
Abstract
Acetogens are anaerobic bacteria that utilise gaseous feedstocks such as carbon monoxide (CO) and carbon dioxide (CO2) to synthesise biomass and various metabolites via the energetically efficient Wood-Ljungdahl pathway. Because of this pathway, acetogens have been considered as a novel platform to produce biochemicals from gaseous feedstocks, potentially replacing the conventional thermochemical processes. Despite their advantages, a lack of systematic understanding of the transcriptional and translational regulation in acetogens during autotrophic growth limits the rational strain design to produce the desired products. To overcome this problem, we presented RNA sequencing and ribosome profiling data of four acetogens cultivated under heterotrophic and autotrophic conditions, providing data on genome-scale transcriptional and translational responses of acetogens during CO2 fixation. These data facilitate the discovery of regulatory elements embedded in their genomes, which could be utilised to engineer strains to achieve better growth and productivity. We anticipate that these data will expand our understanding of the processes of CO2 fixation and will help in the designing of strains for the desired biochemical production.
Measurement(s) | RNA • transcriptome • translatome |
Technology Type(s) | RNA sequencing • ribosomal profiling by sequencing assay |
Factor Type(s) | carbon source • growth conditions |
Sample Characteristic - Organism | Acetobacterium woodii • Clostridium aceticum • Clostridium drakei • Eubacterium limosum |
Sample Characteristic - Environment | anaerobic |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.13625942
Similar content being viewed by others
Background & Summary
Over the past decades, the usage of fossil fuels, which is now responsible for 40% of carbon dioxide (CO2) emission1,2, has rapidly increased, causing damage to the environment through the generation of greenhouse gases. The demand for sustainable replacements for fossil fuels to obtain environmental and economic sustainability has, therefore, also been increasing. Microbial gas fermentation has recently drawn much attention as an alternative method to convert gaseous feedstocks (CO or CO2) into biofuels and biochemicals, owing to its low operation cost compared to the conventional thermochemical process3,4,5,6. Among the organisms capable of gas fermentation, acetogenic bacteria (acetogens) are considered one of the most promising potential platforms7,8.
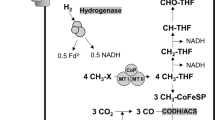
A unique feature of acetogens is the presence of the Wood-Ljungdahl pathway (WLP) coding genes in their genomes9. Among the CO2 fixing pathways discovered to date, the WLP is the most energetically efficient pathway, investing only one molecule of ATP per acetate produced, whereas the other pathways are ATP sinking pathways10,11,12. The WLP reduces CO2 into formate using the electrons derived from the oxidation of molecular hydrogen, then to formyl-tetrahydrofolate (THF) using one molecule of ATP. Subsequently, methenyl-THF, methylene-THF, and methyl-THF are synthesised using two additional reduction powers, which vary according to the acetogen. Subsequently, the methyl group of methyl-THF is transferred to the CO dehydrogenase/acetyl-CoA synthase (CODH/ACS) complex to form acetyl-CoA, combining with the reduced CO and coenzyme A (CoA). The synthesised acetyl-CoA can be converted into acetate to recoup ATP or biomass building blocks.
In addition to the WLP, energy conservation systems play essential roles in acetogens by compensating for the energy required for biomass synthesis. These systems utilise the ferredoxin (Fd): NAD+ oxidoreductase (Rnf) and the Fd: H+ oxidoreductase (Ech) complexes to translocate ions across the membrane to create ion gradient13,14. The established gradient drives the ion back into the cell through the ATP synthase complex, generating the ATP needed for the cell. Along with the ATP synthesis system, electron bifurcation, which oxidises one electron donor and transfers electrons to two different electron acceptors, helps to overcome energetic barriers by reducing low reduction potential Fd via oxidisation of a relatively higher reduction potential hydrogen molecule, which can then reduce CO2 and the ion translocating complex10,15,16.
Along with the WLP and the energy conservation system, acetogens, similar to any other organism, contain intricate regulatory networks to control gene expression under different conditions. To date, a large number of acetogens have been sequenced to identify their genomic features; this further required a systematic understanding of their transcriptional and translational regulatory processes. In contrast to genomic studies, relatively few studies on acetogens’ transcriptomes and translatomes under autotrophic growth conditions have been published. The lack of uniformly generated RNA sequencing (RNA-Seq) and ribosome profiling (Ribo-Seq) data for acetogens has not only limited from obtaining knowledge on the cellular responses but also expanding potential genetic tools for strain engineering. RNA-Seq and Ribo-Seq can determine the strength of promoters and Shine-Dalgarno (SD) sequences in the 5´-untranslated regions, which regulate transcription and translation, respectively.
In this study, we determined changes in the transcriptional and translational responses of acetogens under autotrophic growth condition compared to heterotrophic growth conditions, using RNA-Seq and Ribo-Seq. RNA-Seq and Ribo-Seq were performed on four acetogen species, Acetobacterium woodii, Clostridium aceticum, Clostridium drakei, and Eubacterium limosum cultured under the two conditions. Although studies on E. limosum and a transcriptomic study on C. drakei have been described in previous studies by our group, this study provides a uniformly generated and processed dataset of the additional model acetogens, which allows the comparative analysis of the transcriptome and translatome of CO2 fixing acetogens17,18. The presented RNA-Seq and Ribo-Seq results will provide a fundamental understanding of the responses of the acetogens to autotrophic conditions, and thereby widen genetic tools for strain engineering to produce biochemicals using CO2 as a carbon building block.
Methods
Bacterial culture conditions
For this study, A. woodii DSM 1030, C. aceticum DSM 1496, C. drakei DSM 12750, and E. limosum DSM 20543 were obtained from the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). A. woodii, C. aceticum, and C. drakei were cultured under strict anaerobic conditions at 30 °C and E. limosum was cultured under anaerobic conditions at 37 °C in 150 mL serum bottles filled with 100 mL DSMZ 135 medium (pH 7.0), which is composed of 1 g L−1 NH4Cl, 2 g L−1 yeast extract, 10 g L−1 NaHCO3, 0.1 g L−1 MgSO4 × 7H2O, 0.3 g L−1 cysteine-HCl, 10 mL vitamin solution (4 mg L−1 biotin, 4 mg L−1 folic acid, 20 mg L−1 pyridoxine-HCl, 10 mg L−1 Thiamine-HCl, 10 mg L−1 riboflavin, 10 mg L−1 nicotinic acid, 10 mg L−1 pantothenate, 0.2 mg L−1 Vitamin B12, 10 mg L−1 p-aminobenzoic acid and 10 mg L−1 lipoic acid), 20 mL trace element solution (1.0 g L−1 Nitrilotriacetic acid, 3.0 g L−1 MgSO4 × 7H2O, 0.5 g L−1 MnSO4 × H2O, 1.0 g L−1 NaCl, 0.1 g L−1 FeSO4 × 7H2O, 180 mg L−1 CoSO4 × 7H2O, 0.1 g L−1 CaCl2 × 2H2O, 180 mg L−1 ZnSO4 × 7H2O, 10 mg L−1 CuSO4 × 5H2O, 20 mg L−1 KAI(SO4)2 × 12H2O, 10 mg L−1 H3BO3, 10 mg L−1 Na2MO4 × 2H2O, 30 mg L−1 NiCl2 × 6H2O, 0.3 mg L−1 Na2SeO3 × 5 H2O, 0.4 mg L−1 Na2WO4 × 2H2O), 4.6 mM KH2PO4, 5.4 mM K2HPO4, and 4 µM resazurin. For heterotrophic growth, A. woodii, C. aceticum, and C. drakei were cultivated in the fructose supplemented (5 g L−1) media and E. limosum was cultured in glucose supplemented (5 g L−1) media. For autotrophic growth, all of the strains were cultivated in DSMZ 135 media containing H2/CO2 (80:20) with pressure of 200 kPa in the headspace (50 mL). The media used for culturing A. woodii was supplemented with 2 g L−1 NaCl. For the main culture, the precultured cells were harvested via anaerobic centrifugation, then washed with basal DSMZ 135 media three times and inoculated in 100 mL fresh DSMZ 135 media supplemented with corresponding carbons. All of the strains were cultured in biological duplicates.
RNA-Seq library preparation
Duplicate samples were harvested at the mid-exponential phase by centrifugation at 4,000 g for 15 min at 4 °C. The collected cells were resuspended anaerobically in 500 µL of lysis buffer, comprising 20 mM Tris-HCl (pH 7.4), 140 mM NaCl, 5 mM MgCl2, and 1% Triton X-100. Liquid nitrogen was used to freeze the samples, which were then ground using a mortar and pestle. Lysed cells were thawed on ice, and the debris was removed by centrifugation at 4,000 g for 15 min at 4 °C. Subsequently, the total RNA was isolated using TRIzol (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s instruction. To remove the remaining genomic DNA (gDNA), the RNA was treated with 4 U of rDNase I (Ambion, Austin, TX, USA) for 1 h at 37 °C, then incubated at 75 °C for 10 min to deactivate the enzyme. To remove ribosomal RNAs (rRNA) in the gDNA-depleted RNA, the Ribo-ZeroTM rRNA Removal Kit for Meta-bacteria (Epicentre, Madison, WI, USA) was used according to the manufacturer’s instruction. The quality of the rRNA-depleted RNA was checked using an Agilent 2200 TapeStation system (Agilent Technologies, Santa Clara, CA, USA). To construct the libraries for RNA-seq, the TrueSeq Stranded mRNA Library Prep Kit (Illumina, San Diego, CA, USA) was used on the quality confirmed RNA. The libraries were sequenced using the 150 bp read recipe with an Illumina MiSeqTM system.
Ribo-Seq library preparation
For Ribo-Seq, 100 µM chloramphenicol (CM) was added to the cultures which were then further incubated at 30 °C or 37 °C, corresponding to the duplicate culture conditions, for 10 min. The CM treated cells were subsequently washed using 500 µL polysome buffer composed of 20 mM Tris-HCl (pH 7.4), 140 mM NaCl, 5 mM MgCl2, and 100 µM CM, and resuspended in lysis buffer consisting of 20 mM Tris-HCl (pH 7.4), 140 mM NaCl, 5 mM MgCl2, 100 µM CM, and 1% Triton X-100. The resuspended cells were frozen in liquid nitrogen and ground with a pestle and mortar. The powdered cells were recovered by centrifugation at 4,000 g for 15 min at 4 °C, and the resultant supernatant was additionally centrifuged at 16,000 g for 10 min at 4 °C. To degrade RNA unprotected by ribosomes, 400 U MNase (NEB, Ipswich, MA, USA), 2 µL bovine serum albumin (1 mg mL−1), and 20 µL of 10 × MNase buffer were added and samples were incubated at 37 °C for 2 h with gentle rotation. To inactivate the reaction, 10 µL 0.5 M EGTA (Sigma-Aldrich, St. Louis, MO, USA) was added to the sample. The monosome fraction was recovered using Microspin S-400 HR columns (GE Healthcare Life Sciences, Marlborough, MA, USA). The recovered ribosome-bound RNA was isolated using TRIzol, and the remaining rRNAs were removed with the Ribo-ZeroTM rRNA Removal Kit for Meta-bacteria. For the phosphorylation reaction, samples were denatured at 80 °C for 90 s, equilibrated to 37 °C, and incubated at 37 °C for 1 h with 5 µL 10 × T4 PNK buffer (NEB), 20 U SUPERase-In RNase Inhibitor, and 10 U T4 PNK (NEB). After purification of the RNA samples using RNeasy MinElute Column (Qiagen, Hilden, Germany), the concentration of purified RNA was measured using the Qubit RNA HS assay kit (Invitrogen, Carlsbad, CA, USA). For library construction, the small RNA library prep kit for Illumina (NEB) was used, and the constructed library was sequenced using the 50 bp read recipe on an Illumina Hiseq2500.
Data processing
For RNA-Seq, the adapter sequence of the sequenced reads and quality below Phred score of 20 were trimmed. Trimmed reads shorter than 20 bp were discarded to improve the accuracy of the mapping result. Using CLC Genomics Workbench (CLC Bio, Aarhus, Denmark), the trimmed reads were mapped onto the A. woodii (NC_016894), C. aceticum (NZ_CP009687), C. drakei (NZ_CP020953), and E. limosum (NZ_CP019962) genomes using default parameters (mismatch cost = 2, deletion cost = 3, insertion cost = 3, length fraction = 0.9, and similarity fraction = 0.9) and only the uniquely mapped reads were rescued. The gene expression was calculated from the mapped read count statistics using the DESeq. 2 package in R with default parameters. For Ribo-Seq, the adaptors of the generated reads and quality below Phred score of 20 were removed, then trimmed with the same parameters applied for RNA-Seq, and reads shorter than 20 bp were again removed. The reads were mapped onto the A. woodii (NC_016894), C. aceticum (NZ_CP009687), C. drakei (NZ_CP020953), and E. limosum (NZ_CP019962) genomes using the default parameters (mismatch cost = 2, deletion cost = 3, insertion cost = 3, length fraction = 0.9, and similarity fraction = 0.9) and only the uniquely mapped reads were rescued. Using the obtained reads, the gene expression was normalised using the DESeq. 2 package in R with default parameters.
Data Records
The RNA-Seq and Ribo-Seq datasets for A. woodii were deposited in the European Nucleotide Archive under study accession PRJEB3346019. The RNA-Seq and Ribo-Seq datasets for C. aceticum were deposited in the European Nucleotide Archive under study accession PRJEB3613420. The RNA-Seq and Ribo-Seq datasets for C. drakei, published in previous study18, were deposited in the NCBI Gene Expression Omnibus (GEO) repository with the accession code GSE118519 and the European Nucleotide Archive under study accession PRJEB3613521,22. The RNA-Seq and Ribo-Seq datasets for E. limosum, published in previous study, were deposited in the NCBI Gene Expression Omnibus (GEO) repository with the accession code GSE9761317,23. Detailed information on analysis of RNA-Seq and Ribo-Seq has been deposited in Figshare24.
Technical Validation
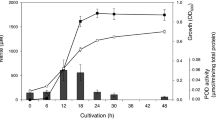
Acetogens have drawn much attention due to their ability to fix CO2 using the efficient WLP and the energy conservation system. To systematically understand this metabolism, four acetogen species, A. woodii, C. aceticum, C. drakei, and E. limosum were cultivated under heterotrophic or autotrophic condition, then sampled at the corresponding mid-exponential point (Fig. 1). RNA-Seq and Ribo-Seq libraries were created and sequenced using the Illumina platforms. For RNA-Seq, total bases of 1,989,686,915 nt, 3,309,840,659 nt, 3,061,597,892 nt, and 4,122,893,029 nt for A. woodii, C. aceticum, C. drakei, and E. limosum, respectively, were generated as raw data (Table 1). After obtaining the raw data, the adaptor sequences and poor-quality reads of lower than 99.9% accuracy were removed, resulting in 1,925,462,358 nt, 3,192,358,774 nt, 2,898,139,861 nt, and 4,033,415,284 nt, for A. woodii, C. aceticum, C. drakei, and E. limosum, with average read lengths of 147.8 nt, 132.8 nt, 141.0 nt, and 137.7 nt, respectively (Table 1). For Ribo-Seq, 30,075,896,511 nt, 15,893,868,174 nt, 26,109,101,619 nt, and 36,178,949,211 nt were produced in total as raw data for A. woodii, C. aceticum, C. drakei, and E. limosum, respectively (Table 2). The reads were then trimmed to remove the adaptor sequence and low-quality sequences, resulting 10,937,323,767 nt, 5,821,740,643 nt, 13,645,439,108 nt, and 5,132,234,720 nt total, for A. woodii, C. aceticum, C. drakei, and E. limosum, with average read lengths of 26.6 nt, 34.6 nt, 32.3 nt, and 36.4 nt, respectively (Table 2). The average length of the trimmed reads for RNA-Seq corresponds to the targeted sequencing length of 150 nt. In contrast, the lengths of the filtered Ribo-Seq reads ranged between 26 nt and 36 nt, which is shorter than the intended length of 50 bp. However, the length of the Ribo-Seq reads correspond to the actual lengths of mRNA protected by the ribosome during translation (between 20 to 40 nt)25,26.

Overall experimental scheme of RNA sequencing (RNA-Seq) and ribosome profiling (Ribo-Seq) of acetogens. General strategy for library construction; under the heterotrophic (fructose or glucose supplemented media) and autotrophic growth conditions, cells were harvested at the mid-exponential phase, and construction of libraries for RNA-Seq and Ribo-Seq was carried out.
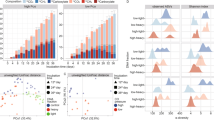
Following the quality control process, the filtered reads were mapped to the corresponding genome sequences (see Method section for detail). For RNA-Seq, the total number of bases mapped to A. woodii, C. aceticum, C. drakei, and E. limosum were 1,656,585,194 nt, 2,177,922,778 nt, 2,751,173,442 nt, and 3,740,112,946 nt, respectively, with a minimum coverage value of 42.8 folds, which is sufficient to determine differential gene expression (Table 1). The mapped RNA-Seq reads were normalised using the DESeq. 2 package in R to determine reproducibility of the samples under the target conditions using hierarchical clustering (Fig. 2a–h) and principal component analysis (Fig. 2i–l), which validates that the biological replicates were reproducible within the same growth conditions. The range of the mapped normalised reads of the acetogens under the conditions were consistent (Fig. 3a–d). Subsequently, to identify differentially expressed genes (DEGs), fold changes between the heterotrophic and the autotrophic conditions were calculated with their associated P-values. Genes with fold changes of >2 or <0.5, respectively, and P-value < 0.01 were defined as up- and down-regulated DEGs, respectively. A total of 685, 793, 600, and 406 upregulated DEGs, and 500, 940, 582, and 505 downregulated DEGs under autotrophic conditions were identified by RNA-seq for A. woodii, C. aceticum, C. drakei, and E. limosum, respectively (Fig. 3e–h).

Quality of RNA sequencing (RNA-Seq) and ribosome profiling (Ribo-Seq) data. Reproducibility of RNA-Seq of A. woodii (a), C. aceticum (b), C. drakei (c), and E. limosum (d) cultivated under heterotrophic (H1 and H2) and autotrophic (A1 and A2) conditions. Pairwise correlation of RNA-Seq of A. woodii (e), C. aceticum (f), C. drakei (g), and E. limosum (h) cultivated under heterotrophic (H1 and H2) and autotrophic (A1 and A2) conditions. Principal components analysis of RNA-Seq and Ribo-Seq of A. woodii (i), C. aceticum (j), C. drakei (k), and E. limosum (l) cultivated under the heterotrophic (H: coloured purple and blue) and autotrophic (A: coloured green and red) conditions for RNA-Seq and Ribo-Seq. Reproducibility of Ribo-seq of A. woodii (m), C. aceticum (n), C. drakei (o), and E. limosum (p) cultivated under heterotrophic (H1 and H2) and autotrophic (A1 and A2) conditions. Pairwise correlation of Ribo-Seq of A. woodii (q), C. aceticum (r), C. drakei (s), and E. limosum (t) cultivated under heterotrophic (H1 and H2) and autotrophic (A1 and A2) conditions.

Normalized distribution of RNA sequencing (RNA-Seq) and ribosome profiling (Ribo-Seq) data. Distribution of normalised reads of RNA-Seq under the heterotrophic (H1 and H2) and the autotrophic (A1 and A2) conditions of A. woodii (a), C. aceticum (b), C. drakei (c), and E. limosum (d). Volcano plots of RNA-Seq of A. woodii (e), C. aceticum (f), C. drakei (g), and E. limosum (h) under the heterotrophic and the autotrophic conditions, with blue and red dots indicating significantly down and upregulated genes, respectively. Distribution of normalised reads of Ribo-Seq result under heterotrophic (H1 and H2) and autotrophic (A1 and A2) conditions of A. woodii (i), C. aceticum (j), C. drakei (k), and E. limosum (l). Volcano plots of A. woodii (m), C. aceticum (n), C. drakei (o), and E. limosum (p) under heterotrophic and autotrophic conditions, with blue and red dots indicating significantly down and upregulated genes, respectively.
Similar to the RNA-Seq data, the Ribo-Seq reads were normalised using DESeq. 2, and the reproducibility of the replicates was confirmed via hierarchical clustering (Fig. 2m–t). Following this, principal component analysis validated that the Ribo-Seq expression patterns of the replicates were highly correlated only within the conditions (Fig. 2i–l). In addition, the range of the normalised Ribo-Seq reads were consistent in all acetogens (Fig. 3i–l). The verified reads were used to determine translational DEGs of the four acetogens under autotrophic conditions. This identified 745, 280, 501, and 854 significantly upregulated genes and 478, 196, 1,417, and 868 significantly downregulated genes from A. woodii, C. aceticum, C. drakei, and E. limosum, respectively (Fig. 3m–p).
Code availability
The version and parameter of all bioinformatics tools used in this work are described in the Methods section.
References
Spigarelli, B. P. & Kawatra, S. K. Opportunities and challenges in carbon dioxide capture. J Co2 Util 1, 69–87, https://doi.org/10.1016/j.jcou.2013.03.002 (2013).
Saeidi, S., Amin, N. A. S. & Rahimpour, M. R. Hydrogenation of CO2 to value-added products—A review and potential future developments. J Co2 Util 5, 66–81, https://doi.org/10.1016/j.jcou.2013.12.005 (2014).
Henstra, A. M., Sipma, J., Rinzema, A. & Stams, A. J. Microbiology of synthesis gas fermentation for biofuel production. Curr. Opin. Biotechnol. 18, 200–206, https://doi.org/10.1016/j.copbio.2007.03.008 (2007).
Bengelsdorf, F. R., Straub, M. & Dürre, P. Bacterial synthesis gas (syngas) fermentation. Environ. Technol. 34, 1639–1651, https://doi.org/10.1080/09593330.2013.827747 (2013).
Bredwell, M. D., Srivastava, P. & Worden, R. M. Reactor Design Issues for Synthesis-Gas Fermentations. Biotechnol Prog 15, 834–844, https://doi.org/10.1021/bp990108m (1999).
Munasinghe, P. C. & Khanal, S. K. Biomass-derived syngas fermentation into biofuels: Opportunities and challenges. Bioresour Technol 101, 5013–5022, https://doi.org/10.1016/j.biortech.2009.12.098 (2010).
Drake, H. L., Gossner, A. S. & Daniel, S. L. Old acetogens, new light. Ann. N. Y. Acad. Sci. 1125, 100–128, https://doi.org/10.1196/annals.1419.016 (2008).
Dürre, P. & Eikmanns, B. J. C1-carbon sources for chemical and fuel production by microbial gas fermentation. Curr. Opin. Biotechnol. 35, 63–72, https://doi.org/10.1016/j.copbio.2015.03.008 (2015).
Shin, J., Song, Y., Jeong, Y. & Cho, B. K. Analysis of the Core Genome and Pan-Genome of Autotrophic Acetogenic Bacteria. Front. Microbiol. 7, 1531, https://doi.org/10.3389/fmicb.2016.01531 (2016).
Schuchmann, K. & Müller, V. Autotrophy at the thermodynamic limit of life: a model for energy conservation in acetogenic bacteria. Nat. Rev. Microbiol. 12, 809–821, https://doi.org/10.1038/nrmicro3365 (2014).
Fuchs, G. Alternative pathways of carbon dioxide fixation: insights into the early evolution of life? Annu. Rev. Microbiol. 65, 631–658, https://doi.org/10.1146/annurev-micro-090110-102801 (2011).
Ragsdale, S. W. Enzymology of the Wood-Ljungdahl pathway of acetogenesis. Ann. N. Y. Acad. Sci. 1125, 129–136, https://doi.org/10.1196/annals.1419.015 (2008).
Müller, V. Energy conservation in acetogenic bacteria. Appl Environ Microbiol 69, 6345–6353 (2003).
Müller, V., Imkamp, F., Biegel, E., Schmidt, S. & Dilling, S. Discovery of a ferredoxin:NAD+-oxidoreductase (Rnf) in Acetobacterium woodii: a novel potential coupling site in acetogens. Ann N Y Acad Sci 1125, 137–146, https://doi.org/10.1196/annals.1419.011 (2008).
Li, F. et al. Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J. Bacteriol. 190, 843–850, https://doi.org/10.1128/JB.01417-07 (2008).
Thauer, R. K., Kaster, A. K., Seedorf, H., Buckel, W. & Hedderich, R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591, https://doi.org/10.1038/nrmicro1931 (2008).
Song, Y. et al. Genome-scale analysis of syngas fermenting acetogenic bacteria reveals the translational regulation for its autotrophic growth. BMC Genom. 19, 837, https://doi.org/10.1186/s12864-018-5238-0 (2018).
Song, Y. et al. Functional cooperation of the glycine synthase-reductase and Wood-Ljungdahl pathways for autotrophic growth of Clostridium drakei. Proc. Natl. Acad. Sci. USA 117, 7516–7523, https://doi.org/10.1073/pnas.1912289117 (2020).
European Nucleotide Archive https://identifiers.org/ena.embl:PRJEB33460 (2019).
European Nucleotide Archive https://identifiers.org/ena.embl:PRJEB36134 (2020).
Song, Y. et al. Functional cooperation of glycine synthase-reductase pathway with Wood-Ljungdahl pathway for autotrophic growth of Clostridium drakei. Gene Expression Omnibus https://identifiers.org/geo:GSE118519 (2019).
European Nucleotide Archive https://identifiers.org/ena.embl:PRJEB36135 (2020).
Song, Y. et al. Genome-scale analysis of syngas fermenting acetogenic bacteria reveals the translational regulation for its autotrophic growth. Gene Expression Omnibus https://identifiers.org/geo:GSE97613 (2018).
Song, Y. et al. Transcriptome and translatome of CO2 fixing acetogens under heterotrophic and autotrophic conditions. figshare https://doi.org/10.6084/m9.figshare.c.4933110 (2021).
O’Connor, P. B., Li, G. W., Weissman, J. S., Atkins, J. F. & Baranov, P. V. rRNA:mRNA pairing alters the length and the symmetry of mRNA-protected fragments in ribosome profiling experiments. Bioinformatics 29, 1488–1491, https://doi.org/10.1093/bioinformatics/btt184 (2013).
Mohammad, F., Woolstenhulme, C. J., Green, R. & Buskirk, A. R. Clarifying the Translational Pausing Landscape in Bacteria by Ribosome Profiling. Cell Rep. 14, 686–694, https://doi.org/10.1016/j.celrep.2015.12.073 (2016).
Acknowledgements
This work was supported by the Intelligent Synthetic Biology Centre of the Global Frontier Project (2011-0031957 to B.-K.C.) and the C1 Gas Refinery Program (2018M3D3A1A01055733 to B.-K.C.) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT).
Author information
Authors and Affiliations
Contributions
B.-K.C. conceived and supervised the study. Y.S., S.C., and B.-K.C. designed the experiments. Y.S., J.S., and S.J. performed experiments. Y.S., J.B., J.S., S.C., and B.-K.C. analysed the data. Y.S., J.B., J.-K.L, S.C.K., S.C., and B.-K.C. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Song, Y., Bae, J., Shin, J. et al. Transcriptome and translatome of CO2 fixing acetogens under heterotrophic and autotrophic conditions. Sci Data 8, 51 (2021). https://doi.org/10.1038/s41597-021-00837-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-021-00837-7
