
Abstract
Skeletonema tropicum is a marine diatom of the genus Skeletonema that also includes many well-known species including S. marinoi. S. tropicum is a high temperature preferring species thriving in tropical ocean regions or temperate ocean regions during summer-autumn. However, mechanisms of ecological adaptation of S. tropicum remain poorly understood due partially to the lack of a high-quality whole genome assembly. Here, we report the first high-quality chromosome-scale genome assembly for S. tropicum, using cutting-edge technologies including PacBio single molecular sequencing and high-throughput chromatin conformation capture. The assembled genome has a size of 78.78 Mb with a scaffold N50 of 3.17 Mb, anchored to 23 pseudo-chromosomes. In total, 20,613 protein-coding genes were predicted, of which 17,757 (86.14%) genes were functionally annotated. Collinearity analysis of the genomes of S. tropicum and S. marinoi revealed that these two genomes were highly homologous. This chromosome-level genome assembly of S. tropicum provides a valuable genomic platform for comparative analysis of mechanisms of ecological adaption.
Similar content being viewed by others


Background & Summary
Diatoms (i.e. Bacillariophyta) are unicellular algae with silicified cell walls that represent one of the most ecologically important phytoplankton groups1,2. Diatoms were estimated to contribute approximately 20% of global primary production on Earth, and up to 40% of marine primary production3. Diatoms are also considered as the most species-rich class of microalgae, with estimates range from 12,000 to 30,000 species4,5,6. To date, genomes of only a handful of diatom species have been constructed chromosome-level assemblies, including Thalassiosira pseudonana7, Phaeodactylum tricornutum8, Fistulifera solaris9,10 and Skeletonema marinoi11. These limited number of high-quality genome assemblies severely hinders in-depth research on the internal phylogeny and evolutionary adaption of diatoms.
Skeletonema is one of the most common diatom genera that dominates most coastal waters, some species of which often form harmful algae blooms (HABs)12,13,14,15. Of the Skeletonema species, S. marinoi is the most dominant phytoplankton species that populates in the colder water (in high-latitude ocean regions and temperate ocean regions during winter-spring seasons)12,16. Interestingly, S. tropicum of the genus Skeletonema has a dramatically different preference to temperature, which appears in tropical ocean regions and summer-autumn seasons in temperate ocean regions12,16,17. Despite of the ecological importance of Skeletonema species, genomic information of the Skeletonema species is rather limited. To date, organelle genomes of some Skeletonema species have been constructed, including mitochondrial genomes (mtDNAs)18, and chloroplast genomes (cpDNAs)19 of five Skeletonema species S. marinoi, S. tropicum, S. grevillei, S. pseudocostatum and S. costatum. The conserved genetic structures of these organelle genomes among Skeletonema species couldn’t explain their mechanisms of ecological adaptation. The chromosome-level genome assembly of the first Skeletonema species, S. marinoi was recently constructed11. The availability of this genome assembly led to the discovery of a substantial expansion of light harvesting genes and photoreceptor gene families, which might help the ecological adaptation of S. marinoi under low light condition during the winter-spring seasons. While the whole genome of S. tropicum was still lacking, hampering the comparative genomics analysis among the Skeletonema species.
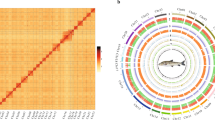
In this study, we report the first chromosome-level genome assembly of the high temperature preferring Skeletonema species S. tropicum (Fig. 1A). The assembled genome size of S. tripicum was 78.69 Mb using PacBio single-molecular DNA sequencing technology20, and the contig N50 was 606.27 Kb. To obtain the high-quality genome assembly at the chromosome level, high-throughput chromatin conformation capture (Hi-C)21 was used and the contigs were clustered into 23 chromosomes, which corresponds to 91.10% of the total contig length. The final assembled genome size of S. tropicum was 78.78 Mb with the scaffold N50 length of 3.17 Mb. A total set of 20,613 putative protein-coding genes (PCGs) were predicted in S. tropicum, among which, 86.14% were annotated to the publicly available database. These chromosome-level genome assemblies of the high temperature preferring Skeletonema species S. tropicum and the low temperature preferring Skeletonema species S. marinoi set up a valuable platform for elucidating mechanisms of temperature adaptation for surviving adverse environments.

Construction of the first chromosome-level genome assembly of S. tropicum. (A). Circos plot of the S. tropicum genome assembly. From outer to inner layers were chromosomes (a), repetitive elements (b), gene densities (c), GC contents (d), respectively. The inner most part layer was the collinear gene pair blocks. (B). Hi-C intra-chromosomal contact map of the genome assembly in S. tropicum.
Methods
Strain isolation and genome sequencing
The S. tropicum strain (CNS00166) analysed in this study was isolated using single-cell capillary from marine water collected in Jiaozhou Bay, China in October 2019. The CNS00166 strain was purified using sterilized seawater for many times. The CNS00166 strain is kept and available in the Key Laboratory of Marine Ecology and Environmental Science from the Institute of Oceanology, Chinese Academy of Science. The axenic cultivation of this strain was maintained in L1 medium22. To ensure low bacterial contamination, penicillin and streptomycin stock solution was added into culture solution. The culture conditions, including culture seawater, temperature, salinity and irradiance intensity, were described previously18. The S. tropicum cells for sequencing were collected by centrifugation and stored in liquid nitrogen. The mtDNA and cpDNA of S. tropicum strain CNS00166 have been reported previously18,19.
High-quality and long-fragment DNA (≥40 Kb) library was prepared by extracting DNA using a magnetic-bead based protocol11. For genome survey analysis, short reads were obtained using MGI short-reads sequencing. The MGI sequencing library (DNBSEQ) was constructed and sequenced using the MGISEQ-2000-PE150 platform. A total of 40.91 Gb (519X sequencing depth) short reads were obtained in this study for genome survey and genome assembly (Table 1). For chromosome-level genome assembly, PacBio continuous long reads (CLR) sequencing library was constructed and sequenced using PacBio Sequel SMRT Cell 1 M. As a result, 10.04 Gb (127X sequencing depth) of PacBio long reads were obtained (Table 1). The N50 length and maximum length of PacBio sequencing reads were 18.18 Kb and 215.08 Kb, respectively. For the Hi-C analysis, algal samples were processed as previously described11 and the Hi-C library was sequenced with MGISEQ-2000-PE150. This process yielded a total of 50.93 Gb of raw data for predicting the spatial proximity of chromatin loci. Three replicates of each RNA sample of S. tropicum in the exponential growth were collected by centrifugation. High-quality RNA was extracted using cetyltrimethylammonium bromide (CTAB) methods11, followed by RNA quality checking using Agilent 2100 Bioanalyzer and NanoDrop. The short-length and full-length transcriptome libraries were sequencing by MGISEQ-2000-PE150 platform and PacBio Sequel SMRT Cell 1 M, respectively.
Genome survey and genome assembly
The genome survey was conducted based on k-mer distribution using the short-length reads using Jellyfish V2.1.423 with k-mer size = 21 and GenomeScope V1.024. The estimated genome size of S. tropicum (CNS00166 strain) was 73.10 Mb with heterozygous ratio was 0.73% and repeat ratio was 48.60% (Table 1).
The PacBio long-read data was used for de novo genome assembly by MECAT225, the primary assembled genome was polished by Arrow (https://github.com/PacificBiosciences/GenomicConsensus) using PacBio long reads and by pilon26 using short reads. Purge Haplotigs27 was used to remove redundancy from the assembled genome. The size of this genome assembly was 78.69 Mb, which was similar to the estimated genome size based on the k-mer analysis. The assembled genome consisted 376 contigs and the N50 was 606.27 Kb. The completeness and quality of this genome assembly was evaluated by BUSCO v5.4.328 against the stramenopiles_odb10 data set. Among the BUSCO orthologous groups, 96.00% were identified as complete in the assembled genome (Table 2).
A total of 50.93 Gb Hi-C sequencing raw data was obtained (Table 1), then was conducted quality control by HiC-Pro v2.5.029. The contigs were mapped onto chromosome-level scaffolds by Juicer v1.630 and 3D-DNA31. As a result, 23 chromosome-level scaffolds were obtained with an anchored rate was 91.10% (Fig. 1), and the length range was from 1558 Kb to 5738 Kb (Table 3). The anchored rate was a little lower probably due to the high heterozygous ratio and repeat content of S. tropicum in this study, the final assembled contigs might contain some highly heterozygosity allelic sequences that are redundant. As only one set of these highly heterozygosity sequences was anchored into the genome assembly with the help of Hi-C data, resulting in relatively lower anchored rate. Finally, the size of genome assembly was 78.78 Mb with the scaffold N50 was 3.17 Mb.
Genome annotation
The genome annotation steps included three parts: repetitive elements annotation, non-coding RNAs annotation and PCGs annotation. The homolog repetitive elements were predicted by RepeatMasker v4.0.732 and RepeatProteinMask v4.0.7 (http://www.repeatmasker.org/cgibin/RepeatProteinMaskRequest) based on the RepBase v21.12 database33. For de novo-based repetitive elements, a de novo repetitive element database was generated by RepeatScout34, Piler35 and LTR_FINDER v1.0736 at first, then de novo-based repetitive elements were predicted by RepeatMasker. Combination of homology-based and de novo-based approaches, a total of 38.73 Mb of transposable elements (TEs) were obtained, contributing 49.17% of assembled genome (Table 4). The DNA, LINE, SINE and LTR account for 5.39%, 5.18%, 0.065% and 25.26% of genome, respectively. In addition, tandem repeats were annotated by Tandem Repeats Finder (TRF v4.09)37, and a total of 6.08 Mb of tandem repeats were obtained accounting for 7.72% of total genome.
Non-coding RNAs are annotated divided into several types, including tRNA, rRNA, snRNA and miRNA. The tRNAs were predicted through tRNAscan-SE38. The rRNA were annotated by Blast v2.2.3139 using the reference sequences of S. marinoi. The snRNAs and miRNAs were identified through INFERNAL in RFAM40.
The PCGs were annotated through integrated approaches, including de novo-, homology- and transcriptome-based information. The de novo prediction were conducted using AUGUSTUS41 and SNAP42, and yielded 24,008 and 31,109 genes, respectively. For the homology-based prediction, the PCG sequences of closely related or model species, including S. marinoi11, T. pseudonana7, Fragilariopsis cylindrus43, Seminavis robusta44, P. tricornutum8 and Arabidopsis thaliana45, were aligned against the S. tropicum genome using Blast v2.2.31, then the gene structures were predicted from these alignments by Exonerate v2.2.046. A total of 84,803 homologous genes were obtained. For the transcriptomic prediction, the RNA-Seq short-read data were aligned to the assembled genome through HISAT2 v 2.1.047 and then assembled and corrected by StringTie v1.3.448 and Pasa_lite (https://github.com/PASApipeline/PASA_Lite). Iso-Seq long-read data were used to get full-length non-chimeric reads by the SMRT Analysis System. A total of 334,554 genes were predicted by the RNA-Seq and Iso-Seq, which contained some redundancy. Finally, gene models from these strategies were merged to form a consensus gene set using MAKER249, and 20,613 PCGs were predicted, with an average gene length of 1675.09 bp and exon length of 750.84 bp (Table 1). The statistics of gene models, including gene length, intron length, exon number and exon length in S. tropicum were comparable to S. marinoi (Fig. 2).

The composition of gene elements in the S. tropicum and other closely related species. (A) Distribution of gene length. (B) Distribution of exon number. (C) Distribution of intron length. (D) Distribution of exon length.
For the functional prediction, these PCGs were annotated to the public databases, including GenBank Nr, SwissProt, Kyoto Encyclopedia of Genes and Genomes (KEGG), eukaryotic orthologous groups (KOG), TrEMBL, InterPro and gene ontology (GO), through Blast v2.2.31 with e-value less than 1e-5. Among all the PCGs, 17,757 genes (86.14%) were functionally annotated to at least one database, and 6544 genes (31.74%) were annotated to at least five databases (Table 5, Fig. 3).

The venn diagram of PCG annotation of S. tropicum to five databases: NR, InterPro, KEGG, SwissProt and KOG.
Data Records
The genome sequencing data (including DNA short-reads sequencing data, DNA PacBio long-reads sequencing data, Hi-C sequencing data, RNA short-reads sequencing data and RNA PacBio long-reads sequencing data) are deposited in the NCBI SRA database under the accession numbers: SRR2685725650, SRR2685725550, SRR2839313951, SRR2685725350, and SRR2685725250. The genomic assembly and annotation results were available at the figshare database52. The genome assembly has also been deposited to NCBI under the accession number of JAWZXG00000000053.
Technical Validation
Low contamination ratio of Bacteria
The low bacteria contamination in the axenic culture of diatom was the critical factor for the high-quality genome assembly. To check low bacteria contamination, 1 Mb of clean short-reads data were selected randomly, and blasted to NCBI NT database. The result showed that the bacteria contamination of S. tropicum was as low as 0.26% (Table 6). The top 20 species of reads annotated to NT database included the Skeletonema species and other closely species, indicating the absence of bacteria comtamination in this project (Table 7). In addition, the short-read DNA data were mapped to the PacBio assembled genome using BWA v. 0.7.1054 to evaluate the GC contents and sequencing depth with 1 Kb window length statistics (Fig. 4), the results showed that the almost all GC points located at the 45%, indicating no exogenous species pollution was found. In addition, the sequencing depth of many points was close to 0, which probably due to its high repeat contents of S. tropicum genome. The reads of repeat content were usually matched to multiple locations of genome assembly in the BWA alignment, resulting in the filtration of the score. Thus, the sequence depth of some locations appeared to 0. The results altogether suggested that genome assembly of S. tropicum was not contaminated by bacteria or other species.

The distribution of GC ratio and sequencing depth. Histograms on the top and right show the frequency distribution of GC ratio and sequencing depth, respectively.
Evaluating genome assembly and annotation completeness
In this study, a total of 519X and 127X of MGI short reads and PacBio reads were used, respectively, which could ensure the quality in the genome assembly. The quality assessments of the genome assembly and annotation were evaluated by BUSCO analysis (Table 2). The results showed that 96.00% and 86.00% were identified as complete orthologs for genome assembly and PCGs annotations, respectively, indicating the high quality of this genome. Although the high heterozygous ratio and repeat content, a high quality genome assembly was obtained in this study. The Hi-C heatmap shows a well-organized interaction pattern within the chromosomal region (Fig. 1), and assembly resulted in 23 chromosome-level scaffolds. Collinearity analysis of amino acid sequences of PCGs between S. tropicum and the same genus species S. marinoi was conducted (Fig. 5A) through Blast v2.2.31 with the evalue less than 1e-05 to identify homologous PCGs, then followed analysed and visualized by WGDI55 and Circos56. The collinearity analysis of DNA sequence (Fig. 5B) was also conducted using mummer 3.057 with minimum alignment length of 1000 bp and many-to-many alignment allowing for rearrangements. The results showed that almost all chromosomes of S. tropicum displayed high homology with the chromosomes of S. marinoi. The clearly strong collinearity between the two close phylogenetic species indicated high quality sequencing and assembly of S. tropicum. Taken together, these confidently confirm the accuracy of the genome assembly and annotation.

Collinearity analysis between S. tropicum and S. marinoi in the view of amino acid sequences (A) and DNA sequences (B).
Code availability
No custom code was used in this study. The data analyses used standard bioinformatic tools specified in the methods.
References
Fu, W. et al. Diatom morphology and adaptation: current progress and potentials for sustainable development. Sustainable Horizons. 2, 100015 (2022).
Falciatore, A., Jaubert, M., Bouly, J.-P., Bailleul, B. & Mock, T. Diatom molecular research comes of age: model species for studying phytoplankton biology and diversity. Plant Cell. 32, 547–572 (2019).
Field, C. B., Behrenfeld, M. J., Randerson, J. T. & Falkowski, P. Primary production of the biosphere: integrating terrestrial and oceanic components. Science. 281, 237–240 (1998).
Malviya, S. et al. Insights into global diatom distribution and diversity in the world’s ocean. PNAS. E1516–E1525 (2016).
Guiry, M. D. How many species of algae are there? J. Phycol. 48, 1057–1063 (2012).
Mann, D. G. & Vanormelingen, P. An inordinate fondness? The number, distributions, and origins of diatom species. J Eukaryot Microbiol. 60, 414–420 (2013).
Armbrust, E. V. et al. The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science. 306, 79–86 (2004).
Bowler, C. et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature. 456, 239–244 (2008).
Maeda, Y. et al. Chromosome-scale genome assembly of the marine oleaginous diatom Fistulifera solaris. Mar Biotechnol. 24, 788–800 (2022).
Tanaka, T. et al. Oil accumulation by the oleaginous diatom Fistulifera solaris as revealed by the genome and transcriptome. Plant Cell. 27, 162–176 (2015).
Liu, S., Xu, Q. & Chen, N. Expansion of photoreception-related gene families may drive ecological adaptation of the dominant diatom species Skeletonema marinoi. Sci Total Environ. 897, 165384 (2023).
Kooistra, W. et al. Global diversity and biogeography of Skeletonema species (Bacillariophyta). Protist. 159, 177–193 (2008).
de Vargas, C. et al. Ocean plankton. eukaryotic plankton diversity in the sunlit ocean. Science. 348, 1261605 (2015).
Ogura, A. et al. Comparative genome and transcriptome analysis of diatom, Skeletonema costatum, reveals evolution of genes for harmful algal bloom. BMC Genomics. 19, (2018).
Gu, H. et al. Emerging harmful algal bloom species over the last four decades in China. Harmful Algae. 102059 (2021).
Liu, S., Cui, Z., Zhao, Y. & Chen, N. Composition and spatial-temporal dynamics of phytoplankton community shaped by environmental selection and interactions in the Jiaozhou Bay. Water Res. 218, 118488 (2022).
Liu, D., Jiang, J., Wang, Y., Zhang, Y. & Di, B. Large scale northward expansion of warm water species Skeletonema tropicum (Bacillariophyceae) in China seas. Chin J Oceanol Limnol. 30, 519–527 (2012).
Liu, S., Wang, Y., Xu, Q., Zhang, M. & Chen, N. Comparative analysis of full-length mitochondrial genomes of five Skeletonema species reveals conserved genome organization and recent speciation. BMC Genomics. 22, 746 (2021).
Liu, S., Xu, Q., Liu, K., Zhao, Y. & Chen, N. Chloroplast genomes for five Skeletonema species: comparative and phylogenetic analysis. Front Plant Sci. 12, (2021).
Eid, J. et al. Real-time DNA sequencing from single polymerase molecules. Science. 323, 133–138 (2009).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326, 289–293 (2009).
Guillard, R. R. L. & Hargraves, P. E. Stichochrysis immobilis is a diatom, not a chrysophyte. Phycologia. 32, 234–236 (1993).
Marçais, G. & Kingsford, C. A Fast, Lock-Free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770 (2011).
Vurture, G. W. et al. Genomescope: fast reference-free genome profiling from short reads. Bioinformatics. 33, 2202–2204 (2017).
Xiao, C.-L. et al. Mecat: fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat Methods. 14, 1072–1074 (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9, e112963 (2014).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics. 19, 460 (2018).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Smit, A. F. A., R. Hubley & Green, P. Repeatmasker, http://www.repeatmasker.org (1996).
Jurka, J. et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 110, 462–467 (2005).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics. 21, i351–i358 (2005).
Edgar, R. C. & Myers, E. W. Piler: identification and classification of genomic repeats. Bioinformatics. 21, i152–i158 (2005).
Xu, Z. & Wang, H. LTR_Finder: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics. 10, 421 (2009).
Griffiths-Jones, S. et al. Rfam: annotating non-coding rnas in complete genomes. Nucleic Acids Res. 33, D121–124 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Johnson, A. D. et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 24, 2938–2939 (2008).
Mock, T. et al. Evolutionary genomics of the cold-adapted diatom Fragilariopsis cylindrus. Nature. 541, 536–540 (2017).
Osuna-Cruz, C. M. et al. The Seminavis robusta genome provides insights into the evolutionary adaptations of benthic diatoms. Nat Commun. 11, 3320 (2020).
The Arabidopsis Genome, I. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 408, 796–815 (2000).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 6, 31 (2005).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 33, 290–295 (2015).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics. 12, 491 (2011).
Ncbi Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP472477 (2023).
Ncbi Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP496561 (2024).
Liu, S. The genomic assembly and annotation results of Skeletonema tropicum. Figshare https://doi.org/10.6084/m9.figshare.24738813 (2023).
Ncbi Sequence Read Archive https://identifiers.org/ncbi/insdc:JAWZXG000000000 (2023).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760 (2009).
Sun, P. et al. WGDI: a user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Mol Plant. 15, 1841–1851 (2022).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Acknowledgements
This research was supported by the Strategic Priority Research Program of Chinese Academy of Sciences (XDB42000000), the Natural Science Foundation of China (42176162), the Chinese Academy of Sciences Pioneer Hundred Talents Program (to Nansheng Chen), the Taishan Scholar Project Special Fund (to Nansheng Chen), the Qingdao Innovation and Creation Plan (Talent Development Program - 5th Annual Pioneer and Innovator Leadership Award to Nansheng Chen, 19-3-2-16-zhc), and the Natural Sciences and Engineering Research Council of Canada (NSERC). Statistical analyses were supported by Oceanographic Data Center, Institute of Oceanology, Chinese Academy of Sciences.
Author information
Authors and Affiliations
Contributions
S. Liu collected the samples, conducted experiments, performed bioinformatics analysis and wrote the manuscript. N. Chen conceived the study and wrote the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, S., Chen, N. Chromosome-level genome assembly of marine diatom Skeletonema tropicum. Sci Data 11, 403 (2024). https://doi.org/10.1038/s41597-024-03238-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03238-8
