
Abstract
Endemic to Australia, jade perch (Scortum barcoo) is a highly profitable freshwater bass species. It has extraordinarily high levels of omega-3 polyunsaturated fatty acids (PUFAs), which detailed genes involved in are largely unclear. Meanwhile, there were four chromosome-level bass species have been previous sequenced, while the bass ancestor genome karyotypes have not been estimated. Therefore, we sequenced, assembled and annotated a genome of jade perch to characterize the detailed genes for biosynthesis of omega-3 PUFAs and to deduce the bass ancestor genome karyotypes. We constructed a chromosome-level genome assembly with 24 pairs of chromosomes, 657.7 Mb in total length, and the contig and the scaffold N50 of 4.8 Mb and 28.6 Mb respectively. We also identified repetitive elements (accounting for 19.7% of the genome assembly) and predicted 26,905 protein-coding genes. Meanwhile, we performed genome-wide localization and characterization of several important genes encoding some key enzymes in the biosynthesis pathway of PUFAs. These genes may contribute to the high concentration of omega-3 in jade perch. Moreover, we conducted a series of comparative genomic analyses among four representative bass species at a chromosome level, resulting in a series of sequences of a deductive bass ancestor genome.
Measurement(s) | whole genome sequencing |
Technology Type(s) | Illumina Sequencing • Oxford Nanopore Sequencing |
Similar content being viewed by others


Background & Summary
Jade perch (Scortum barcoo, NCBI Taxonomy ID: 214431; Fig. 1), also called Barcoo grunter, is a species in the order Perciformes and family Theraponidae. It is native to the Barcoo River of the Lake Eyre Basin, Queensland, Australia1. It was introduced into China since 2001. At present, it has become one of the most economically important aquaculture species in several countries including China and Australia due to its rapid growth, highly efficient feed conversion and strong disease resistance2. Its flesh is sweet, succulent and slightly flaky without intermuscular bones3.

A photo of the collected jade perch (The picture was provided by Hu’s lab3).
Meanwhile, jade perch contains highly unsaturated fatty acids. A previous report from the Australian Common Wealth Scientific and Industrial Research Organization (CSIRO) in 1998 revealed that the jade perch contains the highest concentration of omega-3 among 200 examined sea food species4. Its omega-3 level was approximately three times of that in Atlantic salmon4. Due to these highly nutritional and economical values, jade perch has been widely cultured in many Asian countries, especially in China.
Omega-3 polyunsaturated fatty acids (PUFAs) have a double bond three atoms away from the terminal methyl group. Two representative types of omega-3 PUFAs, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), are both essential fatty acids for the body growth and development of human beings5,6. However, humans cannot synthesize these omega-3 PUFAs. These fatty acids can only be obtained from foods such as fish oils. Most fishes have variable abilities to biosynthesize various omgea-3 PUFAs by using fatty acid precursors, such as α-linolenic acids (ALA, 18 carbons with 3 double bonds). This biosynthesis process is mainly controlled by two key enzymes, fatty acy1 desaturases (encoded by fads) and elongase of very long-chain fatty acid (encoded by elovl)7,8,9.
Mammals have a series of fads1, fads2 and fads3, while most of teleosts have only one fads210,11. The fads2 encodes an enzyme to desaturate fatty acids through a double bond among the defined carbons of the fatty acyl chain. There are several types of fads2 with different desaturated positions, including Δ4 desaturase, Δ5 desaturase, and Δ6 desaturase in various teleosts12,13,14. In addition, the function of Δ8 desaturase of fads2 was also reported in some studies15,16,17. Moreover, a total of seven members of the elovl gene family (elovl1-7) have been reported in mammals18. Among them, elovl1, elovl3, elovl6 and elovl7 encode enzymes to catalyze the elongation of saturated and mono-unsaturated fatty acids, while elovl2, elolv4 and elovl5 encode proteins for elongation of PUFAs19,20,21. The functions of fish elovl2, elovl4 and elovl5 are consistent with those mammalian counterparts9,22,23,24,25,26. Recently, two elovl8 isotypes (elovl8a and elovl8b; not found in mammals) were first reported in rabbitfish (Siganus canaliculatus), and the elovl8b has been shown to play an important role in elongation of PUFAs27. Due to the high omega-3 PUFA content, the jade perch can be used as a good model for detailed exploration of the biosynthesis pathway of the omega-3 PUFAs.
Furthermore, reconstruction of the ancestral teleost karyotype and models of teleost genome evolution have been proposed previously based on teleost genomes of such as Tetraodon and medaka28,29,30. Medaka genome has been reported to have well conserved teleost ancestor karyotypes without major interchromosomal rearrangements30. However, even if genomes of four bass species have been sequenced at a chromosome level, the bass ancestral karyotypes were still not deduced.
Here, we applied an integrated strategy of Illumina, Nanopore and Hi-C (high-through-put chromosome conformation capture) sequencing technologies to yield high-quality and sufficient data for subsequent genome assembly, annotation and chromosome construction. Furthermore, we not only localized several key functional genes related to omega-3 PUFA biosynthesis onto chromosomes, but also predicted some potential mechanisms for the high omega-3 PUFA content in the jade perch flesh. We also established the phylogenetic position of jade perch with seven representative teleosts based on single-copy genes, and deduced the bass ancestral karyotypes with a common chromosome translocation in four examined bass species. This jade perch genome assembly, as an important genetic resource, will support in-depth bass biological studies and practical molecular breeding of this economically important fish.
Methods
Sample collection
A three-year-old jade perch individual (Fig. 1), bred in a local fishery farm in Foshan City, Guangdong Province, China, was sampled for this study. We collected muscle, spleen, head kidney, body kidney and thymus of this individual for genome and transcriptome sequencing. All samples were freshly frozen in liquid nitrogen, and then stored at –80 °C until use. This sampling pipeline was approved by the Institutional Review Board on Bioethics and Biosafety of BGI-Shenzhen, China (No. FT18134).
DNA extraction and genome sequencing
Genomic DNA was extracted from the muscle tissue by using a Nucleic Acid Kit (Qiagen, Germantown, MD, USA) following the manufacturer’s instructions. These DNA samples were used to construct libraries for subsequent sequencing on Illumina (Illumina Inc., San Diego, CA, USA) and Nanopore (Oxford Nanopore Technologies, Oxford, UK) platforms. In brief, three paired-end libraries with different insert sizes (270 bp, 500 bp and 800 bp) were constructed and then sequenced on an Illumina HiSeq X Ten platform. About 118.3 Gb of raw reads with paired-end 150 bp were generated. Approximately 109.9 Gb of clean reads were retained for further assembly after removal of low-quality or duplicated reads and adapter sequences by SOAPfilter v2.231 (parameters: -z -p -g 1 -M 2 -f 0). For Nanopore library, the gDNAs were size-selected (10–50 kb) with a Blue Pippin system (Sage Science, Beverly, MA, USA) and processed using the Ligation Sequencing 1D kit (SQKLSK109, Oxford Nanopore Technologies) according to the manufacturer’s instructions. About 82.6 Gb of raw data with fast5 format were collected. After removal of low-quality reads (mean_qscore < 7), 40.0-Gb reads with a mean length of 21.2 kb passed the quality control. Correction of Nanopore reads was then performed by mapping the Illumina clean reads to the Nanopore sequence data using the LoRDEC32 program with default parameters.
RNA extraction and transcriptome sequencing
Muscle, spleen, head kidney, body kidney and thymus tissues of the jade perch were homogenized in the TRIZOL Reagent (Invitrogen, Carlsbad, CA, USA). Library construction and transcriptome sequencing were performed on an Illumina HiSeq X Ten platform in accordance with the manufacturer’s protocols. A total of 20.0 Gb data (about 4.0 Gb for each tissue) were generated for transcript and genome annotation.
Estimation of the genome size
We estimated the genome size of jade perch by using the routine 17-mer frequency distribution analysis33,34. Cleaned Illumina sequences with the insert size of 800 bp were used as the input file. The genome size was calculated according to the following equation: genome size = k-mer number/the estimated k-mer depth. In the case of sufficient data, the k-mer frequency distribution follows a normal Poisson distribution. The peak of the practical k-mer distribution curve is considered as the expected k-mer depth. The estimated genome size of jade perch is about 692.7 Mb, with 5.8 × 1010 k-mers and a peak k-mer depth of 84.
De novo genome assembly and chromosome construction
For the de novo genome assembly, we adopted a hybrid strategy of combining both the clean Illumina and Nanopore reads. Firstly, we employed Platanus v1.2.435 (parameters: -k 29 -d 0.3 -t 16 -m 450) to generate initial contigs based on the clean Illumina reads. Subsequently, these contigs and error-corrected Nanopore long reads were both used as input files for the DBG2OLC36 assembly pipeline (parameters: LD 1 k 17 KmerCovTh 6 MinOverlap 60 AdaptiveTh 0.012). Finally, a high-quality genome of the jade perch was assembled. The scaffold-level genome of 657.7 Mb was firstly assembled, accounting for 94.9% of the estimated genome size. We also evaluated the completeness of this assembly by using the BUSCO v5.2.2 assessment37. The BUSCO evaluation calculated the genome module benchmark value to be C: 97.9%, including S: 95.6%, D: 2.3%, F: 0.1%, M: 2.0%, and n = 3,640 (C: complete, S: single-copy, D: duplicated, F: fragmental, M: missed, and n: total BUSCO groups of Actinopterygii_ odb9 data set). A total of 3,565 out of the 3,640 (97.9%) from Actinopterygii gene set had been identified completely in the assembled genome of jade perch (Table 1).
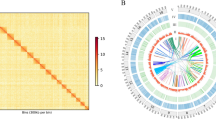
The Hi-C technique was applied to construct a chromosome-level genome of jade perch. In brief, muscle sample was fixed by formaldehyde. The restriction enzyme (Mbo I) was used for digestion of extracted genomic DNAs, followed by repairing of the 5′ overhangs with a biotinylated residue. A paired-end library with an insert size of approximately 300 bp was constructed. About 48.0 Gb of Hi-C reads were generated by sequencing on the HiSeq X ten platform. These Hi-C reads were mapped onto the assembled scaffolds by using Bowtie238 (parameters: --very-sensitive -L 30 --score-min L,-0.6,-0.2 --end-to-end –reorder). We then obtained all valid pair information with scaffold linkage from the results of HiC-Pro v2.8.039 with default parameters. Juicer v1.540 (parameter: chr_num 24) and 3d-DNA v17012341 (parameters: -m haploid -r 2) were applied to anchor these scaffolds into chromosomes. We also utilized Juicebox v1.11.0842 to fix error-joins and removed duplicated contigs. The 3D-DNA pipeline employed the correction file from Juicebox to generate the final chromosome-level genome assembly. By using these Hi-C data, we constructed 24 chromosomes with a total length of 642.9 Mb, accounting for 97.7% of the scaffold-level genome (Fig. 2). The Contig and scaffold N50 values of this final chromosome-level genome assembly are 4.5 Mb and 28.6 Mb, respectively.

Characterization of the assembled genome of jade perch. From the inner to the outer layers: repeat element abundance (violet), GC rate (blue), gene abundance (green), and chromosomes with scale (red).
Repeat annotation
We employed two approaches, including ab initio prediction and homology annotation, to detect repetitive elements in the assembled genome of jade perch. For the ab initio prediction, we applied RepeatModeler v1.0.843 and LTR-FINDER v1.0.644 with default parameters to detect various types of repetitive elements. Subsequently, RepeatMasker v4.0.645 with default parameters was employed to construct a new library based on the Repbase TE v21.0146. Additionally, Tandem Repeats Finder47 (parameters: 2 7 7 80 10 50 2000 -d -h) was used to identify tandem elements. For the homology prediction, RepeatMasker v4.0.645 and RepeatProteinMask v4.0.645 with default parameters were employed to search repeat elements in the genome assembly based on the repeat library from the ab initio prediction. We finally predicted about 129.5 Mb of repetitive sequences, accounting for 19.7% of the assembled jade perch genome. This ratio is at the middle level between European seabass (21.5%)48 and large yellow croaker (18.1%)49, both of which belong to Percomorphaceae.
Gene prediction and functional assignment
To obtain a fully annotated gene set, we combined three methods to predict protein-coding genes, including de novo prediction, homology-based annotation and transcriptome-based annotation. For the de novo prediction, Augustus v2.550 with default parameters was used to predict coding regions on the repeat-masked assembly with default parameters. For the homology-based prediction, protein sequences of seven representative teleosts, including zebrafish (Danio rerio), threespine stickleback (Gasterosteus aculeatus), medaka (Oryzias latipes), Japanese pufferfish (Takifugu rubripes), green-spotted pufferfish (Tetraodon nigroviridis), Southern platyfish (Xiphophorus maculatus), and European seabass (Dicentrarchus labrax), were downloaded from the public NCBI database (release 75) for mapping onto the jade perch genome assembly by TBLASTn51 with an e-value ≤ 10-5. Subsequently, GeneWise v2.2.052 (parameters: --blast_eval 1e-5 --align_rate 0.5 --extend_len 500) was applied to identify gene structures on the basis of the tBLASTn alignments. For the transcriptome-based annotation, pooled RNA-seq reads were mapped onto the jade perch genome by using Tophat v2.1.153. Cufflinks v2.2.1 (http://cole-trapnell-lab.github.io/cufflinks/) was employed to detect gene structures on the RNA-seq alignments. All gene sets from the above-mentioned three approaches were merged by MAKER54 (parameters: max_dna_len = 300000, min_contig = 500, pred_flank = 500, AED_threshold = 1, split_hit = 30000, single_exon = 1, single_length = 250, tries = 2) to generate a final non-redundant gene set. It was functionally annotated by aligning against three public databases, including SwissProt55, TrEMBL56, and KEGG (Kyoto Encyclopedia of Genes and Genomes)57. Gene ontology (GO) annotation was performed by the InterProScan program58.
We predicted a total of 26,905 protein-coding genes (close to the 26,719 of European seabass; Table 2) with a mean length of 15,168.9 bp. Each gene has an average number of 9.9 exons, with a mean length of 179.6 bp. About 97.9% (26,327 genes) of the total predicted genes were assigned with at least function annotation (Table 3).
Ortholog and phylogenetic analyses
Reference protein sequences of eight representative species, including Asian arowana (Scleropages formosus), zebrafish, medaka, Asian seabass (Lates calcarifer), threespine stickleback, European seabass, American black bass (Micropterus salmoides), and Chinese seabass (Lateolabrax maculatus), were downloaded from NCBI database (release 75). These protein sets and our jade perch protein set were filtered by removing those protein sequences with less than 50 amino acids. All to all blast was performed by BLASTP with an e-value ≤ 10-5 to identify homologous sequences51. These protein sets were then clustered into gene families by using OrthoMCL (v2.0.9)59 with default parameters.
To identify the phylogenetic position of jade perch, we employed MUSCLE v3.8.3160 to align single-copy orthologous genes from the used nine species. Subsequently, these protein sequences were converted to their according coding sequences. Alignments were concatenated to form a single supergene for each species. The alignments of supergenes were then used to construct a Maximum Likelihood (ML) tree by PhyML v2.4.461 (parameters: -rates gamma -a e -c 4 -t e). MCMCtree62 (parameters: -model HKY85, -clock independent rates, -seed -1) in the PAML package v4.9 was employed to calculate the divergence times among the selected nine fish species.
A phylogenetic tree was constructed (Fig. 3), and it demonstrated that jade perch and European seabass were clustered into one clade with a bootstrap value of 100. The close relationship between these two bass species is consistent with their traditional morphological classification. Their divergence time was estimated to be 71.7 million years ago (see Fig. 3).

Divergence time tree of nine representative teleosts. Asian arowana was used as the outgroup. The pictures of Asian seabass, European seabass and Chinese seabass were cited from Randall (1997, Wikipedia Commons), Bauchot (1987, Wikipedia Commons) and Chen’s study73, respectively. Orange numbers represent the bootstrap values. Blue numbers represent the divergence times. Red dots represent those confirmed divergence times from the TimeTree (http://www.timetree.org/).
Reconstruction of ancestral chromosomes from four representative bass species
The ancestral teleost karyotype was estimated to contain 13 pairs of potential chromosomes that were marked as Ancestor Chromosomes a~m in previous studies29,63. Each protein set of four representative bass species, including jade perch, Chinese seabass, American black bass and European seabass64, and stickleback was aligned to that of the predicted ancestor chromosomes respectively by using BLASTP (e-value < 1 × 10−10). We then identified the reciprocal best-hit alignments between each of the four bass species and the ancestor chromosomes. Finally, chromosome fissions, fusions and translocations were identified and demonstrated by using SVG in Perl (Fig. 4). We determined that jade perch, European seabass, American black bass and Chinese seabass have well conserved most of the ancestral chromosome karyotype (Fig. 4). A common chromosome translocation appeared in their chromosomes, when compared to the ancestral chromosomes. As to our jade perch genome assembly has more complete sequences than the reported genomes of other bass species (Table 4), it could be the best reference genome assembly for studying the Perciformes evolution.

Evolution of six representative fish chromosome karyotypes. Thirteen ancestral chromosomes are marked with various color bars. Genomic regions from the same ancestral chromosomes were presented in the same color. Green arrows represent translocation, fusion and fission events.
Identification of omega-3 PUFA biosynthesis related genes
Two key gene families (fads and elovl) related to biosynthesis of omega-3 PUFAs were identified in the jade perch. Zebrafish fad and elovl protein sequences (Genbank accession numbers: Fads2, NC_007136.7; Elovl1a, NC_007113.7; Elovl1b, NC_007122.7; Elovl2, NC_007135.7; Elovl4a, NC_007127.7; Elovl4b, NC_007134.7; Elovl5, NC_007124.7; Elovl6, NC_007125.7; Elovl7a, NC_007119.7) were used as the queries to align against the jade perch genome by using TBLASTn51. GeneWise v2.2.052 (parameters: --blast_eval 1e-5 --align_rate 0.5 --extend_len 500) was used to predict gene structures based on these alignments.
We identified the core genes for omega-3 PUFA biosynthesis in the jade perch genome (Fig. 5a). As reported in the majority of teleosts11, elovl2 was absent while one copy of fads2 and one elovl5 were present in jade perch (chr 6 and chr 14, respectively). Two elovl4 genes (elovl4a and elovl4b) were localized in the chr16 and chr13, respectively (Fig. 5a). This number, however, is different from most of the reported fishes, which usually have only one copy of elovl4 gene11,16,25,65,66,67,68. In addition, the elovl4a of jade perch shared a higher identity (82.5%) with its ortholog in African catfish (Clarias gariepinus; Fig. 5b), than that in zebrafish (79.7%). This implies that jade perch, similar to African catfish69, may have an efficient ability to synthesize Omgea-3 PUFAs for their accumulation in the flesh.

Characterization of the core genes for biosynthesis of polyunsaturated fatty acids. (a) Critical fads and evovl genes were localized in the jade perch genome. (b) Elov4a and Elov4b protein sequences from jade perch, African catfish and Zebrafish were aligned for comparison.
Recently, a novel elongase (termed as elovl8) was reported in rabbitfish with two isotypes (elovl8a and elovl8b), and functional experiments revealed that elovl8b, but not elovl8a, has a capacity to elongate C18 and C20 PUFA precursors to produce a longer PUFA27. Similarly, we identified both elovl8a and elovl8b genes in the jade perch genome (chr 3 and chr 8, respectively; see Fig. 5a). At least one of both elongases may participate in the high PUFA accumulation in jade perch flesh.
Previous studies reported that fads2, elovl2, elovl5, and elovl8b were primarily distributed in liver, while elovl4a, elovl4b and elovl8a were mainly present in brain, eye and gonad9,25,27,66,69,70. We therefore speculate that these gene families of both fads and elovl may elevate the biosynthesis content of PUFAs in jade perch, resulting in abundant omega-3 PUFAs in at least its flesh.
Furthermore, these PUFA biosynthesis related genes are individually located on different chromosomes (Fig. 5a). This scattered pattern suggests that these gene copies could not be generated by tandem duplication, instead more possibly by fish-specific genome duplication.
Data Records
The genome assembly and annotation files were deposited in China National GeneBank (CNGB) under the accession number CNP000288971 and NCBI under the accession number SRP37073772. Raw reads of genome and transcriptome generated in the present study were deposited in the NCBI under the accession number SRP37073772.
Technical Validation
The agarose gel electrophoresis was used to check the quality of extracted DNA molecules. The main band is around 20 kb, and the DNA spectrophotometer ratios (260/280) were over 1.8. The quality of purified RNA molecules was examined by Nanodrop ND-1000 spectrophotometer (LabTech, Corinth, MS, USA) as the absorbance > 1.7 at 260 nm/280 nm and 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) as the RIN of 8.0. We further evaluated the completeness of the jade perch genome assembly by using BUSCO v5.2.2, and determined that 97.9% of BUSCO genes were complete.
Code availability
All commands and pipelines used for the genome and transcriptome analyses were performed according to those manuals and protocols of the applied bioinformatics software.
References
Ihwan, M., Syahnon, M., Fakhrul, I., Marina, H. & Ambak, M. New Report on Trichodiniasis (Protozoa: Ciliophora: Peritrichida) in Jade Perch; Scortum barcoo from Peninsular Malaysia. Journal of Fisheries and Aquatic Science 11, 437–443 (2016).
Chen, K., Ma, L., Shi, Y., Zhao, J. & Zhu, X. Genetic diversity analysis of cultured populations of jade perch (Scortum bacoo) in China using AFLP markers. Journal Agricultural and technology 5, 455–461 (2011).
Hu, J., Yan, N., Sun, C., Dong, J. & Ye, X. Biological Characteristics of Jade Perch (Scortum Barcoo). Oceanography & Fisheries Open Access Journal 8, 107–115 (2018).
Iberahim, N. I., Hann, Y., Hamzah, Z. & Syairah, K. Extraction of Omega-3 Fatty Acid from Jade Perch (Scortum barcoo) Using Enzymatic Hydrolysis Technique. Indonesian Journal of Chemistry 20, 282–291, https://doi.org/10.22146/ijc.40903 (2020).
Moertl, D. et al. Dose-dependent effects of omega-3-polyunsaturated fatty acids on systolic left ventricular function, endothelial function, and markers of inflammation in chronic heart failure of nonischemic origin: a double-blind, placebo-controlled, 3-arm study. American heart journal 161, 915. e911–915. e919 (2011).
Campoy, C., Escolano-Margarit, M. V., Anjos, T., Szajewska, H. & Uauy, R. Omega 3 fatty acids on child growth, visual acuity and neurodevelopment. British Journal of Nutrition 107, S85–S106 (2012).
Tocher, D. R. Fatty acid requirements in ontogeny of marine and freshwater fish. Aquaculture Research 41, 717–732 (2010).
Cook, H. W. et al. Alternate pathways in the desaturation and chain elongation of linolenic acid, 18: 3 (n-3), in cultured glioma cells. Journal of lipid research 32, 1265–1273 (1991).
Morais, S., Monroig, O., Zheng, X., Leaver, M. J. & Tocher, D. R. Highly unsaturated fatty acid synthesis in Atlantic salmon: characterization of ELOVL5-and ELOVL2-like elongases. Marine Biotechnology 11, 627–639 (2009).
Castro, L. F. C. et al. Functional desaturase Fads1 (Δ5) and Fads2 (Δ6) orthologues evolved before the origin of jawed vertebrates. PloS one 7, e31950 (2012).
Castro, L. F. C., Tocher, D. R. & Monroig, O. Long-chain polyunsaturated fatty acid biosynthesis in chordates: Insights into the evolution of Fads and Elovl gene repertoire. Progress in lipid research 62, 25–40 (2016).
de Antueno, R. J. et al. Activity of human Δ5 and Δ6 desaturases on multiple n‐3 and n‐6 polyunsaturated fatty acids. FEBS letters 509, 77–80 (2001).
Li, Y. et al. Vertebrate fatty acyl desaturase with Δ4 activity. Proceedings of the National Academy of Sciences 107, 16840–16845 (2010).
Hastings, N. et al. A vertebrate fatty acid desaturase with Δ5 and Δ6 activities. Proceedings of the National Academy of Sciences 98, 14304–14309 (2001).
Lopes-Marques, M. et al. Molecular and functional characterization of a fads2 orthologue in the Amazonian teleost, Arapaima gigas. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology 203, 84–91 (2017).
Monroig, Ó., Webb, K., Ibarra-Castro, L., Holt, G. J. & Tocher, D. R. Biosynthesis of long-chain polyunsaturated fatty acids in marine fish: Characterization of an Elovl4-like elongase from cobia Rachycentron canadum and activation of the pathway during early life stages. Aquaculture 312, 145–153 (2011).
Kabeya, N., Yevzelman, S., Oboh, A., Tocher, D. R. & Monroig, O. Essential fatty acid metabolism and requirements of the cleaner fish, ballan wrasse Labrus bergylta: Defining pathways of long-chain polyunsaturated fatty acid biosynthesis. Aquaculture 488, 199–206 (2018).
Leonard, A. E. et al. Identification and expression of mammalian long-chain PUFA elongation enzymes. Lipids 37, 733–740 (2002).
Tvrdik, P. et al. Role of a new mammalian gene family in the biosynthesis of very long chain fatty acids and sphingolipids. The Journal of cell biology 149, 707–718 (2000).
Jakobsson, A., Westerberg, R. & Jacobsson, A. Fatty acid elongases in mammals: their regulation and roles in metabolism. Progress in lipid research 45, 237–249 (2006).
Guillou, H., Zadravec, D., Martin, P. G. & Jacobsson, A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: Insights from transgenic mice. Progress in lipid research 49, 186–199 (2010).
Agaba, M., Tocher, D. R., Dickson, C. A., Dick, J. R. & Teale, A. J. Zebrafish cDNA encoding multifunctional fatty acid elongase involved in production of eicosapentaenoic (20: 5n-3) and docosahexaenoic (22: 6n-3) acids. Marine Biotechnology 6, 251–261 (2004).
Li, W., Feng, Z., Song, X., Zhu, W. & Hu, Y. Cloning, expression and functional characterization of the polyunsaturated fatty acid elongase (ELOVL5) gene from sea cucumber (Apostichopus japonicus). Gene 593, 217–224 (2016).
Monroig, Ó., Rotllant, J., Sánchez, E., Cerdá-Reverter, J. M. & Tocher, D. R. Expression of long-chain polyunsaturated fatty acid (LC-PUFA) biosynthesis genes during zebrafish Danio rerio early embryogenesis. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1791, 1093–1101 (2009).
Monroig, Ó. et al. Expression and role of Elovl4 elongases in biosynthesis of very long-chain fatty acids during zebrafish Danio rerio early embryonic development. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1801, 1145–1154 (2010).
Gregory, M. K. & James, M. J. Rainbow trout (Oncorhynchus mykiss) Elovl5 and Elovl2 differ in selectivity for elongation of omega-3 docosapentaenoic acid. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1841, 1656–1660 (2014).
Li, Y. et al. Genome wide identification and functional characterization of two LC-PUFA biosynthesis elongase (elovl8) genes in rabbitfish (Siganus canaliculatus). Aquaculture 522, 735127 (2020).
Jaillon, O. et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431, 946–957 (2004).
Kasahara, M. et al. The medaka draft genome and insights into vertebrate genome evolution. Nature 447, 714–719 (2007).
Nakatani, Y., Takeda, H., Kohara, Y. & Morishita, S. Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res 17, 1254–1265 (2007).
Li, R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967 (2009).
Salmela, L. & Rivals, E. LoRDEC: accurate and efficient long read error correction. Bioinformatics 30, 3506–3514 (2014).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
You, X. et al. Mudskipper genomes provide insights into the terrestrial adaptation of amphibious fishes. Nat Commun 5, 1–8 (2014).
Kajitani, R. et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome research 24, 1384–1395 (2014).
Ye, C., Hill, C. M., Wu, S., Ruan, J. & Ma, Z. S. DBG2OLC: efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Scientific reports 6, 1–9 (2016).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome biology 16, 1–11 (2015).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell systems 3, 99–101 (2016).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330 (2009).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic acids research 35, W265–W268 (2007).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 25, 4.10. 11–14.10. 14 (2009).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and genome research 110, 462–467 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic acids research 27, 573–580 (1999).
Tine, M. et al. European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nature communications 5, 1–10 (2014).
Ao, J. et al. Genome sequencing of the perciform fish Larimichthys crocea provides insights into molecular and genetic mechanisms of stress adaptation. PLoS genetics 11, e1005118 (2015).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic acids research 33, W465–W467 (2005).
Mount, D. W. Using the basic local alignment search tool (BLAST). Cold Spring Harbor Protocols 2007, pdb. top17 (2007).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome research 14, 988–995 (2004).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome research 18, 188–196 (2008).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic acids research 31, 365–370 (2003).
Kulikova, T. et al. The EMBL nucleotide sequence database. Nucleic Acids Research 32, D27–D30 (2004).
Ogata, H. et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic acids research 27, 29–34 (1999).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic acids research 37, D211–D215 (2009).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome research 13, 2178–2189 (2003).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research 32, 1792–1797 (2004).
Guindon, S. et al. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Systematic Biology 59, 307–321 (2010).
Yang, Z. & Rannala, B. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Molecular biology and evolution 23, 212–226 (2006).
Bian, C. et al. The Asian arowana (Scleropages formosus) genome provides new insights into the evolution of an early lineage of teleosts. Scientific reports 6, 1–17 (2016).
Tine, M. et al. European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nat Commun 5, 1–10 (2014).
Kabeya, N. et al. Polyunsaturated fatty acid metabolism in a marine teleost, Nibe croaker Nibea mitsukurii: Functional characterization of Fads2 desaturase and Elovl5 and Elovl4 elongases. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology 188, 37–45 (2015).
Carmona-Antoñanzas, G., Monroig, Ó., Dick, J. R., Davie, A. & Tocher, D. R. Biosynthesis of very long-chain fatty acids (C> 24) in Atlantic salmon: Cloning, functional characterisation, and tissue distribution of an Elovl4 elongase. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology 159, 122–129 (2011).
Betancor, M. B. et al. Molecular and functional characterisation of a putative elovl4 gene and its expression in response to dietary fatty acid profile in Atlantic bluefin tuna (Thunnus thynnus). Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology 240, 110372 (2020).
Li, S. et al. Functional characterization and differential nutritional regulation of putative Elovl5 and Elovl4 elongases in large yellow croaker (Larimichthys crocea). Scientific reports 7, 1–15 (2017).
Oboh, A., Navarro, J. C., Tocher, D. R. & Monroig, O. Elongation of very long-chain (> C 24) fatty acids in Clarias gariepinus: cloning, functional characterization and tissue expression of elovl4 elongases. Lipids 52, 837–848 (2017).
Hamid, N. K. A. et al. Isolation and functional characterisation of a fads2 in rainbow trout (Oncorhynchus mykiss) with Δ5 desaturase activity. PLoS One 11, e0150770 (2016).
China National GeneBank, https://db.cngb.org/search/project/CNP0002889/ (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP370737 (2022).
Chen, B. et al. Chromosome-Level Assembly of the Chinese Seabass (Lateolabrax maculatus) Genome. Front Genet 10, 275 (2019).
Acknowledgements
This study was supported by Guangdong Provincial Special Fund for Modern Agriculture Industry Technology Innovation Teams (no. 2019KJ141 to Y.L) and Shenzhen Science and Technology Innovation Program for International Cooperation (no. GJHZ20190819152407214 to Q.S.).
Author information
Authors and Affiliations
Contributions
These authors contributed equally: Yishan Lu, Ruihan Li, Liqun Xia, Jun Cheng; Y.L., Q.S. and C.B. conceived this project; L.X., J.C., H.X. and D.Y. participated in data analysis; Y.X., R.G. and J.X. collected the samples; C.B. and Q.Z. assembled the genome; R.L. generated the annotation set; C.B. constructed the synteny blocks and ancestor sequences; C.B., Q.S., Q.Z, R.L. and Y.L. wrote the manuscript. Q.S. and Y.L. revised the manuscript. All authors have read and approved the final manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lu, Y., Li, R., Xia, L. et al. A chromosome-level genome assembly of the jade perch (Scortum barcoo). Sci Data 9, 408 (2022). https://doi.org/10.1038/s41597-022-01523-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-022-01523-y
