnes, P. J. Chronic Obstructive Pulmonary Disease: A Growing but Neglected Global Epidemic. PLoS Med. 4, e112 (2007).
van Eeden, S. F. & Hogg, J. C. The response of human bone marrow to chronic cigarette smoking. Eur. Respir. J. 15, 915–21 (2000).
Wood, A. M. & Stockley, R. A. The genetics of chronic obstructive pulmonary disease. Respir. Res. 7, 130 (2006).
MacNee, W. & Tuder, R. M. New paradigms in the pathogenesis of chronic obstructive pulmonary disease I. Proc Am Thorac Soc, https://doi.org/10.1513/pats.200905-027DS (2009).
Sakao, S., Voelkel, N. F. & Tatsumi, K. The vascular bed in COPD: Pulmonary hypertension and pulmonary vascular alterations. European Respiratory Review, https://doi.org/10.1183/09059180.00007913 (2014).
Peinado, V. I. et al. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am. J. Physiol. (1998).
Santos, S. et al. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur. Respir. J., https://doi.org/10.1183/09031936.02.00245902 (2002).
Peinado, V. I. et al. Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med, https://doi.org/10.1164/ajrccm.159.5.9807059 (1999).
Muñoz-Esquerre, M. et al. Systemic and pulmonary vascular remodelling in chronic obstructive pulmonary disease. PLoS One, https://doi.org/10.1371/journal.pone.0152987 (2016).
Burnstock, G., Brouns, I., Adriaensen, D. & Timmermans, J.-P. Purinergic Signaling in the Airways. Pharmacol. Rev., https://doi.org/10.1124/pr.111.005389 (2012).
Pelleg, A., Schulman, E. S. & Barnes, P. J. Extracellular Adenosine 5′-Triphosphate in Obstructive Airway Diseases. Chest, https://doi.org/10.1016/j.chest.2016.06.045 (2016).
Mohsenin, A. & Blackburn, M. R. Adenosine signaling in asthma and chronic obstructive pulmonary disease. Current Opinion in Pulmonary Medicine, https://doi.org/10.1097/01.mcp.0000199002.46038.cb (2006).
Della Latta, V., Cabiati, M., Rocchiccioli, S., Del Ry, S. & Morales, M. A. The role of the adenosinergic system in lung fibrosis. Pharmacol. Res., https://doi.org/10.1016/j.phrs.2013.08.004 (2013).
Burnstock, G. Purine and pyrimidine receptors. Cellular and Molecular Life Sciences, https://doi.org/10.1007/s00018-007-6497-0 (2007).
Mortaz, E., Folkerts, G., Nijkamp, F. P. & Henricks, P. A. J. ATP and the pathogenesis of COPD. Eur. J. Pharmacol., https://doi.org/10.1016/j.ejphar.2010.04.019 (2010).
Esther, C. R., Lazaar, A. L., Bordonali, E., Qaqish, B. & Boucher, R. C. Elevated airway purines in COPD. Chest, https://doi.org/10.1378/chest.10-2471 (2011).
Yegutkin, G. G. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: Functional implications and measurement of activities. Critical Reviews in Biochemistry and Molecular Biology, https://doi.org/10.3109/10409238.2014.953627 (2014).
Faas, M. M., Sáez, T. & de Vos, P. Extracellular ATP and adenosine: The Yin and Yang in immune responses? Molecular Aspects of Medicine, https://doi.org/10.1016/j.mam.2017.01.002 (2017).
Lommatzsch, M. et al. Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med., https://doi.org/10.1164/rccm.200910-1506OC (2010).
Zhou, Y., Murthy, J. N., Zeng, D., Belardinelli, L. & Blackburn, M. R. Alterations in adenosine metabolism and signaling in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. PLoS One, https://doi.org/10.1371/journal.pone.0009224 (2010).
Goodarzi, M. T., Abdi, M., Tavilani, H., Nadi, E. & Rashidi, M. Adenosine deaminase activity in COPD patients and healthy subjects. Iran. J. Allergy, Asthma Immunol., 09.01/ijaai.712 (2010).
Polosa, R. & Blackburn, M. R. Adenosine receptors as targets for therapeutic intervention in asthma and chronic obstructive pulmonary disease. Trends in Pharmacological Sciences, https://doi.org/10.1016/j.tips.2009.07.005 (2009).
Haskó, G. & Pacher, P. A 2A receptors in inflammation and injury: lessons learned from transgenic animals. J. Leukoc. Biol., https://doi.org/10.1189/jlb.0607359 (2008).
Cronstein, B. N. Adenosine, an endogenous anti-inflammatory agent. J. Appl. Physiol. (1994).
Blackburn, M. R. Too much of a good thing: Adenosine overload in adenosine-deaminase-deficient mice. Trends in Pharmacological Sciences, https://doi.org/10.1016/S0165-6147(02)00045-7 (2003).
Zhou, Y. et al. Distinct Roles for the A2B Adenosine Receptor in Acute and Chronic Stages of Bleomycin-Induced Lung Injury. J. Immunol., https://doi.org/10.4049/jimmunol.1002907 (2011).
Aliagas, E. et al. Is the purinergic pathway involved in the pathology of COPD? Decreased lung CD39 expression at initial stages of COPD. Respir. Res., https://doi.org/10.1186/s12931-018-0793-0 (2018).
Quast, C., Alter, C., Ding, Z., Borg, N. & Schrader, J. Adenosine Formed by CD73 on T Cells Inhibits Cardiac Inflammation and Fibrosis and Preserves Contractile Function in Transverse Aortic Constriction-Induced Heart Failure. Circ. Hear. Fail., https://doi.org/10.1161/CIRCHEARTFAILURE.116.003346 (2017).
Saha, S. & Brightling, C. E. Eosinophilic airway inflammation in COPD. International journal of chronic obstructive pulmonary disease, https://doi.org/10.2147/copd.2006.1.1.39 (2006).
Saetta, M. et al. Airway eosinophilia in chronic bronchitis during exacerbations. Am. J. Respir. Crit. Care Med., https://doi.org/10.1164/ajrccm.150.6.7952628 (1994).
Muñoz-Esquerre, M. et al. Vascular disease in COPD: Systemic and pulmonary expression of PARC (Pulmonary and Activation-Regulated Chemokine). PLoS One, https://doi.org/10.1371/journal.pone.0177218 (2017).
Llinàs, L. et al. Similar gene expression profiles in smokers and patients with moderate COPD. Pulm. Pharmacol. Ther., https://doi.org/10.1016/j.pupt.2010.10.010 (2011).
GOLD. Gold 2017. Glob. Inititiative Chronic Obstr. Lung Dis., https://doi.org/10.1164/rccm.201701-0218PP (2017).
Smyth, G. K., Yang, Y. H. & Speed, T. Statistical Issues in cDNA Microarray Data Analysis. Funct. genomics Methods Protoc., https://doi.org/10.1385/1-59259-364-X:111 (2003).
Acknowledgements
We thank Ms. María José Manuel and Ms. Montserrat Navarro from the COPD Unit of Bellvitge University Hospital for their help with subject recruitment. We also thank the thoracic surgery team and pathologists of Bellvitge University Hospital for their assistance in sample collection, the Centres Científics i Tecnològics at the University of Barcelona for their technical assistance, and Anna Carrera-Salinas for her assistance in developing the heat maps. We acknowledge the CERCA Programme/Generalitat de Catalunya for institutional support. The research was funded by Instituto de Salud Carlos III through the grant PI16/00193 (co-funded by the European Regional Development Fund (ERDF) ‘A Way to Build Europe’), as well as by grants from Menarini and SEPAR (Grant SEPAR 067/2015). E. Cuevas received a post-MIR C. Recerca grant from Bellvitge University Hospital.
Author information
Authors and Affiliations
Contributions
O.C. participated in sample processing, TLDA experiments, data analysis, statistical analysis and figure preparation. E.C. participated in subject recruitment and data analysis. M.M.-E. performed the subject recruitment, participated in sample collection and performed sample processing. M.L.-S. participated in subject recruitment, sample collection and sample processing. Y.P.-G. participated in subject recruitment and data analysis. J.D. participated in subject recruitment, interpreting the results and providing a critical review of the manuscript for important intellectual content. E.A. is the co-corresponding author who contributed to the design of the study, sample processing and TLDA experiments, as well as being involved in obtaining funding, data interpretation, manuscript drafting, and critically reviewing the manuscript for important intellectual content. S.S. is the corresponding author who contributed to the study concept and design, as well as being involved in obtaining funding, study supervision, data interpretation, manuscript drafting, and critically reviewing the manuscript for important intellectual content. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Careta, O., Cuevas, E., Muñoz-Esquerre, M. et al. Imbalance in the Expression of Genes Associated with Purinergic Signalling in the Lung and Systemic Arteries of COPD Patients. Sci Rep 9, 2796 (2019). https://doi.org/10.1038/s41598-019-39233-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-39233-y
This article is cited by
-
Purinergic regulation of pulmonary vascular tone
Purinergic Signalling (2024)
-
NT5E gene and CD38 protein as potential prognostic biomarkers for childhood B-acute lymphoblastic leukemia
Purinergic Signalling (2022)
-
Extracellular adenosine triphosphate is associated with airflow limitation severity and symptoms burden in patients with chronic obstructive pulmonary disease
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
