
Abstract
The Antarctic whitefin plunderfish Pogonophryne albipinna belongs to the family Artedidraconidae, a key component of Antarctic benthic ecosystems within the order Perciformes and the suborder Notothenioidei. While genome research on P. albipinna using short-read sequencing is available, high-quality genome assembly and annotation employing long-read sequencing have yet to be performed. This study presents a chromosome-scale genome assembly and annotation for P. albipinna, utilizing a combination of Illumina short-read, PacBio long-read, and Hi-C sequencing technologies. The resulting genome assembly spans approximately 1.07 Gb, with a longest scaffold measuring 59.39 Mb and an N50 length of 41.76 Mb. Of the 1,111 Hi-C scaffolds, 23 exceeded 10 Mb and were thus classified as chromosome-level. BUSCO completeness was assessed at 95.6%. The assembled genome comprises 50.68% repeat sequences, and a total of 31,128 protein-coding genes were predicted. This study will enhance our understanding of the genomic characteristics of cryonotothenioids and facilitate comparative analyses of their adaptation and evolution in extreme environments.
Similar content being viewed by others


Background & Summary
The Artedidraconidae family, part of the suborder Notothenioidei within the order Perciformes, plays a significant role in Antarctic benthic ecosystems. It accounts for a substantial portion of fish species diversity in the high Antarctic Zone, Weddell Sea, and Ross Sea1,2,3,4,5. Comprising four genera—Artedidraco, Dolloidraco, Histiodraco, and Pogonophryne—Artedidraconids feature a mental barbel with species-specific morphology6,7,8,9,10,11,12. Traditional taxonomy identifies 27 species within the genus Pogonophryne, the most diverse among Antarctic notothenioids13. However, recent research suggests that this species diversity may be overestimated14,15. Specifically, Parker et al.14 proposed condensing the majority of Pogonophryne species into five (or six, if new species are included) based on comprehensive analyses of phylogenomic data and morphological traits. Eastman and Eakin15 further organized the 27 Pogonophryne species into five groups within three categories: the P. albipinna group (unspotted), and the P. barsukovi, P. marmorata, P. mentella groups (dorsally spotted), as well as the P. scotti group (dorsally unspotted).
Among these, P. albipinna, also known as the whitefin plunderfish, is a representative species of the P. albipinna group. It is distinguished not only by a lack of dark spots on its head and trunk but also by its predominantly white fins and its habitat in water depths exceeding 1,500 meters10,15,16,17. Although genome studies on P. albipinna have been published, such as a complete mitochondrial genome report18 and a preliminary genome survey19, research employing state-of-the-art technologies for high-quality genome assembly and gene annotation has not been conducted. Furthermore, while the chromosome number for other Pogonophryne species, such as P. barsukovi, P. marmorata, P. mentella, and P. scotti, has been established through cytogenetic studies as 2n = 4620,21, the chromosome number for P. albipinna remains unidentified.
Recent research has focused on the genomic characteristics of Antarctic fish species, revealing whole genome sequence and assembly data. These studies also provide genomic insights into adaptations to low-temperature environments, including genes associated with freeze resistance, oxygen-binding, and oxidative stress22,23,24,25,26,27,28,29. The genus Pogonophryne is hypothesized to exhibit specific features for cold-water adaptation, such as functional alterations in hemoglobin or the presence of antifreeze glycoprotein (AFGP). For example, P. favosa possesses a specialized structure, convexitas superaxillaris, located beneath the base of the pectoral fin, which secretes antifreeze proteins30. In a separate study, the amino acid sequences and ligand-binding properties of hemoglobin were examined in two species of Artedidraconidae (Artedidraco orianae and P. scotti). These species demonstrated unexpectedly high oxygen affinity, contrasting with the hemoglobin deficiency observed in channichthyid icefish31.
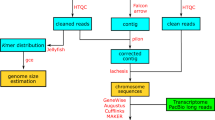
In this study, we performed a chromosome-level genome assembly and annotation of P. albipinna, utilizing PacBio long-read sequencing and high-throughput chromosome conformation capture (Hi-C) technology. This work aims to elucidate the genomic characteristics of Antarctic fish and may serve as a basis for further investigations into their adaptation and evolutionary responses to extreme environments.
Methods
Sampling and DNA extraction
Samples of P. albipinna were collected from the Ross Sea, Antarctica (77°05′S, 170°30′E in CCAMLR Subarea 88.1) and subsequently transported to the Korea Polar Research Institute (KOPRI) in a frozen state. Muscle tissues were excised from these frozen specimens for the extraction of high molecular weight (HMW) DNA using a conventional phenol/chloroform-based method. Molecular identification of the species was carried out using a primer set (FishF2 and FishR2) specifically designed to amplify the mitochondrial cytochrome c oxidase I (COI) gene region32.
Long-read sequencing and assembly
The extracted HMW DNA was utilized to construct 20 kb size-selected PacBio Sequel libraries, following the manufacturer’s protocol and employing the BluePippin size-selection system (Sage Science, Beverly, MA, USA). Specifically, the SMRTbell library was prepared using the SMRTbell Template Prep Kit 1.0, and the SMRTbell-polymerase complex was generated using the Sequel Binding Kit 3.0 (Pacific Biosciences, Menlo Park, CA, USA). This complex was then loaded into SMRT cells 1 M v3 and sequenced with the Sequel Sequencing Kit 3.0 (Pacific Biosciences, Menlo Park, CA, USA) for a 600-min movie time per cell. The genome of P. albipinna was sequenced using six PacBio SMRT cells, generating 7,776,779 raw reads with a total bases of approximately 81.11 Gb (Table 1). De novo genome assembly was performed using FALCON-Unzip assembler v0.433, with parameter settings of length_cutoff = 12,000 and length_cutoff_pr = 10,000. Subsequently, the draft genome assembly was polished using Pilon v1.2334 to enhance its accuracy; this utilized a BAM file generated by BWA-MEM35 based on short-read sequencing data obtained in a prior genome survey19. Lastly, Purge Haplotigs36 was employed to identify and deduplicate haplotigs in the assembled genome.
Hi-C sequencing and chromosome scaffolding
Muscle tissue was frozen and ground in liquid nitrogen for the construction of the Dovetail™ Hi-C library, following the instructions in the Dovetail™ Hi-C kit manual (Dovetail Genomics, Scotts Valley, CA, USA). Sequencing of the Hi-C library was performed on an Illumina NovaSeq. 6000 platform with a 2 × 150 bp paired-end run configuration. A total of 733,064,394 Hi-C reads, with an aggregate length of approximately 110.69 Gb (Table 1), were aligned to the draft genome assembly using Juicer v1.5.737. Subsequently, a candidate assembly was produced using the 3D de novo assembly (3D-DNA) pipeline38. This candidate assembly underwent manual review, modification, and visualization via Juicebox v1.539 to finalize both the genome assembly and the Hi-C contact map.
Our finalized genome assembly measured approximately 1.07 Gb with a maximum scaffold length of 59.39 Mb. We identified 1,111 Hi-C scaffolds, 23 of which exceeded 10 Mb in length, ranging between 13.61 Mb and 59.39 Mb (Table 2 and Table 3). These 23 pseudo-chromosomes in the P. albipinna genome aligned well with the 21 chromosomes of the G. aculeatus genome (Fig. 1). Notably, chromosomes from Group 1 and Group 4 of G. aculeatus corresponded to two chromosomes in P. albipinna each (HiC_scaffold_11 + 27 and HiC_scaffold_5 + 14). Karyotype studies have indicated that four out of the five species groups in the Pogonophryne genus possess 23 chromosome pairs20,21. This study was the first to identify these 23 scaffolds as chromosomes in P. albipinna, affirming that all groups within the Pogonophryne genus have a chromosomal count of 2n = 46.

Chromosome-level genome assembly of Pogonophryne albipinna. (a) Hi-C interaction heat map for P. albipinna. The blue boxes represent the chromosomes. (b) Collinear relationship between P. albipinna and Gasterosteus aculeatus. Connections within the circle represent alignments between the two assemblies.
Transcriptome sequencing
RNA was extracted from muscle tissue using the RNeasy Plus Mini kit (Qiagen, Hilden, Germany), in accordance with the manufacturer’s guidelines. Owing to the quality constraints of the RNA, different specimens were used for DNA and RNA isolation. For Iso-seq library construction, first-strand cDNA was synthesized using a SMARTer PCR cDNA synthesis kit (Clontech, Palo Alto, CA, USA). The SMRTbell library was then prepared as per the manufacturer’s protocol. Sequencing was conducted on a Sequel system (Pacific Biosciences, Menlo Park, CA, USA) using two SMRT cells 1 M v3 LR and Sequel sequencing chemistry 3.0. Iso-seq produced 37,596,041 subreads with a total of 62.65 Gb of nucleotides (Table 1). Analysis of Iso-seq data was performed using the Iso-seq 3 pipeline in SMRT Link v6.0.0 with default settings.
Repeat analysis and masking
A de novo repeat library was generated using RepeatModeler v1.0.340, incorporating the utilities RECON v1.0841, RepeatScout v1.0.542 and Tandem Repeats Finder v4.0943, all of which operated with default parameters. All repeats identified by RepeatModeler, except for transposons, were cross-referenced with the UniProt/SwissProt database44. To specifically identify long terminal repeat retrotransposons (LTR-RTs), LTR_retriever was executed45, utilizing raw LTR data sourced from LTRharvest46 and LTR_FINDER47. The assembled repeat library was then utilized to mask repetitive elements via RepeatMasker v4.0.9, accessed on November 24, 2020, from https://www.repeatmasker.org/. Analysis revealed that the P. albipinna genome comprises 50.68% repetitive sequences, of which 48.03% were transposable elements (TEs), including short interspersed nuclear elements (SINEs, 0.29%), long interspersed nuclear elements (LINEs, 5.50%), long terminal repeats (LTRs, 17.91%), and DNA transposons (15.38%) (Table 4). Kimura divergence values for each alignment were calculated, and the interspersed repeat landscape was plotted using the scripts “calcDivergenceFromAlign.pl” and “createRepeatLandscape.pl”. The Kimura distances for all TE copies indicated that the P. albipinna genome harbored a greater number of recent TE copies with Kimura divergence K-values ≤ 5, primarily influenced by Gypsy LTR and hAT DNA elements (Fig. 2).

Kimura distance-based copy divergence analysis of transposable elements in teleost genomes. The graphs depict genome coverage (Y-axis) for each type of TE in the Pogonophryne albipinna genome.
Gene prediction and functional annotation
Gene structure annotation was conducted using EVidenceModeler (EVM) v1.1.148, integrating multiple types of evidence for gene prediction. Initially, the Program to Assemble Spliced Alignments (PASA) pipeline v2.5.149 was applied to Iso-seq data to generate transcript evidence. Ab initio gene prediction on the repeat-masked genome assembly was then performed using GeneMark-ES v4.6850. Protein hints were generated using Actinopterygii protein sequences from the SwissProt database44 using ProtHint v2.6.051. These hints were employed to produce protein-based evidence via GeneMark-EP+ v4.6851 and for ab initio gene prediction with Augustus v3.4.052. EVM combined all gene models, assigning weight values to each type of evidence (ABINITIO_PREDICTION, 1; PROTEIN, 50; TRANSCRIPT, 50) to produce a consensus gene structure. The consensus gene prediction was further refined using the PASA pipeline49 to include untranslated regions (UTRs) and alternatively spliced isoforms, based on Iso-seq data. In the P. albipinna genome assembly, EVM pipeline predicted a total of 31,128 protein-coding genes (Table 5). The cumulative lengths of exons and coding sequences were 48.20 Mb and 43.33 Mb, respectively, averaging 8.46 exons per gene (Table 5). Functional annotation of the predicted genes was performed by aligning them to the NCBI non-redundant protein (nr) database53 using BLASTP v2.9.054, with an e-value cutoff set at 1e-5. Protein functions were predicted using InterProScan v5.44.7955 on the translated protein sequences from the transcripts. Gene Ontology (GO) terms were assigned to the sequences using the Blast2GO56 module in OmicsBox v1.3.1157. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotation was accomplished using the KEGG Automatic Annotation Server (KAAS)58 and KEGG Mapper59. Trinotate v3.2.060 provided a comprehensive functional annotation of the transcriptome sequences. Specifically, coding regions were identified using TransDecoder v5.5.0, followed by sequence homology searches using BLAST54 against the UniProt/SwissProt database44. Protein domain identification was performed using HMMER61 via the Pfam database62, while protein signal peptides were predicted with SignalP v5.063 and transmembrane domains with TMHMM v2.064. Consequently, 30,992 genes (99.56%) were annotated in at least one database (Table 5). Among these, 26,292 genes (84.5%) received annotations in the GO database (Table 5), and the distribution of GO terms is presented in Fig. 3.

Gene ontology (GO) annotations of the predicted genes in the Pogonophryne albipinna genome. The horizontal axis indicates the number of genes in each class, while the vertical axis indicates the classes in the 2-level GO-annotation.
Gene family identification and phylogenetic analysis
Protein sequences from sixteen teleost species were obtained, with only the longest transcript variant of each gene being selected for further analysis (Table S1). Orthogroups for 17 teleost species were determined based on protein sequence similarity using OrthoFinder v2.4.065 with default parameters. The analysis revealed that 6,727 orthogroups were shared across all 17 species, while 186 orthogroups, encompassing 766 genes, were specific to P. albipinna (Fig. 4a, Table S2). A maximum-likelihood (ML) phylogenetic tree was constructed using the concatenated protein sequences of 1,092 single-copy orthologous genes common to the 17 teleost species, employing MEGA X software66. Divergence times were estimated using TimeTree67, with median estimates for Gadus morhua and Danio rerio set at 224 million years ago. In the resulting tree, P. albipinna clustered with five other Antarctic fish species, diverging from a common ancestor with G. aculeatus approximately 84.24 million years ago (Fig. 5). The divergence time between P. albipinna and N. coriiceps was estimated to be around 22.82 million years ago, followed by a separation from the C. aceratus/P. charcoti clade about 19.59 million years ago (Fig. 5). Gene family expansions and contractions were analyzed using CAFE v4.2.168, with the parameters -p 0.05 and -filter. The analysis revealed that the P. albipinna genome had 208 significantly expanded and 127 significantly contracted gene families (Fig. 5). Expanded gene families in P. albipinna were enriched in telomere-related biological process GO terms (Table S3). GO enrichment analysis results for genes in expanded, contracted, and P. albipinna-specific gene families are presented in Tables S3–5. Comparative analysis of orthologous gene clusters among six Antarctic fish species (P. albipinna, C. aceratus, D. mawsoni, N. coriiceps, P. charcoti, and T. loennbergii) was conducted and visualized using OrthoVenn369. In these analyses, 11,420 orthologous gene families were commonly identified among the six Antarctic species, while 256 gene families were unique to the P. albipinna genome (Fig. 4b).

Gene family comparison. (a) Orthologous gene families between Pogonophryne albipinna and other fish species. (b) Venn diagram showing orthologous gene families among P. albipinna and five other Antarctic fish species.

Phylogenetic analysis of Pogonophryne albipinna within the teleost lineage and analysis of gene family gains and losses, including the number of gained gene families (+) and lost gene families (−). Each branch site number indicates divergence times between lineages.
Data Records
The final genome assembly of Pogonophryne albipinna has been deposited in GenBank with the accession number JAPTMU00000000070. The PacBio (SRR26989350), Hi-C (SRR26989351), and Iso-seq (SRR26989352) reads have been deposited in the NCBI Sequence Read Archive (SRA) database under study accession number of SRP30445471.
Technical Validation
Quality control of nucleic acids and libraries
The quality and quantity of the extracted DNA were assessed using a Qubit 2.0 fluorometer (Invitrogen, Life Technologies, Carlsbad, CA, USA) and a Fragment Analyzer (Agilent Technologies, Santa Clara, CA, USA). The main peak of the input genomic DNA was 28 kb and the final size of the SMRTbell library for long-read sequencing was ~24 kb. The size distribution of Hi-C fragments was centered around 200 bp and the final size-selected Hi-C library was distributed a size range of 200 bp to 1 kb. The RNA quality and quantity were assessed using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and a Qubit 2.0 fluorometer (Invitrogen, Life Technologies, CA, USA), respectively. The RNA integrity number (RIN) value of the total RNA was 8.8 and the average library size for Iso-seq was ~2,800 bp.
Evaluation of genome assembly and annotation
To evaluate the assembly’s completeness, we used Benchmarking Universal Single-Copy Orthologs (BUSCO) v4.1.272 in genome assessment mode, employing the Actinopterygii_odb10 dataset. The assembly showed 95.6% (3,479) complete and 1.2% (42) fragmented genes among 3,640 Actinopterygii single-copy orthologs (Table 6). Additionally, BUSCO v4.1.272 in transcriptome assessment mode represented 85.4% (3,109) of completed and 3.1% (112) of fragmented BUSCOs in actinopterygii_odb10 dataset. The assembly’s contiguity was assessed using the N50 value, defined as the length of the shortest contig or scaffold constituting 50% of the total genome length. The N50 value for the P. albipinna genome assembly was 41.76 Mb (Table 2). Quality value (QV) and k-mer completeness were estimated using Merqury v1.373, resulting in a QV of 39.15 and completeness of 93.48% (Table 7). These metrics indicate high base-level accuracy and completeness for the assembly.
Code availability
All bioinformatic software and pipeline used in this study were implemented according to the protocols provided by the software developers. The versions and parameters for each software can be found in the Methods section. Unless otherwise stated, default parameters were employed.
References
La Mesa, M., Cattaneo-Vietti, R. & Vacchi, M. Species composition and distribution of the Antarctic plunderfishes (Pisces, Artedidraconidae) from the Ross Sea off Victoria Land. Deep Sea Res. II: Top. Stud. Oceanogr. 53, 1061–1070 (2006).
Olaso, I., Rauschert, M. & De Broyer, C. Trophic ecology of the family Artedidraconidae (Pisces: Osteichthyes) and its impact on the eastern Weddell Sea benthic system. Mar. Ecol. Prog. Ser. 194, 143–158 (2000).
Eastman, J. T. & Hubold, G. The fish fauna of the Ross Sea, Antarctica. Antarct. Sci. 11, 293–304 (1999).
Kock, K.-H. Antarctic fish and fisheries. (Cambridge University Press, 1992).
Hubold, G. Ecology of Weddell Sea fishes. Ber. Polarforsch. 103 (1992).
Hureau, J. C. Vol. 2 (eds Fischer, W. & Hureau, J. C.) Ch. Artedidraconidae, 245–251 (FAO, 1985).
Eastman, J. T. & Eakin, R. R. Fishes of the genus Artedidraco (Pisces, Artedidraconidae) from the Ross Sea, Antarctica, with the description of a new species and a colour morph. Antarct. Sci. 11, 13–22 (1999).
Eakin, R. R., Eastman, J. T. & Jones, C. D. Mental barbel variation in Pogonophryne scotti Regan (Pisces: Perciformes: Artedidraconidae). Antarct. Sci. 13, 363–370 (2001).
Lombarte, A., Olaso, I. & Bozzano, A. Ecomorphological trends in the Artedidraconidae (Pisces: Perciformes: Notothenioidei) of the Weddell Sea. Antarct. Sci. 15, 211–218 (2003).
Eakin, R. in Fishes of the Southern Ocean (eds Gon, O. & Heemstra, P. C.) 332–356 (JLB Smith Institute of Ichthyology, 1990).
Eastman, J. T. Evolution and diversification of Antarctic notothenioid fishes. Am. Zool. 31, 93–110 (1991).
Balushkin, A. & Eakin, R. A new toad plunderfish Pogonophryne fusca sp. nova (Fam. Artedidraconidae: Notothenioidei) with notes on species composition and species groups in the genus Pogonophryne Regan. J. Ichthyol. 38, 574–579 (1998).
Eastman, J. T. & Eakin, R. R. Checklist of the species of notothenioid fishes. Antarct. Sci. 33, 273–280 (2021).
Parker, E., Dornburg, A., Struthers, C. D., Jones, C. D. & Near, T. J. Phylogenomic species delimitation dramatically reduces species diversity in an Antarctic adaptive radiation. Syst. Biol. 71, 58–77 (2022).
Eastman, J. T. & Eakin, R. R. Decomplicating and identifying species in the radiation of the Antarctic fish genus Pogonophryne (Artedidraconidae). Polar Biol. 45, 825–832 (2022).
Eastman, J. T. Bathymetric distributions of notothenioid fishes. Polar Biol. 40, 2077–2095 (2017).
Miller, R. G. History and atlas of the fishes of the Antarctic Ocean. (Foresta Institute for Ocean and Mountain Studies, 1993).
Tabassum, N. et al. Characterization of complete mitochondrial genome of Pogonophryne albipinna (Perciformes: Artedidraconidae). Mitochondrial DNA B: Resour. 5, 156–157 (2020).
Jo, E. et al. Genome survey and microsatellite motif identification of Pogonophryne albipinna. Biosci. Rep. 41 (2021).
Morescalchi, A., Morescalchi, M., Odierna, G., Sitingo, V. & Capriglione, T. Karyotype and genome size of zoarcids and notothenioids (Taleostei, Perciformes) from the Ross Sea: cytotaxonomic implications. Polar Biol. 16, 559–564 (1996).
Ozouf-Costaz, C., Hureau, J. & Beaunier, M. Chromosome studies on fish of the suborder Notothenioidei collected in the Weddell Sea during EPOS 3 cruise. Cybium 15, 271–289 (1991).
Ahn, D.-H. et al. Draft genome of the Antarctic dragonfish, Parachaenichthys charcoti. Gigascience 6, gix060 (2017).
Lee, S. J. et al. Chromosomal assembly of the Antarctic toothfish (Dissostichus mawsoni) genome using third-generation DNA sequencing and Hi-C technology. Zool. Res. 42, 124 (2021).
Chen, L. et al. The genomic basis for colonizing the freezing Southern Ocean revealed by Antarctic toothfish and Patagonian robalo genomes. GigaScience 8, giz016 (2019).
Kim, B.-M. et al. Antarctic blackfin icefish genome reveals adaptations to extreme environments. Nat. Ecol. Evol. 3, 469–478 (2019).
Shin, S. C. et al. The genome sequence of the Antarctic bullhead notothen reveals evolutionary adaptations to a cold environment. Genome Biol. 15, 1–14 (2014).
Jo, E. et al. Chromosomal-Level Assembly of Antarctic Scaly Rockcod, Trematomus loennbergii Genome Using Long-Read Sequencing and Chromosome Conformation Capture (Hi-C) Technologies. Diversity 13, 668 (2021).
Bista, I. et al. Genomics of cold adaptations in the Antarctic notothenioid fish radiation. Nat. Commun. 14, 3412 (2023).
Rivera-Colón, A. G. et al. Genomics of secondarily temperate adaptation in the only non-Antarctic icefish. Mol. Biol. Evol. 40, msad029 (2023).
Balushkin, A. & Korolkova, E. New species of plunderfish Pogonophryne favosa sp. n.(Artedidraconidae, Notothenioidei, Perciformes) from the Cosmonauts Sea (Antarctica) with description in artedidraconids of unusual anatomical structures-convexitas superaxillaris. J. Ichthyol. 53, 562–574 (2013).
Tamburrini, M. et al. The hemoglobins of the Antarctic fishes Artedidraco orianae and Pogonophryne scotti: amino acid sequence, lack of cooperativity, and ligand binding properties. J. Biol. Chem. 273, 32452–32459 (1998).
Ward, R. D., Zemlak, T. S., Innes, B. H., Last, P. R. & Hebert, P. D. DNA barcoding Australia’s fish species. Philos. Trans. R. Soc. B: Biol. Sci. 360, 1847–1857 (2005).
Chin, C.-S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v2 [q-bio.GN] (2013).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 19, 1–10 (2018).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Hubley, R. & Smit, A. F. RepeatModeler Open-1.0. (2008).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489 (2021).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 9, 1–14 (2008).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, 1–22 (2008).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O. & Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506 (2005).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom. Bioinform. 2, lqaa026 (2020).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Marchler-Bauer, A. et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229 (2010).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Götz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435 (2008).
BioBam Bioinformatics. OmicsBox-Bioinformatics made easy. (2019).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185 (2007).
Kanehisa, M. & Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 29, 28–35 (2020).
Bryant, D. M. et al. A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 18, 762–776 (2017).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Punta, M. et al. The Pfam protein families database. Nucleic Acids Res. 40, D290–D301 (2012).
Almagro Armenteros, J. J. et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423 (2019).
Krogh, A., Larsson, B., Von Heijne, G. & Sonnhammer, E. L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14 (2019).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547 (2018).
Hedges, S. B., Dudley, J. & Kumar, S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22, 2971–2972 (2006).
Han, M. V., Thomas, G. W., Lugo-Martinez, J. & Hahn, M. W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997 (2013).
Sun, J. et al. OrthoVenn3: an integrated platform for exploring and visualizing orthologous data across genomes. Nucleic Acids Res., gkad313 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc:JAPTMU000000000 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP304454 (2023).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 1–27 (2020).
Acknowledgements
This research received support from the Korea Institute of Marine Science & Technology Promotion (KIMST) grant funded by the Ministry of Oceans and Fisheries (KIMST 20220547), the National Institute of Fisheries Science (NIFS; R2023003), and a grant from Korea University.
Author information
Authors and Affiliations
Contributions
H.P. and J.-H.K. designed the study. E.J., S.C., S.J.L., J.K., E.K.C., M.C., J.K., S.C. and J.L. carried out genome sequencing and assembly. E.J. and S.C. drafted the manuscript. All authors participated in manuscript writing and editing, as well as in compiling the supplementary information and preparing the figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jo, E., Choi, S., Lee, S.J. et al. Chromosome-level genome assembly and annotation of the Antarctica whitefin plunderfish Pogonophryne albipinna. Sci Data 10, 891 (2023). https://doi.org/10.1038/s41597-023-02811-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02811-x
