
Abstract
Advances in tissue labeling and clearing methods include improvement of tissue transparency, better preservation of fluorescence signal, compatibility with immunostaining and large sample volumes. However, as existing methods share the common limitation that they can only be applied to human tissue slices, rendering intact human organs transparent remains a challenge. Here, we describe experimental details of the small-micelle-mediated human organ efficient clearing and labeling (SHANEL) pipeline, which can be applied for cellular mapping of intact human organs. We have successfully cleared multiple human organs, including kidney, pancreas, heart, lung, spleen and brain, as well as hard tissue like skull. We also describe an advanced volumetric imaging system using a commercial light-sheet fluorescence microscope that can accommodate most human organs and a pipeline for whole-organ imaging and visualization. The complete experimental process of labeling and clearing whole human organs takes months and the analysis process takes several weeks, depending on the organ types and sizes.
Similar content being viewed by others


Introduction
Three-dimensional (3D) mapping of the human organs at cellular resolution and generating reference maps of organs across ages or diseases represent the common objectives of diverse consortia including Human Biomolecular Atlas Program (HuBMAP)1,2, Human Cell Atlas3, Human Tumour Atlas4 and LungMap5. Traditional histological techniques of slicing, staining, imaging and 3D reconstruction of the cellular details of biological tissue pieces are challenging to scale up to large human organs. For example, mapping one intact adult human brain using such traditional methods took years of tedious work, and the lengthy process resulted in mechanical distortion and accidental loss of sections of tissue6. While improvements are constantly being made in the instruments and methods for iterative image analysis of multimodal imaging of human organs using PET/MRI, these modalities are still limited by low resolution and the inability to probe cellular and molecular parameters.
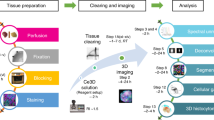
In recent years, extensive biomedical research in neuroscience7,8,9,10,11,12, development13 and cancer14,15 has substantially benefited from the optical tissue labeling and clearing methods that bypass the major problems of histology. Especially, clearing and imaging of whole adult mouse bodies has opened a holistic examination window into physiological and pathological systems in an unbiased way9,14. However, human organs obviously are much larger in size and, owing to the effects of aging, contain more complex tissue components, which limit the post-staining methods that can be applied to human organs compared with organs from months-to-year-old rodents. Therefore, achieving whole human organ transparency has been challenging, despite numerous trials on slices11,16,17,18,19. Hence, we developed a full pipeline for whole human organ labeling, clearing, imaging and 3D map reconstruction at cellular level, which implemented a new tissue labeling and clearing technology termed SHANEL and an advanced volumetric imaging system using a commercial light-sheet fluorescence microscope (LSFM)20. This pipeline can easily be adopted in laboratories, within routinely available reagents and equipment (Fig. 1).

SHANEL is composed of seven parts. After human organs and tissue are collected (Part 1), they are prepared for blood removal and fixation (Part 2). The next step is performed by active pumping (Part 3a) or passive incubation (Part 3b), based on whether the organs are intact or not. Samples are then subjected to permeabilization, delipidation, ECM loosening and labeling with chemical probes. If antibodies compatible with the SHANEL protocol are used, the organ of interest should be dissected into slices less than 1.5 cm thick for immunolabeling (Part 4). Samples are then dehydrated, delipidated and matched for RI, until they become transparent (Part 5). Imaging of whole human organs is performed in an UltraMicroscope Blaze LSFM, and images of tissue slices are captured with an UltraMicroscope II LSFM at high resolution (Part 6). The data are stitched, volume fused, and rendered in a 3D visualization (Part 7).
Development of the SHANEL protocol
Clearing, labeling and imaging of human organs is difficult to implement in practice owing to several challenges. In Table 1, we delineate the key challenges encountered during the time-consuming endeavor of human organ processing and the ways in which these problems could be addressed.
Perfusion and fixation of intact organs
To preserve human organs rapidly and uniformly in a life-like state, we introduced an active perfusion system to deliver 0.01 M phosphate-buffered saline (PBS)/heparin and 4% paraformaldehyde (PFA) solutions into whole organs through the vascular network before the organs are harvested if major blood vessels are available (Fig. 2 and Extended Data Fig. 1a). The advantages of this step include washing out as much remaining blood as possible, preventing clot formation by maintaining circulation of the vascular system and achieving faster tissue fixation than through passive immersion. The organs are dissected carefully to preserve intact anatomical shapes and connect the main arteries with exogenous tubes for later experiments. Alternatively, human samples could be passively fixed in 4% PFA or 10% formalin buffer to covalently crosslink proteins. It is worth mentioning that human organ and tissue donation organizations such as the International Institute for the Advancement of Medicine are reliable resources that provide transplantable organs with intact vascular systems and detailed donor information.

a, Photo of dissected human kidney with inserted tubing (yellow arrows). Scale bar, 1 cm. b,c, The original tubing was replaced with a PTFE (chemical-resistant, anti-adhesive, biocompatible) tube (b) and tightly fixed by double rope-fastening (c; red rectangle in b). d, The inserted nozzle of the PTFE tube was cut at an angle to ensure smooth insertion into the organ. e, A set of connecting tubing containing a black pump reference tube in the middle, two colorless PVC tubes at both ends, three tube connectors and a pipette tip. Switch between position 1 and position 2 to avoid breakage of the reference tubing. The red arrows indicate the flow circulation direction. f, Blood vessel labeling with dextran solution using a pump perfusion system. g, Rotation of the wheel enables solution flow into the kidney. h, The correct way to fasten the black pump reference tube to the cassette is shown. i, Photos of human kidney after dextran labeling. j, The dextran-labeled human kidney was sealed in a plastic bag.
Organ clearing strategy
Among the hydrophilic reagent-based21,22,23,24, hydrogel-embedding19,25,26,27 and hydrophobic reagent-based9,10,13,14 tissue clearing methods, we chose to work with hydrophobic reagents in the SHANEL pipeline. An important advantage of hydrophobic tissue clearing is sample shrinkage, enabling us to accommodate and image large organs using an LSFM. Ethanol is employed to remove the water inside of human tissue, by increasing ethanol concentration in a stepwise manner. Dichloromethane (DCM) is used to extract the remaining lipids and ethanol, after which the tissue is mostly composed of fixed proteins. In the end, the relatively homogeneous human tissue is rendered transparent by immersing it in BABB solution (benzyl benzoate:benzyl alcohol 2:1, v/v). BABB solution has a refractive index (RI) of 1.56, which is the same as that of the cross-linked proteins.
Imaging system
In general, organs cleared with hydrophobic reagents will shrink ~30% in volume10,20. However, commercially available microscopes such as the LaVision UltraMicroscope II system (chamber size of 72 × 74 × 35 mm, sample traveling range of 10 × 10 × 10 mm in X, Y, Z) or ZEISS Lightsheet 7 (sample size of 10 × 10 × 20 mm) cannot hold large cleared human organs such as an intact eye (size of 30 × 30 × 30 mm). To overcome this limitation, together with Miltenyi Biotec we developed a prototype UltraMicroscope (chamber size of 250 × 90 × 70 mm) (Fig. 3). This microscope is now commercialized by Miltenyi Biotec as UltraMicroscope Blaze (chamber size of 129 × 51 × 64 mm, sample travel range of 50 × 24 × 23 mm)—a fully automated light-sheet microscope for imaging large cleared samples covering the range from an entire mouse to most human organs.

a, Photo of human pancreas before clearing. Scale bar, 1 cm. b, Photo of transparent human pancreas after PI cell nuclei labeling and clearing, showing mesenteric artery (white arrow) and lymph nodes (yellow asterisks). Scale bar, 1 cm. c, The mounting of an organ on a sample holder requires glue and black tape. The red arrows show the boundaries of the moving range of the holder in the Y direction. d, The sample holder was wrapped with four sections of tape, and a drop of glue was placed on top of each section. e, An example of sample positioning with alignment to one end of the holder to cover one sample edge. f, Another example of sample positioning with alignment to the other end to cover the other sample edge. g, The mounted human pancreas was illuminated by light-sheet microscopy.
Permeabilization and decolorization reagents
Efforts to label and clear human brain tissue by screening thousands of delipidation and decoloring chemicals28 or chemicals that increase tissue permeability17,19 have already shown the difficulties related to incomplete tissue transparency, time-consuming procedures and limited antibody penetration. Given that aged human tissue is composed of dense and intricate hydrophobic and hydrophilic molecules, we hypothesized that efficient detergent permeabilization is necessary to render the human tissue accessible to reagents traveling end-to-end through it. Detergents are amphiphilic, possessing both hydrophilic and hydrophobic properties, and form micelles in solution that can interact with molecules of the tissue. Traditional detergents such as ionic sodium dodecyl sulfate (SDS) or non-ionic Triton X-100 (4-(1,1,3,3-tetramethylbutyl) phenyl-polyethylene glycol), carrying typical ‘head-to-tail’ chemical structures, are inefficient at permeabilizing sturdy human tissues because the micelle sizes of these detergents are too big to enter deep inside the tissue. We identified CHAPS (3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate) as a powerful candidate to completely and quickly permeabilize intact human organs, as it forms much smaller micelles with molecular weight of 6,150 Da. CHAPS renders the cellular and extracellular matrix of aged tissue within intact human organs more permeable, thus improving the accessibility of reagents. To remove the red color of remaining blood clots, we screened diverse heme-eluting chemical analogs that are compatible with CHAPS. Analogs bearing ethanolamine structures have been shown to improve decolorization effects in the presence of CHAPS20. Potential candidates such as N-butyldiethanolamine28 and N,N,N′,N′-tetrakis (2-hydroxypropyl) ethylenediamine (Quadrol)24 have been proven to be efficient in decolorizing tissues, but they are at least twice as expensive as N-methyldiethanolamine (NMDEA) (the price of NMDEA is ~30€/L). Considering that intact human organs require large amounts of decolorization chemical agents, we decided to use a NMDEA and CHAPS mixture to achieve cost-efficient permeabilization and decolorization. Still, the volume of the reagents consumed in a single step of handling an intact human brain is ~5–6 L, and the total cost of CHAPS/NMDEA solution is ~1,800€ (ref. 20).
Labeling
Small molecular dyes (<1–2 kDa) or large antibodies (~150 kDa) provide fluorescence contrast of sufficient signal-to-background ratio to identify specific structures of human tissue. DNA or RNA chemical probes show high binding specificities and affinities to the nucleic acid of cells across a wide range of fluorescence spectra (e.g., DAPI, Hoechst in the blue–green range; JO-PRO-1, propidium iodide (PI) in green–red range; TO-PRO3, short-wavelength infrared (SIR) in the infrared range)29. However, primary or secondary antibodies conjugated to chemical fluorochromes cannot penetrate and label more than 1 cm deep into adult human organs due to the probes’ big sizes. Hence, we introduced chemical pretreatments to loosen the cellular and extracellular matrix. First, a solvent mixture of DCM:methanol (2:1, v/v) was used to extract the hydrophobic lipids inside the tissue, which would prevent the free movement of hydrophilic labeling reagents dissolved in buffer30. Second, the tissue was subjected to acetic acid for partial hydrolysis of intertwined collagen by cleavage of the noncovalent intra- and intermolecular bonds31. This process maintains the collagen chains intact, but the cross-links are cleaved. Third, guanidine hydrochloride buffer was employed to extract the proteoglycans of the tissue in dissociative conditions32. After these chemical extraction steps, the tissue matrix became accessible to both small-molecule dyes and large antibodies at a depth range of several centimeters. Cell nuclei in intact human pancreas were perfused and labeled with PI (Fig. 4). Vasculature in multiple human organs was labeled with dextran solution (Fig. 5). SHANEL is also compatible with passive incubation of dye and antibodies to stain PFA-fixed human tissue up to several centimeters size range (Fig. 6). As we have previously demonstrated, 1.5 cm cubic human brain pieces were successfully labeled with primary and secondary antibodies to reveal microglia (e.g., Iba1) and dopaminergic cells (e.g., tyrosine hydroxylase)20. Antibodies conjugated with large protein dyes (e.g., phycoerythrin) also fully labeled more than 1 cm cubic human kidney and lung pieces to reveal cytoplasmic (e.g., catalase), extracellular (e.g., collagen IV) and membranous (e.g., cytokeratin 19) structures of tissue (Extended Data Fig. 2 and Table 2).

a, 3D reconstruction of human pancreas imaged by light-sheet fluorescence microscopy showing the PI-labeled cell nuclei (green) and autofluorescence at 785 nm (magenta). Scale bar, 1 cm. b, Zoomed 3D reconstruction showing the blood vessels and lymph nodes (red rectangle). Scale bars, 1 mm and 200 µm, respectively. c, Higher magnification of the endocrine portion region in the human pancreas is zoomed in using a 12× light-sheet microscopy objective and reconstructed in 3D by Imaris software. Scale bar, 200 µm. d, Various pancreas islets can be easily identified in the 3D-reconstructed endocrine portion region on the basis of their specific shapes (red asterisks). Intralobular ducts (yellow arrows) can be located near the islets. Scale bars, 50 µm, 50 µm and 30 µm, respectively.

a, Human heart after performing the SHANEL protocol. 3D reconstruction shows blood vessels of the coronary artery (red) and myocardium that can be imaged in autofluorescence at 488 nm (AF, green). Scale bars, 1 cm and 2 mm, respectively. b, Human kidney after SHANEL. The structure of glomeruli can be seen in dextran-labeled tissue. TO-PRO3-labeled cell nuclei are shown in blue. Scale bars, 1 cm and 2 mm, respectively. c, One lobe of human lung was cleared and stained with dextran. Bronchus can be seen in AF. Scale bars, 1 cm and 2 mm, respectively. d, Image of human pancreas. Ductal structures are shown with AF, and vasculature is labeled with dextran after SHANEL. Scale bars, 1 cm and 2 mm, respectively. e, Human spleen after SHANEL. The fine structure of the splenic artery is marked with dextran, and the other blood vessel structures are visualized with AF in Imaris. Scale bars, 1 cm and 2 mm, respectively.

a, Skull labeled with lectin (blood vessels, yellow) and PI (nuclei, blue); details of blood vessels and cell nuclei can be seen in XY view. Scale bars, 1 mm, 1 mm and 100 µm, respectively. b, Brain slice labeled with lectin (blood vessels) and Neurotrace Nissl stain. Scale bars, 2 mm and 300 µm, respectively. c, Human brain tissue labeled with Iba1 antibody. Penetration of antibody can be verified in the YZ and XZ view cross-sections, details of microglia can be seen in the magnified XY view. Scale bars, 1,000 µm, 700 µm, 500 µm and 40 µm, respectively. d, Lung tissue is labeled with α-SMA antibody, and AF shows morphometry of the bronchial tree and acinar structure. YZ and XZ views, and magnified XY view are also shown. Scale bars, 700 µm, 500 µm, 500 µm and 150 µm, respectively.
Image analysis
With the prototype or commercial UltraMicroscope Blaze, it is possible to scan the intact human eye, kidney, thyroid and pancreas by mosaic imaging. The scanning time depends on the size of the sample, the percent overlap between images of the mosaic, the number of scanning channels and the size of the Z step (Extended Data Fig. 3). Consequently, terabytes of large data can be generated from a single organ. Software such as Fiji, Arivis, Imaris and Photoshop can be used to handle the large datasets for 3D reconstruction and movie generation with a standard lab workstation (e.g., >256 gigabytes of RAM and terabytes of storage space) (Extended Data Figs. 4–6). However, quantitative analysis of such large datasets can be difficult and imprecise using such software, which in general relies on simple strategies such as filter-based normalization, thresholding or watershed algorithms. Recently, deep learning approaches14,20,33 have shown superior performance in quantification of large-scale data in terms of processing accuracy and speed. It is anticipated that 3D human organ mapping could be greatly advanced with a full exploitation of tissue clearing and imaging combined with deep learning technologies.
Applications of SHANEL
Mammalian skeletal bones protect delicate internal organs, contain most of our body’s calcium supply and are especially difficult to label and clear. Expanding deep tissue labeling, clearing and imaging of bones using SHANEL would greatly benefit investigations of the 3D geometric features of bone volume and cells. In addition to soft tissues such as bone marrow, bones contain hard mineral-dense regions that are deposited with calcium-bearing hydroxyapatite crystals embedded in a collagen matrix. The calcium content considerably increases the optical scattering of bone34. EDTA has been demonstrated as an effective decalcification reagent in previous studies35,36. Similarly, Tainaka et al. developed a carbonated hydroxyapatite-based screening system to identify potent decalcification chemicals compatible with tissue clearing and found EDTA combined with imidazole showed superior effects28. Hence, we conducted decalcification of bones using 20% EDTA at 37 °C before SHANEL tissue labeling and clearing, as shown with examples of human skull pieces and pulvinar soft tissue inside the joint cartilage surface surrounded by bones (Fig. 7). As human bones are much thicker and harder than mouse ones, a much longer decalcification time—in the range of weeks to months—is needed to achieve the desired softness. Alternatively, reagents composed of strong, mild or weak acids (e.g., nitric acid, formic acid, hydrochloric acid, chromic acid, etc.) can be used for faster decalcification. For example, 5% nitric acid is an option for rapid decalcification yielding acceptable tissue integrity and antigenicity37,38.

a, Photo of transparent human pulvinar after PI cell nuclei labeling and clearing by passive incubation. Scale bar, 1 cm. b, 3D reconstruction of human pulvinar imaged by light-sheet fluorescence microscopy showing autofluorescence (AF) at 488 nm (gray) and PI-labeled cell nuclei (green). Scale bar, 1 cm. c, XY section view of pulvinar showing the connection between bone tissue and the pulvinar fibrofatty tissue. Scale bar, 150 µm. d, Human skull bone after PI cell nuclei labeling and clearing. Scale bar, 1 cm. e, The human skull bone imaged by light-sheet microscopy (left) and confocal microscopy (right). Scale bars, 500 µm and 200 µm, respectively.
SHANEL tissue clearing technology has been successfully applied to organs from other mammalian species such as pig brain and pancreas, and is compatible with vDISCO immunostaining20. This enables imaging large mammalian organs in which fluorescent proteins such as GFP, YFP, mCherry and tdTomato are expressed. Since transgenic zebrafish, rat, mouse, pig and macaque with fluorescent protein expression in specific organs have been developed, SHANEL could readily be adopted to clear and image organs from diverse organisms. To combine vDISCO immunostaining with SHANEL organ clearing, we recommend that the organs are first actively perfused or passively incubated with the mixture of CHAPS and NMDEA to permeabilize and decolorize tissue. Afterwards, vDISCO immunostaining process is used via perfusion/incubation with the chosen nanobody in a solution of 1.5 vol% goat serum, 0.5 vol% Triton X-100, 0.5 mM of methyl-β-cyclodextrin, 0.2 wt/vol% trans-1-acetyl-4-hydroxy-l-proline and 0.05 wt/vol% sodium azide in PBS. Finally, the organs are ready to be cleared by SHANEL reagents. We anticipate that, after CHAPS and NMDEA treatment, nanobody/antibody immunolabeling and SHANEL tissue clearing could be applied to diverse mammalian species to investigate broad biological questions. In cases where antibody-based tissue labeling fails owing to inability of large antibodies to cross through whole organs, nanobodies that are ten times smaller than conventional antibodies could provide a more viable alternative.
Although the SHANEL tissue labeling and clearing method was developed for intact human organs, it also works for small tissue pieces, for example, on human biopsies20. In general, biopsy samples are small in size and suitable for the application of the passive SHANEL labeling and clearing method.
Comparison with other methods
In the last years, a diverse array of methods for labeling and clearing of human organ pieces has been developed, including CLARITY17,39, OPTIClear16, MASH18, CUBIC28, SWITCH40, SHIELD11 and ELAST19. CLARITY and OPTIClear take months to clear fixed human brain tissue pieces (<5 mm thickness). MASH explored the small-molecule fluorescent dye labeling and clearing of human brain cortex (<5 mm thickness) by modifying the iDISCO protocol. Other methods are applicable to 3D imaging of human myocardial tissue41, lymph node and lung pieces (<1 cm3) (ref. 42). SWITCH allowed multiple rounds of antibody labeling in 100-µm-thick human brain sections. On the basis of the SWITCH method, SHIELD used a tissue transformation strategy to stabilize a 2-mm-thick human brain slice via intramolecular epoxide linkages to prevent degradation. ELAST enabled human brain tissue (<5 mm thickness) antibody labeling and clearing by mechanically stretching tissue–hydrogel hybrids.
For the first time, SHANEL technology achieved the labeling and clearing of intact adult mammalian organs as large as several cubic centimeters, including human brain, pig brain, pig pancreas, human kidney, human thyroid20, human heart, human pancreas, human lung and spleen (Fig. 5). The key step of the SHANEL technology relies on the permeabilization and decolorization by CHAPS/NMDEA solution. The whole process can be conducted by passive incubation or active perfusion, depending on the availability of major vessels for external connection to commercialized pumps. Active perfusion permits scaling up the process to multiple organs, with general lab equipment available in most molecular biology labs, and no special expertise or training required to implement. It is preferred to perform active perfusion for large adult organs, in order to speed up the process to a reasonable period (e.g., 1.5 months for human kidney, 4 months for human brain). The timeline for each step varies case by case depending on the specific organ. Even a pair of kidneys from the same donor could be different in size, hardness, amount of blood clots and pigments accumulation. It is important to ensure that each step—permeabilization and decolorization, delipidation, cellular and extracellular matrix loosening and labeling, dehydration and RI matching—has been fully completed.
Overview of the protocol
SHANEL provides a flexible platform to achieve whole or partial organ labeling, clearing, and imaging at a cellular level in diverse human and animal organs (Fig.1, Extended Data Fig. 7). The pipeline includes the following main stages: (i) preparation of fixed organs by perfusion or passive incubation, depending on the donor (Steps 1–5); (ii) sample pretreatment, including optional steps such as decalcification and blood vessel labeling (Steps 6–10); (iii) labeling with chemical probes or antibodies (Steps 11–18); and (iv) tissue clearing and imaging with light-sheet microscopy (Steps 19–39).
Expertise needed to implement the protocol
We would recommend researchers with a biological, medical or human anatomy background or with previous experience in handling commercial light-sheet microscopes to implement this protocol. Experience using Fiji, Imaris and Arivis software, will aid in mastering the image analysis pipeline more quickly.
Limitations of the current SHANEL pipeline
Like most of the tissue clearing methods, the SHANEL tissue labeling and clearing protocol includes a delipidation step, which is not compatible with labeling using fluorescent lipophilic dyes, such as DiI. One possibility to overcome this limitation is using modified DiI analogs, such as CM-DiI, SP-DiI or FM 1-43FX, that could be covalently attached to tissue proteins during aldehyde fixation. These dyes would adhere to the cellular membranes and proteins, even after lipid clearing43.
Another issue that we have to take into consideration is the fact that blood clots and lipofuscin cause strong autofluorescence in human samples. The accumulation of lipofuscin is associated with the process of aging44, and its presence complicates the analyses of specific fluorescence labels. It has been reported that chemical treatment45,46,47 (e.g., CuSO4, Sudan Black B and NaBH4) or photobleaching19 can reduce or eliminate autofluorescence, without adversely affecting the targeted labels. We found that CuSO4 was very effective in reducing the lipofusin autofluorescence while being compatible with labeling. However, owing to the natural blue color of CuSO4 solution, the sample could turn light blue after treatment depending on the concentration of CuSO4 solution and would partially absorb the laser light energy used during imaging. Therefore, there is a compromise of fluorescence signal-to-background ratio when using CuSO4 to decrease the autofluorescence signal. In the future, it would be valuable to identify a colorless chemical for effective removal of autofluorescence in the SHANEL protocol.
Furthermore, the validation of new antibodies, nanobodies or dyes before their application within large organs is an additional necessary step. Human organs have to be pretreated with diverse chemicals to loosen the extracellular matrix (ECM) to allow the antibody or nanobody to travel inside. These chemicals could potentially affect the binding of the antibody or nanobody to its target antigen epitopes. Nevertheless, positive staining after performing the SHANEL protocol indicates that the antibody target is well preserved, and specificity is confirmed by comparing the staining pattern with previous results obtained using traditional immunostaining techniques (e.g., in tissue slices). A well-accepted way of quickly selecting an antibody that is sensitive, specific and compatible with SHANEL is to choose an antibody listed in the top-cited ranks in a searchable antibody database48 (https://www.citeab.com/). Even commercially available antibodies/nanobodies that have been subjected to rigorous in-house validation for other applications need to be tested for compatibility with SHANEL. To test these antibodies, human organs firstly go through all pretreatment steps of CHAPS/NMDEA, delipidation and ECM loosening in SHANEL, then are subjected to a traditional slicing technique (e.g., cryostat) to generate lots of thin sections (e.g., 10–50 µm). We use these sections to test the product lot, and optimize the working concentration and staining conditions. An antibody that provides a good signal in tissue sections has a good chance of being compatible with SHANEL in intact organs. For a list of labeling reagents confirmed in our lab, see Table 2.
Although the SHANEL tissue labeling and clearing technology can turn human organs as large as the intact brain transparent, current LSFMs are limited in the size of organs they can accommodate. Owing to imaging chamber size limitations, the LaVision light-sheet microscopy can image a maximum sample size of 4 × 12 × 1.7 cm, in X, Y, Z, respectively. For volumetric imaging of whole organs with long-working-distance objectives, in particular, the current commercial light-sheet microscopes have poor resolution in Z (available resolution 5 μm), which inevitably leads to non-isotropic volumetric images. To reduce the influence of non-isotropic volumetric images in light-sheet microscopy, recently developed light-sheet microscopes such as MesoSPIM are integrated with a moving-waist beam that increases resolution, especially in the Z dimension49.
Finally, The SHANEL method does not include a step to eliminate the signal from antibody staining, and we have not tried multiple rounds of antibody relabeling. In the future, we hope to optimize SHANEL for multiple-round antibody labeling.
Materials
Human organ materials
Caution
Procedures involving human organs or tissues must conform to relevant institutional and national regulations. Informed consent must be obtained from each donor. Human organs were taken from different human body donors. All donors gave their informed and written consent to explore their cadavers for research and educational purposes, when still alive and well. The signed consents are kept at the Anatomy Institute, University of Leipzig, Germany. Institutional approval was obtained in accordance with the Saxonian Death and Funeral Act of 1994. The signed body donor consents are available on request.
-
Human kidney, pancreas, pulvinar and human brain with skull dissected from an 88-year-old female donor
-
Human heart, lung, spleen and pancreas for vessel labeling from a 79-year-old female donor
-
Brain slices for immunolabeling from an 86-year-old female donor and lung and kidney tissue for immunolabeling from a 97-year-old female donor
Reagents
-
0.01 M PBS (diluted from 0.1 M PBS stock solution, which contains 1.37 M sodium chloride, 0.027 M potassium chloride, 0.08 M disodium hydrogen phosphate and 0.02 M potassium dihydrogen phosphate, pH 7.4, Apotheke Klinikum der Universität Munchen, cat. no. P32799)
-
Deionized water (diH2O; in-house purification system)
-
4% PFA in 1× PBS solution, 4 wt/vol%, pH 7.4 (Morphisto, cat. no. 11762.01000)
Caution
Toxic reagent; use only in a well-ventilated fume hood.
Critical
Store at 4 °C.
-
Heparin (Braun, cat. no. 2047217, 25000 IE/5ml)
-
Tetramethylrhodamine isothiocyanate–dextran (mw 500000, Sigma-Aldrich, cat. no. 52194)
-
p-Maleimidophenyl isocyanate (PMPI; Thermo Fisher, cat. no. 28100)
Caution
Eye and skin irritation; wear suitable protective clothing (gloves, lab coat and safety glasses).
-
Dithiothreitol (DTT; Sigma-Aldrich, cat. no. 43815)
Caution
Eye, skin and respiratory irritation; wear suitable protective clothing (gloves, lab coat and safety glasses).
-
PI (Thermo Fisher, cat. no. P1304MP)
-
CHAPS (Carl Roth, cat. no.1479.4)
Caution
Light powder; avoid contact and inhalation of dust.
Critical
Store at 4 °C.
-
NMDEA (Sigma-Aldrich, cat. no. 471828)
Caution
Eye irritation; wear suitable protective clothing (gloves, lab coat and safety glasses).
-
Guanidine hydrochloride (Carl Roth, cat. no. 6069.3)
Caution
Skin and eye irritation; wear suitable protective clothing (gloves, lab coat and safety glasses).
-
Acetic acid (Carl Roth, cat. no. T179.1)
Caution
Flammable and corrosive; keep bottle in a storage cabinet with extreme fire resistance. Skin and eye damage; wear suitable protective clothing (gloves, lab coat and safety glasses).
-
Sodium acetate (Sigma-Aldrich, cat. no. S2889)
Critical
Store separated from strong acids and strong oxidants.
-
Triton X-100 (PanReac Applichem, cat. no. A4975,1000)
Caution
Corrosive, irritant and environmental hazard. Skin and eye damage; wear suitable protective clothing (gloves, lab coat and safety glasses).
-
2-Hydroxypropyl-β-cyclodextrin (HPCD; PanReac Applichem, cat. no. A0367,0100)
-
DMSO (Carl Roth, cat. no. A994.2)
Caution
Skin and eye irritation; wear suitable protective clothing (gloves, lab coat and safety glasses).
-
NaOH (Carl Roth, cat. no. 6771.1)
Caution
Corrosive hazard. Skin and eye damage; wear suitable protective clothing (gloves, lab coat and safety glasses).
-
Ethanol (Merck, cat. no. 10098535000)
Caution
Flammable; keep bottle in a storage cabinet with extreme fire resistance.
-
Methanol (Carl Roth, cat. no. 4627.6)
Caution
Flammable; keep bottle in a storage cabinet with extreme fire resistance. Acute toxic and health hazard; handle with care. Wear suitable protective clothing (gloves, lab coat and safety glasses).
-
DCM (Carl Roth, cat. no. KK47.1)
Caution
Health hazard. Volatile; use only in a well-ventilated fume hood.
-
BABB (benzyl benzoate:benzyl alcohol 2:1, Sigma-Aldrich, cat. no. W213802, cat. no. 24122)
Caution
Irritant and environmental hazard; recycle hazard waste.
-
Sodium azide (Sigma-Aldrich, cat. no. 71290)
Caution
Acute toxic and environmental hazard; recycle hazardous waste according to institutional regulations.
-
Goat serum (GIBCO, cat. no. 16210072)
-
Copper sulfate (CuSO4; Carl Roth, cat. no. CP86.1)
-
Ammonium chloride (Carl Roth, cat. no. P726.1)
Equipment
-
Surgery scissors (FST, cat. no. 14958-11)
-
Big metal tweezers (FST, cat. no. 11000-20)
-
Fine metal tweezers (FST, cat. no. 11252-40)
-
Peristaltic pump (ISMATEC, REGLO Digital MS-4/8 ISM 834)
-
Polytetrafluoroethylene (PTFE) tubing (VWR, cat. no. 228-0735)
-
Reference tubing for the ISMATEC peristaltic pump (ISMATEC, cat. no. SC0026)
-
PVC tubing (Omnilab, cat. no. 5437920)
-
Tubing connector (Omnilab, cat. no. 5434482)
-
Glass chamber (Omnilab, cat. no. 5163279)
-
Glass chamber (LABICAT, cat. nos. 40070360, 40070180)
-
Shaking rocker (IKA, 2D digital)
-
Shaker (IKA, model KS 260 basic)
-
5 ml tubes (Eppendorf, cat. no. 0030 119.401)
Critical
Using these tubes is highly recommended owing to their resistance to clearing solutions.
-
15 ml tubes (Thermo Fisher Scientific, cat. no. 339651)
Critical
Using these tubes is highly recommended owing to their resistance to clearing solutions.
-
50 ml tubes (Thermo Fisher Scientific, cat. no. 339653)
Critical
Using these tubes is highly recommended owing to their resistance to clearing solutions.
-
1 L glass beakers (any; we used the ones from DURAN)
-
1 L glass bottles (any; we used the ones from DURAN)
-
2 L glass bottles (any; we used the ones from DURAN)
-
500 ml glass bottles (any; we used the ones from DURAN)
-
PTFE covered magnetic stirring bars (any)
-
Hot magnetic stirrer (IKA, model RCT basic B-5000, cat no. 0003810000)
-
Aluminum foil (any)
-
Plastic wrap (any)
-
Disposable underpads (Medimex, cat. no. E1911804, or any)
-
Syringes of 1 ml, 5 ml, 10 ml, 50 ml (any)
-
Tapes, black and transparent ones (any)
-
Parafilm (Bemis, cat. no. PM-992)
-
pH meter (WTW, model pH7110)
-
Shaking rocker (IKA, model 2D digital)
-
Incubator (Memmert, model UN160, although any incubator that reaches and maintains 37 °C and has a shaker inside is suitable)
-
Fume hood
Imaging systems
-
Computer (coupled with a custom light-sheet microscope) and equipped with the following characteristics. Processors: Intel Core i7-7800X CPU @ 3.50 GHz; graphic card: NVIDIA Quadro K420; random access memory: 32 GB; solid state drive: Samsung SSD 860 PRO 1 TB; hard disk drive: Seagate Enterprise Capacity SATA 12 TB (ST12000NM0007); operating system: Windows 10 Pro, 64 bit
-
Miltenyi Biotec UltraMicroscope II light-sheet microscope coupled with:
-
SuperK EXTREME/FIANIUM supercontinuum white light laser (NKT Photonics, model SuperK EXTREME EXW-12)
-
Andor sCMOS camera Neo 5.5 (Andor, mod. no DC-152Q-C00-FI)
-
Filter sets: excitation (ex) 470/40 nm, emmission (em) 535/50 nm; ex 545/25 nm, em 605/70 nm; ex 560/30 nm, em 609/54 nm; ex 580/25 nm, em 625/30 nm; ex 640/40 nm, em 690/50 nm
-
Olympus MVX10 zoom body (zoom range 0.63–6.3×)
-
Olympus revolving zoom body unit (U-TVCAC)
-
1× air objective (Olympus MV PLAPO ×1/0.25 NA, working distance (WD) 65 mm)
-
2× immersion objective (Olympus MVPLAPO2XC/0.5 NA, WD 6 mm)
-
4× immersion objective (Olympus XLFLUOR ×4 corrected/0.28 NA, WD 10 mm)
-
12× immersion objective (LaVision MI PLAN 12×/0.53 NA, WD 10 mm)
-
20× immersion objective (Zeiss ×20 Clr Plan-Neofluar/0.1 NA, WD 5.6 mm)
-
-
Miltenyi Biotec prototype UltraMicroscope light-sheet microscope for large samples coupled with:
-
LASOS Multi Color System Series for the laser box (LASOS, model MCS5 F2-01)
-
Andor sCMOS camera Zyla 5.5 (Andor, mod. no ZYLA-4.2P-CL10)
-
Filter sets: ex 488 nm, em 525/50 nm; ex 561 nm, em 595/40 nm; ex 640 nm, em 680/30 nm; ex 785 nm, em 845/55 nm
-
Olympus revolving zoom body that can switch between 1× and 2× with an autofocus-Cube AFC-UM2-UBG
-
1.1× objective (LaVision MI PLAN ×1.1/0.1 NA, WD 17 mm)
-
-
Zeiss LSM 880 inverted laser-scanning confocal microscope coupled with: 25× water-immersion objective (Leica, ×25/0.95 NA, WD 2.5 mm) mounted with a custom mounting thread
Critical
Check the compatibility of the microscope and objectives of different brands with the vendors.
Computer and data processing and analysis tools
-
Computer (HP Z840 workstation; Windows 10 Pro, 64 bit; CPU: Intel Xeon E5-2640 v3; installed memory: 256 GB; GPU: NVIDIA Quadro M5000)
-
Fiji (ImageJ2, https://fiji.sc/) and the Stitch Sequence of Grids of Images plugin50 for stitching original mosaic tiffs from light-sheet microscope
-
Total Commander (v. 8.52a x64, https://www.ghisler.com/) for renaming the files of stitched tiffs
-
ImageMagick (v. 7.0.5-4, https://imagemagick.org/script/index.php) for Lempel–Ziv–Welch (LZW) TIFF compression
-
Vision4D (v.3.0.1 ×64, Arivis) for stitching different organ parts from multiple scans
-
Arivis converter (v.2.12.6, Arivis)
-
Imaris (Bitplane) for visualization of 3D images
-
Photoshop CS6 (v. 13.0, Adobe)
-
The source code of the ‘Stitching.py’ Fiji macro for stitching sequences of images is available at http://discotechnologies.org/ or Zenodo (https://doi.org/10.5281/zenodo.5383535)
Reagent setup
Dextran blood vessel labeling solution
Tetramethylrhodamine isothiocyanate–dextran is a commonly used dye to trace blood vessels in vivo. To chemically fix dextran inside the blood vessels, we used PMPI and DTT to crosslink the hydroxyl group of dextran to the surrounding sulfhydryl-containing proteins. The working solution of dextran contains 5 mg/ml tetramethylrhodamine isothiocyabate–dextran, 0.4 mM PMPI and 1 mM DTT in PBS. As shown in the table below, each reagent is prepared separately as follows: (1) dissolve 8.6 mg PMPI in 2 ml DMSO; (2) dissolve 15.4 mg DTT in 5 ml PBS; (3) dissolve 0.5 g dextran in 93 ml PBS. Mix all solutions together before the perfusion.
PMPI | DTT | Dextran | PBS | |
|---|---|---|---|---|
MW (g/mol) | 214.2 | 154.2 | 500,000 | – |
Stock concentration (mM) | 20 | 20 | – | – |
Stock solution | DMSO | PBS | – | – |
Weight (mg) | 8.6 | 15.4 | 500 | |
Volume of solution (ml) | 2 | 5 | – | 93 |
Critical
Prepare fresh solutions before starting the experiment.
Caution
Prepare the solutions in a hood and wear a mask.
EDTA solution
Prepare EDTA decalcification solution by dissolving 20% (w/v) EDTA in diH2O, adjusting the pH to 8.0–8.5 using NaOH or HCl. In a 2 L bottle with a stirring bar, dissolve NaOH continuously to keep 1.5 L diH2O alkaline, then slowly add 400 g EDTA powder. Replenish diH2O, adding more NaOH or HCl until reaching 2 L with a pH of 8.0–8.5.
Caution
If EDTA precipitation is observed during storage, add more NaOH until the solution becomes transparent with a pH of 8.0–8.5. EDTA solution can be stored at 4 °C for several months.
CHAPS/NMDEA solution
CHAPS/NMDEA mixture is composed of 10% (wt/v) CHAPS and 25% (wt/v) NMDEA in diH2O. In a 2 L bottle, mix 200 g of CHAPS and 500 g of NMDEA in diH2O with a stirrer at room temperature (22–25 °C) and set the final volume to 2 L. The reagent can be stored at 4 °C for several months.
Caution
CHAPS is a light powder; it is suggested to wear a mask to avoid inhalation during the process.
Acetic acid solution
Acetic acid solution is 0.5 M in diH2O. In a 1 L bottle, mix 30 mL acetic acid in diH2O at room temperature and adjust the final volume to 1 L. The reagent can be stored at room temperature for several months.
Caution
Prepare the solution in a hood and wear a mask owing to the smell of acetic acid.
Guanidine solution
Guanidine solution is a mixture of 4 M guanidine hydrochloride, 0.05 M sodium acetate and 2% (wt/vol) Triton X-100 in PBS, pH 6.0. In a 2 L bottle, mix 764.2 g guanidine hydrochloride, 8.2 g sodium acetate and 40 g Triton X-100 in PBS with a stirrer at room temperature and set the final volume to 2 L. Adjust the pH to 6.0 with 4 M NaOH solution and 30% hydrochloric acid solution. The reagent can be stored at room temperature for several months.
Dehydration and rehydration solutions
Prepare a series of dehydration solutions by mixing ethanol with diH2O to obtain 50%, 70% and 100% (v/v) ethanol solutions. Prepare the solutions fresh before using.
Delipidation solutions
The delipidation solution could be a DCM/methanol mixture or pure DCM solution. For tissue pretreatment before labeling, prepare a mixture of two volumes of DCM and one volume of methanol. For tissue clearing after labeling, use pure DCM.
Caution
DCM is a health hazard and highly volatile. It should be handled in a fume hood, wearing safety goggles and double-layer nitrile gloves to avoid inhalation and contact with skin/eyes.
Critical
Prepare fresh solution before use.
Sodium azide stock solution
The concentration of sodium azide stock solution is 0.05% (wt/v) dissolved in 1× PBS. This solution can be stored at 4 °C for several months.
Caution
Avoid contact with skin and eyes.
Blocking buffer for antibody staining
The blocking buffer contains 0.2% (v/v) Triton X-100, 10% (v/v) DMSO and 10% (v/v) goat serum in 1× PBS. In a 500 ml bottle, mix 1 ml Triton X-100, 50 ml DMSO and 50 ml goat serum in PBS with a stirrer at room temperature.
Critical
Prepare fresh solution before use. The pretreated tissue samples can be stored in blocking buffer at 4 °C for several weeks before antibody staining if adding 0.01% (w/v) sodium azide.
Antibody incubation buffer
The antibody incubation buffer is composed of 3% (v/v) goat serum, 3% (v/v) DMSO, 0.2% (v/v) Tween-20 and 10 mg/L heparin in 1× PBS. If antibody solubility in water is poor, prepare an alternative antibody incubation buffer composed of 10% (w/v) HPCD, 3% (v/v) goat serum and 10 mg/L heparin in 1× PBS.
Critical
Prepare solution fresh before use.
Antibody washing buffer
Prepare the antibody washing buffer by combining 0.2% (v/v) Tween-20 and 10 mg/L heparin in 1× PBS. This solution can be stored at 4 °C for several months.
CuSO4 solution
The CuSO4 solution can be used to eliminate tissue autofluorescence. Dissolve 1.6 g CuSO4 and 2.67 g ammonium chloride in diH2O, adjust pH to 5.0 with NaOH or HCl and set the final volume to 1 L.
RI matching solution (BABB)
BABB is the RI matching solution. Prepare by mixing benzyl alcohol and benzyl benzoate in a ratio of 1:2 by volume. BABB solution should be prepared in a glass container in a fume hood and stored with a tight bottle cap at room temperature for several months in a laboratory cabinet.
Caution
Benzyl benzoate is harmful if swallowed, and causes skin and eye irritation. Benzyl alcohol is harmful if swallowed and inhaled. Handle wearing safety goggles, nitrile gloves (preferably double layer) and a lab coat.
Equipment setup
Pump system
To set up an active perfusion system, connect an Ismatec peristaltic pump to a set of tubing: one black pump reference tube, two colorless PVC tubes, three tube connectors and a pipette tip as shown in Fig. 2 and Extended Data Fig. 1. This pump allows connecting up to four channels.
Procedure
Perfusion, fixation and washing of human organs
Timing 1–2 d
-
1
Select plastic tubes matching the diameter of the opened arteries and veins of the human organs, and use these tubes to stitch in the opened arteries and veins.
Caution
Identify and mitigate any risk factors for infectious disease transmission before human organ handling.
-
2
At room temperature, using a gravitation irrigator system, set up the irrigator height as 0.5 m to perfuse the human organs. Mark the start level of PBS/heparin (2.5 U heparin per milliliter) solution of the irrigator and perfuse via the plastic tubes with PBS/heparin solution for 3–5 h until the blood is flushed out, which is signaled by the specimens becoming brighter. It takes 50 L of PBS/heparin solution to flush the blood out from a heart–lung block and 20 L from smaller organs such as kidney, spleen or brain.
-
3
Swap the PBS/heparin solution with 4% PFA solution and continue to perfuse for 15 minutes. Control the perfusion speed by setting up the irrigator height to 0.5 m. Twenty liters are needed per lung–heart block and 10 L for organs such as kidney, spleen or brain.
Caution
If there is no noticeable flow from the human organs, lift the irrigator to increase the perfusion speed (maximal 1.2 m height). A fume hood is necessary when working with formalin solution, which is toxic and has a strong, pungent odor.
-
4
Post-fix the human organs with ten times the volume of 4% PFA solution used in Step 3 for 7 d.
Caution
The duration of fixation can affect sample integrity. Optimal fixation conditions should be assessed for each organ and tissue to prevent underfixation or overfixation.
-
5
Wash the PFA-fixed human organ(s) to prepare them for subsequent experiments. If perfusion is applicable, under a fume hood, set up the pump perfusion system with connecting tubing on a layer of disposable pads. Put the PFA-fixed human organs in an appropriate glass container, and actively perfuse with enough PBS by covering the surface of the organ at room temperature. Wash three times with PBS solution for 2 h each. Set the speed of the pump to 45–46 rpm (160–170 mmHg), and the flow rate to 12.5 ml/min. If perfusion is not applicable, under a fume hood, put a layer of disposable pads on the shaker. Put the PFA-fixed human organs in an appropriate glass container or falcon tubes, then passively incubate with enough PBS to cover the sample at room temperature for 4 h each time and at least three buffer changes. The speed of shaker is 50 rpm.
Caution
Stretch the parafilm tight to seal the connection point for protection if needed.
Pause point
The fixed human organs can be stored at 4 °C in PBS with 0.01% (wt/vol) sodium azide for several months.
Organ pretreatment according to the experimental needs
Timing 1–14 d
-
6
In this step, we describe two methods that are suitable for different kinds of human organs. If the organ vasculature is well preserved, we can perfuse it with dextran blood vessel labeling solution (option A). EDTA decalcification is needed in case there is bone tissue to be labeled and cleared (option B).
-
A
Blood vessel labeling with dextran by perfusion
Timing 1 d
-
(i)
Set up the pump perfusion system with connecting tubing on a layer of disposable pads (Fig. 2). Put the fixed human organ in an empty, clean glass container, then perfuse it with fresh dextran blood vessel labeling solution (see ‘Reagent setup’). Ensure that the solution is flowing out from each tube inserted in the organ, which means the blood vessel is fully filled with dextran solution. Set the speed of the pump to 45–46 rpm (160–170 mmHg), and the flow rate to 12.5 ml/min. The leaking solution can be collected in a container and reused for another round of perfusion labeling.
Critical step
Ensure that the organ turns red after dextran injection.
-
(ii)
After completing vessel labeling, wash the surface of the organ with running water to get rid of the unspecific contaminations from the leaking solution.
Caution
If there is extra dye solution coming out from the organ, wipe the organ dry with paper to keep the whole organ surface clean from dye solution.
-
(iii)
Put the labeled organ in a plastic bag, and seal with tape.
-
(iv)
Put the plastic bag with labeled organ in a container, and place it in a 37 °C incubator overnight.
-
(v)
The next day, take the organ out and cover it with enough PBS at room temperature and place on the shaker to wash. Wash three times with PBS solution for 2 h each.
-
(i)
-
B
Decalcification with EDTA
Timing 7–20 d
-
(i)
Incubate human organs with bone tissue in 20% (wt/vol) EDTA (pH ~8.0) with 0.01% (wt/vol) sodium azide solution at 37 °C. Refresh the solution every 3–5 d until the bone part becomes soft and pliable (test by bending the bone), then wash with PBS three times for 2 h each time.
Caution
If the container is not sealed well, the solution will evaporate, leading to formation of EDTA crystals.
-
(i)
-
A
Permeabilization and decolorization with CHAPS/NMDEA
Timing 3–20 d
-
7
If perfusion is applicable, continue using the same pump perfusion system with enough CHAPS/NMDEA solution to cover the organ for days to 1 month at room temperature for decolorization. A successful decolorization is achieved when the solution turns green or dark green. Refresh the solution two to three times until it does not change color anymore. Place the glass chamber in a fume hood, and cover with plastic wrap and aluminum foil to avoid the evaporation of water and exposure to light (Extended Data Fig. 1c,d).
-
8
If the sample is not suitable for perfusion, continue with passive incubation on the shaker. Incubate the organs with enough CHAPS/NMDEA solution to cover the organs for days at 37 °C, refreshing the solution when it turns green or dark green, until the solution does not change color anymore. This should take two to three solution changes.
Caution
The reference tubing should be displaced back and forth between position 1 and position 2 (Fig. 2e) every 2 d to avoid the tubing attachment becoming loose after continuous long-term abrasion. Check the tubing quality carefully from time to time, as the perfusion solution can occasionally leak from cracks in tubing if there is accumulation of abrasion from continuous running of the pump, especially in the evening when the system is unsupervised. The disposable pads are useful for cleaning up any leaking accidents.
Delipidation
Timing 7–50 d
-
9
Perform the incubations outlined below at room temperature under the fume hood. If perfusion is applicable, continue with same pump perfusion system, if not, continue with passive incubation on the shaker.
-
PBS wash twice for 6 h each time
-
Dehydrate with 50% EtOH/H2O solution for 1–7 d
-
Dehydrate with 70% EtOH/H2O solution for 1–7 d
-
Dehydrate with 100% EtOH solution for 1–7 d
-
Dehydrate with 100% EtOH solution for 1–7 d, until the organ does not shrink anymore
-
Delipidate with DCM/MeOH (2:1, v/v) solution for 1–7 d, refresh the solution two to three times
-
Rehydrate with 100% EtOH solution for 1–7 d
-
Rehydrate with 70% EtOH/H2O solution for 1–7 d
-
Rehydrate with 50% EtOH/H2O solution for 1–7 d
-
Rehydrate with diH2O for 1–7 d
Caution
The glass chamber should be placed in a fume hood and covered with plastic wrap and aluminum foil to avoid evaporation of the solution and light exposure. EtOH, MeOH and DCM are highly volatile liquids, especially under the fume hood. The chamber should be carefully sealed with several layers of plastic wrap and tape to prevent evaporation. Replenish the desired solutions if needed to ensure the whole sample is covered during the perfusion. As DCM could erode some types of tubing (e.g., polystyrene), the recommended tubing is PTFE or polypropylene (PP). Replace the reference tubing every 2 d.
-
ECM loosening
Timing 2–15 d
-
10
Perform the following incubations at room temperature, under the fume hood. If perfusion is applicable, continue using the same pump perfusion system; if not, continue with passive incubation on the shaker.
-
Collagen hydrolysis with acetic acid solution for 1–7 d
-
DiH2O wash twice for 6 h each time
-
Glycoprotein extraction with guanidine solution for 1–7 d
Caution
The glass chamber should be covered with plastic wrap and aluminum foil to avoid evaporation and light exposure.
-
(Optional) Labeling
Timing 7–15 d
Critical
If perfusion is applicable, continue using the same pump perfusion system. If it is not applicable, continue with passive incubation on the shaker. The whole organ can be stained with a cell nuclei dye, for example using a PI or TO-PRO-3 solution in PBS (1 µg/ml and 1 µM final concentration, respectively) at room temperature for 2–7 d. The solution should cover the whole organ.
Critical
Caution
Antibodies are macromolecules, and their large size may limit their ability to penetrate and stain thick organs. Thus, if perfusion is not available, big organs should be dissected into sections no more than 1.5 cm thick to reach full antibody penetration. Actively pumped antibody solutions to stain intact human organ have not yet been tested, while nanobodies have been proven to stain intact pig organs in active pumping set up.
-
11
Sample preparation for antibody/dye/nanobody screening: slice parts of the targeted tissue to 40-µm-thin sections on glass slides using a cryostat following standard protocols or cut into 0.3–1 mm floating slices by vibratome following standard protocols. Use the sections on glass if storing samples for several months in a −80 °C freezer and floating slices for quick antibody screening performed within 1 week.
-
12
SHANEL pretreatment for antibody screening: at room temperature, treat the floating slices with CHAPS/NMDEA solution two times for 12 h each. Wash three times with PBS for 20 min each. Gradually dehydrate in 50%, 70% and 100% EtOH/H2O, 2 h per step. Change to DCM/MeOH (2:1, v/v) solution overnight. Rehydrate in 100%, 70%, 50% EtOH/H2O and diH2O, 2 h per step. Treat with acetic acid solution overnight, wash twice for 20 min in diH2O, treat with guanidine solution for 6 h and wash twice for 20 min in diH2O.
-
13
Block the floating slices with blocking buffer at 37 °C overnight, then incubate with antibodies in antibody incubation buffer at 37 °C for 2 d.
-
14
The concentration of antibody tested should be according to the manufacturer’s suggestions. If needed, incubate the tissue with appropriate secondary antibody at 37 °C for 2 d. Wash the samples with washing buffer three times for 1 h at room temperature.
-
15
Gradually dehydrate in 50%, 70% and 100% EtOH/H2O, 2 h per step. Change to DCM for 1 h, then change to BABB until the slices become transparent.
-
16
Lastly, examine the results with a confocal microscope to find the best conditions for strong and specific antibody staining.
Critical step
We strongly suggest using Alexa or Atto dye as the fluorophores conjugated to antibodies, because these dyes are stable and bright in clearing solution.
-
17
After obtaining good-quality staining results from antibody screening, dissect the region of interest from the human organ and treat the tissue slice or block following the timing guidelines in Table 3.
Critical step
Autofluoresecnt lipofusin accumulates in aged human samples. Incubate the slice with CuSO4 solution for 1–7 d before clearing to eliminate tissue autofluorescence.
Clearing
Timing 7–40 d
-
18
If perfusion is applicable, continue using the same pump perfusion system at room temperature, under the fume hood for the following incubations. If not applicable, continue with passive incubation on the shaker.
-
PBS wash twice for 6 h each time
-
Dehydrate with 50% EtOH/H2O solution for 1–7 d
-
Dehydrate with 70% EtOH/H2O solution for 1–7 d
-
Dehydrate with 100% EtOH solution for 1–7 d
-
Dehydrate with 100% EtOH solution for 1–7 d, until the organ does not shrink anymore
-
Delipidate with DCM solution for 1–7 d, until the sample sinks to the bottom of the container
-
Perform RI matching with BABB solution for 1–30 d until the organ becomes completely transparent
Caution
The glass chamber should be covered with plastic wrap and aluminum foil to avoid evaporation and light exposure. EtOH and DCM are highly volatile liquids, especially under the fume hood. The chamber should be carefully sealed with several layers of plastic wrap and tape to prevent the volatility of solutions. Replenish the desired solutions to cover the whole sample if needed during the circulation. DCM can erode some types of tubing (e.g., polystyrene), therefore, the recommended tubing is from PTFE or PP. Replace the reference tubing every 2 d (Supplementary Video 1).
Pause point
The cleared human organs in BABB solution can be stored covered with aluminum foil at room temperature for months to years before imaging.
-
Imaging with light-sheet fluorescent microscopy
Timing 1–8 d
Critical
Here we use the prototype light-sheet microscope from Miltenyi Biotec to image the human pancreas (Fig. 3). The commercialized UltraMicroscope Blaze can also be applied for high-resolution imaging of large human organs. Both microscopes are controlled by the same software named ‘Imspector’.
-
19
Check if there are bubbles inside the organ in BABB solution. If yes, carefully suck out the bubbles using a 1 ml syringe (Supplementary Video 2).
-
20
Use one piece of tissue paper soaked with 80% EtOH/H2O solution to clean the sample holder, and wait for the sample holder to dry.
-
21
Cut three pieces of tape, and stick them on the top, middle and bottom parts of the sample holder.
-
22
Add glue on each piece of tape on the sample holder. To stick the sample durably, the glue should cover the full area of the tape.
Caution
If the glue is not sufficient, the sample will not be stable on the holder and may shift or fall down during the 2–3 d of scanning.
-
23
Use tissue paper to absorb BABB solution from one side of sample several times.
-
24
The glue would contact the dry side of the sample. Carefully put the sample on the sample holder, and try to keep the center of sample aligning with the holder. One end of sample should be close to one end of the moving range of holder (e.g., top to top).
-
25
Wait for 30–60 s until the glue solidifies.
-
26
Put the sample holder back into the light-sheet microscope, and fix it.
-
27
Check again if there are bubbles inside of organs in BABB solution. If yes, carefully suck out the bubbles using 1 ml syringe.
Caution
Gently touch the sample when sucking out bubbles; otherwise, the glue bonding would be destroyed mechanically and the sample would shift or fall down.
-
28
To set imaging parameters in Imspector software (Extended Data Fig. 3) go to ‘Settings’ → ‘Hardware’ → ‘Ultra3 Settings’ → ‘Objective’ to choose the right objective, for example ‘LVBT 1x’.
-
29
Start the View settings of the software with the following settings:
Measurement mode
Multicolor mosaic acquisition
Devices 1
Ultra3 filter
Devices 2
xyz-Table Z
Devices 3
xyz-Table X
Devices 4
xyz-Table Y
AS (autosave)
‘abled’ in green
Split
‘abled for Devices 1’
Channel setup
Click the targeted scanning channels according to the labeling, Excitation 561, Emission 595/40 and Excitation 785, Emission empty here
Lightsheet properties
Set ‘Sheet NA’ to ‘0.035’, and ‘Thickness’ to ‘21 µm’
Sheet width
100%
Zoom
1.0×
Measurement → Liquid
BABB
Measurement → Mount
None
Exposure time
90.000–100.000 ms
Gain
16-bit low noise
Step (the step size in µm between each two images in Z axis)
8.00 (for human pancreas)
Critical step
The smaller the step size, the more images will be acquired, and the longer scanning will take, but the resolution in XZ and YZ direction will increase.
-
30
Set ‘LightSheet Selection’ to be only ‘left’ or only ‘right’ when checking the signal and position of the sample.
-
31
Select the top surface of the sample for the focus by adjusting the sample stage in the Z direction (Supplementary Video 3).
Caution
When adjusting the focus, be careful to avoid collision of the moving objective with the sample. The sample should only be illuminated on the surface in the starting position step.
-
32
Mount the 1.1× objective, and then click the ‘Focus → down’ button in the software until the lens gets immersed in the BABB solution inside the chamber holding the sample, then adjust the movement of the objective to get a clear and bright signal. Then lower the sample holder until the sample is just lower than the light-sheet illumination region, click ‘Current Position → Set as Zero’ and ‘Scan Range → Start’ to ‘0.000’.
-
33
Next lift the sample holder. The signal gets blurry when the imaging plane gets deeper in the sample. When this occurs, click ‘Scan Range → End’. Since with light-sheet microscopy using a 1.1× objective, the working distance is ~17 mm in z direction, the maximum imaging depth would be −14 mm.
-
34
Adjust ‘Mosaic → Set Parameters’ to find the proper ‘X Axis, Y Axis’ and ‘Overlap’ to cover the scanning volume of the samples from all edges. In general, for a 1.1× objective, ‘3×8’ with 30% overlap or ‘4×7’ with 33% overlap would cover the human pancreas or human kidney. For a 12× objective, ‘10×10’ with 30–40% overlap is possible.
Caution
The following image stitching will be less accurate if the overlap value is set too small. When setting the scanning volume, do not modify the travelling limitations of the stage in X, Y and Z direction. Otherwise, the software would report an error and fail.
-
35
Choose ‘Autosave Settings’ to fill in the ‘path’ and ‘name’ for saving, for example ‘humanpancreas_1x_4×7_33o_90ex_8um_56130_78540_forwardup’.
-
36
Stop the live ‘View’, and click the ‘Lightsheet Selection’ to be ‘both’.
-
37
Start the scanning, and keep the room in the dark for ~2–3 d.
-
38
When the scanning has finished, take the sample holder out and cut the tape to remove the sample from the holder. Put the sample in BABB solution and peel off the glue and tape at the same time. Wash the sample holder with 100% EtOH, and clean with tissue paper.
-
39
Repeat Steps 19–38 for scanning different parts of the sample, changing the position of the sample, or flipping the sample in Step 31 to make sure the scanning covers both ends and both sides of the sample. For example, the human pancreas took four rounds of scanning to image the whole organ covering the forward-up, forward-down, backward-up and backward-down orientations.
Data stitching, renaming and compression
Timing 1–2 d
-
40
Scanning with the light-sheet microscope generates a series of grid images in the auto save folder. Inside the auto save folder, prepare new folders named C00, C01, etc. corresponding to the channels used for imaging.
-
41
Begin by stitching a preview image. Start Fiji, and go to the plugin ‘Stitch Sequence of Grids of Images’.
-
42
In the ‘Stitch Sequence of Grids of Images’ dialog, set the following parameters as specified (Extended Data Fig. 4a) and keep the rest as default:
‘Grid size in X’ as the number of tiles per row.
‘Grid size in Y’ as the number of tiles per column.
‘Grid size in Z’ as 1.
‘overlap’ as the overlap defined when scanning.
‘Input directory’ as the folder path containing the grid images.
‘file names’: copy one of the file names of the grid images from the auto save folder, and replace the specific numbers of Ultra3[00 ×00] to be the variable numbers Ultra3[{yy} x {xx}]; Z0000 should be changed to Z{zzzz}.
‘Output directory’ as the folder where the resulting stitched images will be saved.
‘Start x’ and ‘start y’ as ‘0’.
‘Start z’ as any number roughly in the middle of the z-stack
‘Start i’ as ‘1’.
-
43
Click ‘OK’, and wait for the preview image to be stitched.
-
44
Open the preview stitched image in the output folder using Fiji, and check if the image aligned properly without any mismatched images. When the preview stitching is completed, a file called ‘TileConfiguration.text.registered’ will be generated in ‘Input folder’. This file contains the grid image layout parameters.
Critical step
It is possible to use other alternative algorithms for stitching, for example, TeraStitcher (https://abria.github.io/TeraStitcher/).
-
45
Copy the ‘TileConfiguration.text.registered’ file into the different channels folder (C00, C01, etc.), and change the file format to.txt.
-
46
In one channel, open the ‘TileConfiguration.txt’ file and change the Z panel number back to 0000 in all tiles using the replace function, for example, ‘Zxxxx’ to ‘Z0000’.
-
47
If there is more than one channel, copy this ‘TileConfiguration.txt’ file to the other channel folders, and replace the channel number to the corresponding channel number, for example, ‘C00’ to ‘C01’ in folder ‘C01’.
-
48
Open the plugin file ‘Stitching.py’ with Fiji.
-
49
Run the ‘TileConfiguration.txt’ file, and enter the total number of sections in one round of scanning.
-
50
Repeat Steps 48–49 for each ‘TileConfiguration.txt’ file in the other channel folders to stitch the rest of the images. All stitched images will be separately saved in corresponding channel folders, ‘C00’, ‘C01’, etc.
-
51
The stitched images will have the same name in the different channel folders ‘Manual_StitchedZxxxx’. Use ‘Total Commander’ software to rename the images. Open the software, find the corresponding channel folder (e.g., C00) and select all of the images in the folder. Then click ‘Files’ → ‘Multi-Rename Tool’, and fill the information as follows:
‘file name’ set as ‘C00_Z’. Then click ‘[C] Counter’.
Define the counter number as ‘Start at 0’, ‘Step by 1’ and ‘Digits 4’.
-
52
Repeat Step 51 for all other channel folders.
-
53
Combine all stitched images from different channel folders into one folder so that they can be compressed.
-
54
Open the ‘LZW compression’ of ‘ImageMagick’. Input the file path of the folder with all the stitched images and output to the same path. Press ‘Enter’ to compress all the stitched images, and wait for Arivis volumetric fusion.
Arivis fusion
Timing 1–6 d
Critical
Whole organ scanning, covering different parts of the organ, will be completed after multiple rounds of light-sheet microscopy scaning. Following organ stitching by Fiji, use ‘Arivis Vision 4D’ to fuse the 3D organ volumes.
-
55
Arivis Vision 4D software requires a .sis file format to visualize and analyze the image. To convert the image into a .sis file, open ‘Arivis SIS Converter’ and select ‘Add Files’ to load the stitched images composed of all channels from one folder, for example, ‘C00’ folder. If the dialog ‘Assume same structure for all files?’ appears, click ‘Yes’.
Critical step
All stitched images from different channels must be transferred to the same folder in advance.
-
56
In the ‘Arivis SIS Converter’ tab, select ‘Custom import’ as file import style and define the name of .sis Arivis file in the export folder (e.g., ‘Human pancreas_forward up.sis’).
-
57
To define the imported image pixel size, select ‘more options’ and select the right image pixel type, then click ‘OK’.
-
58
A new window ‘Manual import mapper’ will appear; click ‘Selection’, and choose ‘Pattern matching’.
-
59
Click ‘Start’ to begin the conversion.
Caution
The .sis Arivis file could be several terabytes after fusion; for example, human pancreas Arivis file is 2.5 TB. It is recommended to check the drive capacity before starting the conversion.
-
60
Repeat Steps 56–59 to convert volumetric scans of the different parts of the organ to.sis files. For example, four rounds of scanning were performed on the human pancreas to cover different parts of the organ, and the results should be converted into four individual .sis Arivis files, corresponding to the forward-up, forward-down, backward-up and backward-down scans.
-
61
After all different parts of the organ image set are converted to Arivis file (.sis), open the first .sis file in Arivis Vision 4D. Correct the parameters of pixel size in X, Y and Z coordinates by clicking ‘Data’ → ‘Pixel Size’.
-
62
To fuse the second part of the organ with the first part, load the second .sis file as a new image set: click ‘File’ → ‘Import’ to choose the second .sis file, then click ‘Import Files’ → ‘more options’ → ‘Import as New Image set’ and set ‘Target Pixel Type’ to ‘16 Bit integer’.
Critical step
The pixel type should be matched for all loaded volumetric image sets.
-
63
If the two volumetric scans are from different orientations, correct the second image set to the same orientation as the first image set by clicking ‘Flipping’ and click ‘OK’ to run the transformation (Extended Data Fig. 5a).
-
64
After transformation, a new image set called ‘flipped’ will appear in the ‘Image Set’. Click the ‘Vertical Split’ on the menu. Then click ‘Navigator’ → ‘Vertical Split’ to load two volumetric scans side by side (one is the first image set, the other is the flipped second image set) (Extended Data Fig. 5b). Check the loaded image set quality by scrolling the 2D images to zoom in and out, and adjust the color bar of each channel for brightness and contrast.
-
65
If satisfied with the image quality of these two volumetric scans, prepare the matching landmarks for volume fusion in the Arivis Vision 4D software. Find at least three landmarks in both overlapping volumes from the two image sets (Extended Data Fig. 5c,d).
Critical step
The landmarks should distribute in the overlapping volume as much as possible. The landmarks could be the same cell, the same vessel structure or the same junctions.
-
66
To label the landmarks in both image sets, click the icon ‘Place New Object’ → ‘Marker’ to label the target landmark region.
-
67
Click ‘Show Objects Table’ to view the marker information.
-
68
After collecting enough landmarks from the two image sets, volume fusion can be initiated. Click ‘Data’ → ‘Volume Fusion’ → ‘Image Sets’ to choose one image set as ‘Base Image Set’ and the other image set as ‘Moving Image Set’. Before running the full function of volume fusion, add all landmarks in the ‘Transformation’ → ‘Landmark Registration’ → ‘Base Image Set’ → ‘Add All Objects as Landmarks’ by right clicking the set icon. Do the same for ‘Moving Image Set’. Then select the ‘Scale’ to be ‘10%’ for generating a new preview image set (Extended Data Fig. 5e).
Critical step
The sequence of landmarks should be the same in both image sets. The highlighted locations of landmarks will be displayed in the right window.
-
69
If not satisfied with the fusion in the new preview image set, for example, if the new preview image set shows mismatching or gaps after the volume fusion, try different landmarks to get the best fusion quality in both 2D and 3D view.
-
70
If the preview fusion doesn’t show mismatching or gaps, change the ‘Scale’ to ‘100%’ in Step 68 and run the final fusion to generate the new .sis file. Repeat Steps 62–69 to fuse each of the other parts of the volumetric scan into the new fused .sis file. For example, repeat the process three times to generate the final 3D fusion .sis file for the human pancreas, which contains four Arivis Vision4D image sets.
-
71
After finishing the whole organ fusion (Extended Data Fig. 5f), export a series of .tiff files by clicking ‘File’ → ‘Export’ → ‘TIFF Exporter’ and defining the file name in a new folder.
-
72
As the Arivis Vision4D exports all the .tiff files with different channels into the same folder, run ‘Total Commander’ software to rename each image and separate the channels. Click the ‘Multiple rename tool’ in the Total Commander, and repeat Step 51.
-
73
After all the images are renamed, run ‘LZW TIFF 16bit’ to compress all fused images in the saved folder.
Imaris 3D visualization
Timing ~0.5–2 d
Critical
We describe below the essential steps to perform visualization with Imaris 3D. For a more detailed overview, please check the user guide on the Imaris website.
-
74
Convert the image set .tiff files to the .ims file format. To perform the file conversion, open the ‘Imaris File Converter’, and click ‘Add Files’ to load all .tiff images from the saved folder (Extended Data Fig. 6a).
-
75
When the .tiff files contain different channels, save the channel information during the conversion process by using the ‘Settings’ window. Select ‘File name with delimiter’, confirm the format as C“C”_Z“Z”.tif, and click ‘Start All’ (Extended Data Fig. 6b).
-
76
Double click to open the converted .ims file, and proceed to adjust the voxel size by performing the following steps: ‘Edit’ → ‘Image Properties’ → ‘Geomertry’ → ‘Voxel Size’; X = 6.5/objective, Y = 6.5/objective and Z = ‘z’, where ‘z’ is the step size from the imaging microscopy setting.
Example: When choosing the 1× objective with a z step of 8 µm, the voxel size would be X = 6.5 µm, Y = 6.5 µm, Z = 8 µm; when choosing the 12× objective with a z step of 6 µm, the voxel size would be X = 0.542 µm, Y = 0.542 µm, Z = 6 µm (Extended Data Fig. 6c).
-
77
Once the voxel size is adjusted, proceed to change the channel colors and adjust the contrast: click ‘Edit’ → ‘Show Display Adjustment’ (Extended Data Fig. 6c).
-
78
Before rendering the 3D view, click ‘Imaging Processing’ → ‘Camera Function’ → ‘Set The Angle Of The Camera’.
-
79
Select the region of interest, and capture an image in the ‘Snapshot’ menu. Click ‘Snapshot’ → ‘Preferences’ to set the size with DPI of 300 and save as Type (TIFF images) in the Image Output Directory.
Timing
-
Steps 1–5, perfusion, fixation and washing of human organs: 1–2 d
-
Step 6, organ pretreatment according to the experimental needs: 1–14 d
-
Steps 7–8, permeabilization and decolorization with CHAPS/NMDEA: 3–20 d
-
Step 9, delipidation: 7–50 d
-
Step 10, ECM loosening: 2–15 d
-
Steps 11–17, (optional) labeling: 7–15 d
-
Steps 18, clearing: 7–40 d
-
Steps 19–39, imaging with light-sheet fluorescent microscopy: 1–8 d
-
Steps 40–54, data stitching, renaming and compression: 1–2 d
-
Steps 55–73, Arivis fusion: 1–6 d
-
Steps 74–79, Imaris 3D visualization: ~0.5–2 d
-
See Table 3 for timing guidelines for treatment of various human organs.
Troubleshooting
Troubleshooting advice can be found in Table 4.
Anticipated results
We described SHANEL in this protocol as a scalable tissue labeling and clearing method for a variety of human organs (Supplementary Video 4), including pancreas (Figs. 3–5) and kidney, and show additional vessel labeling of spleen, heart and lung (Fig. 5). Compared with our original publication20, we also extended the original method to be compatible with hard human tissue with bone (Fig. 7). SHANEL allows imaging of intact human organs at cellular resolution, which paves the way for human organ mapping, and potentially extends our knowledge in human organ anatomy. The technique can be used to study vascular morphology and pathology, as the fine details of capillaries can be visualized throughout whole organ (Fig. 5).
We previously used SHANEL to label and quantify the pancreas islets in INS-EGFP transgenic pig20, demonstrating the scalability of this method to study large-scale biological samples. SHANEL is also applicable to antibody staining in human tissue or biopsies (Fig. 6), increasing its adaptability in biomedical research.
After using the recently developed LSFM to image transparent human organs or tissues and to therefore acquire spatial information of vessels and fluorescent protein signal (Fig. 3, Extended Data Fig. 3), the whole dataset can be efficiently stitched with an easily operated software (Extended Data Fig. 4). Here we show an example of how raw images were processed and visualization of the entire 3D field and cellular states information was rapidly achieved (Extended Data Figs. 5, 6). Note that, owing to the size of human organs, terabytes of the image data will be generated. Consequently, we recommend combining our methodology with deep learning approaches to achieve accuracy and speed in quantification and further analysis. We summarize a workflow (Extended Data Fig. 7) that will be useful for researchers to understand the main steps of this protocol.
Data availability
References
Snyder, M. P. et al. The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature 574, 187–192 (2019).
Rood, J. E. et al. Toward a common coordinate framework for the human body. Cell 179, 1455–1467 (2019).
Rozenblatt-Rosen, O., Stubbington, M. J. T., Regev, A. & Teichmann, S. A. The Human Cell Atlas: from vision to reality. Nature 550, 451–453 (2017).
Srivastava, S., Ghosh, S., Kagan, J. & Mazurchuk, R. The PreCancer Atlas (PCA). Trends Cancer 4, 513–514 (2018).
Ardini-Poleske, M. E. et al. LungMAP: The Molecular Atlas of Lung Development Program. Am. J. Physiol. Lung Cell Mol. Physiol. 313, L733–L740 (2017).
Amunts, K. et al. BigBrain: an ultrahigh-resolution 3D human brain model. Science 340, 1472–1475 (2013).
Deverman, B. E. et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 34, 204–209 (2016).
Erturk, A. et al. Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nat. Protoc. 7, 1983–1995 (2012).
Cai, R. et al. Panoptic imaging of transparent mice reveals whole-body neuronal projections and skull–meninges connections. Nat. Neurosci. 22, 317–327 (2019).
Pan, C. et al. Shrinkage-mediated imaging of entire organs and organisms using uDISCO. Nat. Methods 13, 859–867 (2016).
Park, Y.-G. et al. Protection of tissue physicochemical properties using polyfunctional crosslinkers. Nat. Biotechnol. 37, 73–83 (2019).
Renier, N. et al. Mapping of brain activity by automated volume analysis of immediate early genes. Cell 165, 1789–1802 (2016).
Belle, M. et al. Tridimensional visualization and analysis of early human development. Cell 169, 161–173 e112 (2017).
Pan, C. et al. Deep learning reveals cancer metastasis and therapeutic antibody targeting in the entire body. Cell 179, 1661–1676.e19 (2019).
Kubota, S. I. et al. Whole-body profiling of cancer metastasis with single-cell resolution. Cell Rep. 20, 236–250 (2017).
Lai, H. M. et al. Next generation histology methods for three-dimensional imaging of fresh and archival human brain tissues. Nat. Commun. 9, 1066 (2018).
Morawski, M. et al. Developing 3D microscopy with CLARITY on human brain tissue: Towards a tool for informing and validating MRI-based histology. Neuroimage 182, 417–428 (2018).
Hildebrand, S., Schueth, A., Herrler, A., Galuske, R. & Roebroeck, A. Scalable labeling for cytoarchitectonic characterization of large optically cleared human neocortex samples. Sci. Rep. 9, 10880 (2019).
Ku, T. et al. Elasticizing tissues for reversible shape transformation and accelerated molecular labeling. Nat. Methods 17, 609–613 (2020).
Zhao, S. et al. Cellular and molecular probing of intact human organs. Cell 180, 796–812 e719 (2020).
Murakami, T. C. et al. A three-dimensional single-cell-resolution whole-brain atlas using CUBIC-X expansion microscopy and tissue clearing. Nat. Neurosci. 21, 625–637 (2018).
Matsumoto, K. et al. Advanced CUBIC tissue clearing for whole-organ cell profiling. Nat. Protoc. 14, 3506–3537 (2019).
Susaki, E. A. et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell 157, 726–739 (2014).
Tainaka, K. et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell 159, 911–924 (2014).
Chung, K. et al. Structural and molecular interrogation of intact biological systems. Nature 497, 332–337 (2013).
Ku, T. et al. Multiplexed and scalable super-resolution imaging of three-dimensional protein localization in size-adjustable tissues. Nat. Biotechnol. 34, 973–981 (2016).
Treweek, J. B. et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nat. Protoc. 10, 1860–1896 (2015).
Tainaka, K. et al. Chemical landscape for tissue clearing based on hydrophilic reagents. Cell Rep. 24, 2196–2210 e2199 (2018).
Lai, H. M., Ng, W. L., Gentleman, S. M. & Wu, W. Chemical probes for visualizing intact animal and human brain tissue. Cell Chem. Biol. 24, 659–672 (2017).
Cequier-Sánchez, E., Rodríguez, C., Ravelo, A. G. & Zárate, R. Dichloromethane as a solvent for lipid extraction and assessment of lipid classes and fatty acids from samples of different natures. J. Agric. Food Chem. 56, 4297–4303 (2008).
Schmidt, M. M. et al. Collagen extraction process. Int. Food Res. J. 23, 913–922 (2016).
Yanagishita, M., Podyma-Inoue, K. A. & Yokoyama, M. Extraction and separation of proteoglycans. Glycoconj. J. 26, 953–959 (2009).
Todorov, M. I. et al. Machine learning analysis of whole mouse brain vasculature. Nat. Methods 17, 442–449 (2020).
Ugryumova, N., Matcher, S. J. & Attenburrow, D. P. Measurement of bone mineral density via light scattering. Phys. Med. Biol. 49, 469–483 (2004).
Greenbaum, A. et al. Bone CLARITY: clearing, imaging, and computational analysis of osteoprogenitors within intact bone marrow. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aah6518 (2017).
Grüneboom, A. et al. A network of trans-cortical capillaries as mainstay for blood circulation in long bones. Nat. Metab. 1, 236–250 (2019).
Gonzalez-Chavez, S. A., Pacheco-Tena, C., Macias-Vazquez, C. E. & Luevano-Flores, E. Assessment of different decalcifying protocols on osteopontin and osteocalcin immunostaining in whole bone specimens of arthritis rat model by confocal immunofluorescence. Int. J. Clin. Exp. Pathol. 6, 1972–1983 (2013).
Savi, F. M., Brierly, G. I., Baldwin, J., Theodoropoulos, C. & Woodruff, M. A. Comparison of different decalcification methods using rat mandibles as a model. J. Histochem. Cytochem. 65, 705–722 (2017).
Liu, A. K. et al. Bringing CLARITY to the human brain: visualization of Lewy pathology in three dimensions. Neuropathol. Appl. Neurobiol. 42, 573–587 (2016).
Murray, E. et al. Simple, scalable proteomic imaging for high-dimensional profiling of intact systems. Cell 163, 1500–1514 (2015).
Perbellini, F. et al. Free-of-Acrylamide SDS-based Tissue Clearing (FASTClear) for three dimensional visualization of myocardial tissue. Sci. Rep. 7, 5188 (2017).
Nojima, S. et al. CUBIC pathology: three-dimensional imaging for pathological diagnosis. Sci. Rep. 7, 9269 (2017).
Jensen, K. H. & Berg, R. W. CLARITY-compatible lipophilic dyes for electrode marking and neuronal tracing. Sci. Rep. 6, 32674 (2016).
Mann, D. M., Yates, P. O. & Stamp, J. E. The relationship between lipofuscin pigment and ageing in the human nervous system. J. Neurol. Sci. 37, 83–93 (1978).
Schnell, S. A., Staines, W. A. & Wessendorf, M. W. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J. Histochem. Cytochem. 47, 719–730 (1999).
Neumann, M. & Gabel, D. Simple method for reduction of autofluorescence in fluorescence microscopy. J. Histochem. Cytochem. 50, 437–439 (2002).
Yang, J. et al. Quenching autofluorescence in tissue immunofluorescence [version 1; peer review: 2 approved with reservations, 1 not approved]. Wellcome Open Res. https://doi.org/10.12688/wellcomeopenres.12251.1 (2017).
Helsby, M. A. et al. CiteAb: a searchable antibody database that ranks antibodies by the number of times they have been cited. BMC Cell Biol. 15, 6 (2014).
Voigt, F. F. et al. The mesoSPIM initiative: open-source light-sheet microscopes for imaging cleared tissue. Nat. Methods 16, 1105–1108 (2019).
Preibisch, S., Saalfeld, S. & Tomancak, P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25, 1463–1465 (2009).
Acknowledgements
We thank A. Ghasemi Mag for developing the Python script ‘Stitching.py’ to stitch sequences of images. We thank I. Horvath, K. Joseph, K. Biniossek and E. Süheda for revising the manuscript. We thank Miltenyi Biotec for providing the PE-conjugated antibodies. A schematic of the SHANEL pipeline (Fig. 1) was created with BioRender.com. This work was supported by the Vascular Dementia Research Foundation, Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy, ID 390857198) and DFG (SFB 1052, project A9; TR 296 project 03), H.M. and Z.R. would like to thank the China Scholarship Council (CSC) for the financial support (no. 201806780034 and no. 201806310110).
Author information
Authors and Affiliations
Contributions
A.E. conceived and led the project. S.Z. developed the original SHANEL protocol. H.M., Z.R. and S.Z. performed the experiments and wrote the manuscript. R.C. performed the antibody screening. H.S. and I.B. dissected and provided the human organs. All authors commented on the manuscript text.
Corresponding author
Ethics declarations
Competing interests
A.E. and S.Z. have filed a patent on the SHANEL technology described in this protocol.
Peer review
Peer review information
Nature Protocols thanks Etsuo A. Susaki and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key reference using this protocol
Zhao, S. et al. Cell 180, 796–812 (2020): https://doi.org/10.1016/j.cell.2020.01.030
Extended data
Extended Data Fig. 1 Human brain clearing by perfusion system.
a, The dissection of human brain with CL and CR (left and right carotids, respectively), VL and VR (left and right vertebral arteries, respectively). b, The human brain after dissection and fixation. c, The setup to clear a human brain by perfusion system under the fume hood. The human brain was put in a glass container and connected with input and output tubing controlled by a peristaltic pump. There are four channels controlling the four connecting tubes as indicated in c (i). Two output tubes are connected to the carotid arteries as shown in c (ii), and two output tubes are connected to the vertebral arteries as shown in c (iii). The input tubes are protected with gauze to avoid solid impurities entering and blocking the tubing (see c (iv)). c (v) shows the decolorization effects of CHAPS/NMDEA that manifest as the blooming of dark-green color from the organ. d, The glass container was sealed with several layers of plastic wrap to prevent evaporation of the running solutions, especially EtOH, DCM and MeOH. e, Photo of human brain after SHANEL pretreatment and before clearing. Remaining blood in the vessels (b) was decolorized.
Extended Data Fig. 2 Passively stained conjugated antibody in centimeter-size human kidney and lung tissue.
a,b, Collagen IV, catalase and cytokeratin 19 antibody staining (green) in human kidney (a) and lung (b). Top panel in a and b shows immunofluorescent staining images obtained by confocal microscopy to verify antibody compatibility with SHANEL. Bottom panel shows 3D reconstruction of tissue with antibody labeling in Imaris. Scale bars, 20 µm and 500 µm, respectively.
Extended Data Fig. 3 Imaging software settings.
The user interface of LaVision Imspector software. a, Settings of the objective lens and magnification. b, Names of the devices should be listed in the correct order. c, One example for settings of the tiling scan with 4 × 7 tiles and 33% overlap.
Extended Data Fig. 4 Key steps for data stitching, renaming and compression.
a, One example for correcting information in ‘Stitching Image Grid Sequence’; a1 highlights the replacement of red letters in Step 42 about ‘file names’. b, The key information of ‘TileConfiguration’ txt file; b1 highlights the responding channel number that should be corrected and saved in the respective separate folder, and b2 highlights that the starting Z number should be ‘0000’ for all ‘TileConfiguration’ files. c, The information of ‘Stitching.py’. d, One example for settings of ‘Multi-Rename Tool. e, One example of compressing tiff files with ‘LZW TIFF’. Panel a produced with software from ref. 50, Oxford University Press.
Extended Data Fig. 5 Key steps for Arivis fusion of 3D images.
a, An example of flipping one set of volumetric dataset in X, Y or Z direction to match with another volumetric dataset. b, Overview of two volumetric datasets oriented in the same XYZ direction after flipping. c,d, Examples of identical structural markers from two volumetric datasets for fusion. e, Loading of three key markers for optimal fusion. f, 3D fused image of pancreas from two volumetric datasets.
Extended Data Fig. 6 Key steps for Imaris data loading and visualization.
a, An example of converting a stitched image sequence data to the .ims format using Imaris File Converter. b, The settings for file names with a delimiter should be changed to ‘C _ Z.tif’. c, 3D image properties including XYZ voxel size and channel color can be set in Imaris for analysis.
Extended Data Fig. 7 Overview of SHANEL workflow.
This workflow summarizes the main steps after organ collection: experimental setup (Steps 1–5), pretreatments (Steps 6–10), labeling and clearing (Steps 11–18), imaging (Steps 19–39) and data processing (Steps 40–79). We describe two ways of handling samples: active perfusion with a pump, if the blood vessels can be connected with external tubes, and passive incubation on a shaker. We also outline the differences in the procedure that are dependent on the organ components, organ size, targeted labeling and imaging equipment.
Supplementary information
Supplementary Information
Supplementary Table 1.
Supplementary Video 1
Change position of tubing to prevent breakage during active pumping.
Supplementary Video 2
Use a syringe to remove bubbles in the cleared sample.
Supplementary Video 3
Set the start point and end point in the Z direction during the light-sheet microscope imaging.
Supplementary Video 4
Human organs with vessel labeling.
Rights and permissions
About this article

Cite this article
Mai, H., Rong, Z., Zhao, S. et al. Scalable tissue labeling and clearing of intact human organs. Nat Protoc 17, 2188–2215 (2022). https://doi.org/10.1038/s41596-022-00712-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-022-00712-8
This article is cited by
-
Spatial landmark detection and tissue registration with deep learning
Nature Methods (2024)
-
Current and future applications of light-sheet imaging for identifying molecular and developmental processes in autism spectrum disorders
Molecular Psychiatry (2024)
-
Three dimensional evaluation of cerebrovascular density and branching in chronic traumatic encephalopathy
Acta Neuropathologica Communications (2023)
-
Light sheet fluorescence microscopy of cleared human eyes
Communications Biology (2023)
-
Efficient 3D light-sheet imaging of very large-scale optically cleared human brain and prostate tissue samples
Communications Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
