
Abstract
Structure-based docking screens of large compound libraries have become common in early drug and probe discovery. As computer efficiency has improved and compound libraries have grown, the ability to screen hundreds of millions, and even billions, of compounds has become feasible for modest-sized computer clusters. This allows the rapid and cost-effective exploration and categorization of vast chemical space into a subset enriched with potential hits for a given target. To accomplish this goal at speed, approximations are used that result in undersampling of possible configurations and inaccurate predictions of absolute binding energies. Accordingly, it is important to establish controls, as are common in other fields, to enhance the likelihood of success in spite of these challenges. Here we outline best practices and control docking calculations that help evaluate docking parameters for a given target prior to undertaking a large-scale prospective screen, with exemplification in one particular target, the melatonin receptor, where following this procedure led to direct docking hits with activities in the subnanomolar range. Additional controls are suggested to ensure specific activity for experimentally validated hit compounds. These guidelines should be useful regardless of the docking software used. Docking software described in the outlined protocol (DOCK3.7) is made freely available for academic research to explore new hits for a range of targets.
Similar content being viewed by others

Introduction
Screening chemical libraries using biophysical assays has long been the dominant approach to discover new chemotypes for chemical biology and drug discovery. High-throughput screening (HTS) of libraries of 500,000 to 3 million molecules has been used since the 1990s1, and multiple drugs have had their origins in this technique2. While the libraries physically screened in HTS were an enormous expansion on those used by classical, pre-molecular pharmacology3, they nevertheless represent only a tiny fraction of possible ‘drug-like’ molecules4. DNA-encoded libraries5, where molecules are synthesized on DNA that encodes their chemistry, begin to address this problem by offering investigators libraries of 108 molecules, sometimes more, in a single, highly compact format; and multiple such libraries can be used in a single campaign. However, as DNA-encoded libraries are restricted to reactions on DNA, reaction chemistries are limited to aqueous solutions, thereby limiting the type of chemical reactions and subsequent chemical libraries available with this technology6.
Computational approaches using virtual libraries are an attractive way to explore a much larger chemical space7. Large numbers of molecules—certainly into the tens of billions, and likely many more—may be enumerated in a virtual library. Naturally, very few of these compounds can ever be actually synthesized because of time, cost and storage limitations, but one can imagine a computational method to prioritize those that should be pursued. In practice, this idea has had two limitations that have prevented wide-scale adoption: the virtual libraries have rarely been carefully curated for true synthetic accessibility8, and there were well-founded concerns that computational methods, such as molecular docking, were not accurate enough to prioritize true hits within this large space9.
In the last several years, however, two advances have at least partly addressed these problems:
First, several vendors and academic laboratories have introduced ‘make-on-demand’ libraries based on relatively simple two- or three-component reactions where the final product is readily purified in high yields10. At Enamine, a pioneer in this area, >140 reactions may be used to synthesize products from among >120,000 distinct and often highly stereogenic building blocks, leading to a remarkably diverse and, critically, pragmatically accessible library of currently over 29 billion molecules11.
Second, structure-based molecular docking, for all of its problems, has proven able to prioritize among these ultra-large libraries, if not at the tens of billion molecule level, then at the 0.1–1.4 billion molecule level12,13,14, finding unusually potent and selective molecules against several unrelated targets (Table 1). Indeed, simulations and proof-of-concept experiments suggest that, at least for now, as the libraries get bigger, docking results and experimental molecular efficacies improve12.
If docking ultra-large libraries brings new opportunities, it also brings new challenges. Docking tests the fit of each library molecule in a protein binding site in a process that often involves sampling hundreds-of-thousands to millions of possible configurations. Each molecule is scored for fit using one of several different scoring functions15,16,17,18. To be feasible for a billion-molecule library on moderately sized computer clusters (e.g., 500–1,000 cores), this calculation must consume not much more than 1 s/molecule/core (1 ms/configuration).
This need for speed means that the calculation cannot afford the level of detail and number of interaction terms that would be necessary to achieve chemical accuracy. For instance, docking typically undersamples conformational states, ignores important terms (e.g., ligand strain) and approximates terms that it does include (e.g., fixed potentials)19,20.
Owing to these approximations and neglected terms, docking energies have known errors, and the method cannot even reliably rank order molecules from a large library screen21,22. What it can hope to do, however, is separate a tiny fraction of plausible ligands from the much larger number of library molecules that are unlikely to bind a target. This level of prioritization is enhanced with the careful implementation of best-practice guidelines and controls. It is the goal of this essay to provide investigators with such best practices and control calculations for ultra-large library docking, though they equally apply to modest-sized library docking (Table 1). These will not ensure the success of a prospective docking campaign—the only true test of the method—but they may eliminate some of the more common reasons for failure.
We begin by describing protocols and controls that can be used across docking programs and that are general to the field (Fig. 1). There are by now multiple widely used and effective docking programs23,24,25,26,27,28,29,30,31, employing different strategies for sampling ligand orientations and ligand conformations in the protein site, for handling protein flexibility, and for scoring ligand fit once they have been docked. Notwithstanding these differences, there are strategies and controls that may be used across docking programs, including how to prepare protein sites for docking calculations, benchmarking controls to investigate whether one can identify known ligands from among a large library, and controls to investigate whether one’s calculations contain biases towards particular types of interactions. Since the true success of a virtual screen is the experimental confirmation of docking hits, we also propose a set of control assays to validate initial in vitro results. We then turn to protocols that are specific to the docking program we use in our own laboratory, DOCK3.7—these necessarily get into fine details, and will be of most interest to investigators wanting to use this particular program.

The two required inputs for such a screen are the target structure and a screening database. Prior to using the database, the target structure must be converted into a representation used by the docking software and the pocket should be optimized with control calculations using retrospective analysis on known actives. After the prospective library has been docked, top-ranked hits can be filtered and selected for experiment. Multiple assays and controls are typically necessary to confirm activity.
General guidelines for virtual structure-based drug discovery
Structure preparation and suitability for docking
Any structure-based campaign begins with a suitable target site
The most promising starting point for a virtual screening campaign is typically a high-resolution ligand-bound structure. Ligand-bound (holo) structures usually outperform ligand-free (apo) structures as the geometries of the binding pocket are better defined in the bound state than in the unbound state32,33. If there is no available holo structure, tools such as SphGen34, SiteMap35 and FTMap36 can be used to identify potential ligand binding sites.
Generally, small, enclosed binding pockets that well complement a ligand perform better than large, flat and solvent-exposed binding sites typical of protein–peptide or protein–protein interactions. For instance, neurotransmitter G-protein-coupled receptor (GPCR) orthosteric sites, such as the β2 adrenergic, D4 dopamine, histamine H1 and A2a adenosine receptors37,38,39,40 typically have higher hit rates and more potent docking hits than do peptide receptors like the CXCR4 receptor41, and these often perform better than the more open sites typical of soluble enzymes like β-lactamase12,42. In most cases, targeting protein–protein interaction surfaces, outside of a few that are well defined43, often leads to disappointing results.
Modifying the high-resolution protein structure
It is not always a good idea to use the structure exactly as it was found in the database.
-
Dealing with mutations. For stability, crystallization and other biochemical reasons, high-resolution protein structures are sometimes determined in a mutant form; such mutations should be reverted to the wild type especially if they are within the ligand site to be targeted. Missing side chains and loops in the experimental structures should be added as well if they are close to the binding site, while those that are weakly defined by the experimental observables (e.g., low occupancy, high displacement parameters (B values), poor electron density) should be examined critically
-
What about water molecules? When the resolution permits, water molecules can also be included in the target preparation, often treating them as nondisplaceable parts of the protein structure. Typically, water molecules enclosed in the targeted binding pocket or involved in interactions between the co-crystallized ligand should be considered as they may determine side-chain conformations or offer additional hydrogen-bonding sites. Some docking programs will allow water to be switched on or off during the docking44 or include other implicit solvent terms45
-
Buffer components. Buffer components such as PEG and salts are likely specific to the crystallization conditions, and should be removed
-
Cofactors. Cofactors like heme or metal ions should be considered if they are involved in ligand recognition
-
Hydrogen atoms. Due to the resolution limits of most experimentally determined structures, hydrogen atoms are often unresolved. The position of many hydrogen atoms can be readily modeled according to holonomic constraints (e.g., backbone amide hydrogen atoms). For those that are not, such as hydroxyl protons on serine and tyrosine residues, imidazole hydrogens on histidine, and the adjustment of frequently erroneous terminal amide groups for glutamine and asparagine residues46, programs like Reduce47 (default in DOCK3.7), Maestro (Schrödinger)48, PropKa49 or Chimera50 can be used to protonate the target of interest. While these automated protocols usually produce reasonable protonation states, special care should be taken for the residues within the ligand binding pocket or residues that form a catalytic/enzymatic site. Lastly, although less frequently encountered, glutamic and aspartic acids can adopt protonated forms under specific circumstances51
-
In summary, the protonation of the protein structure is critical for the success of docking to more accurately depict the Van der Waals (VDW) surface and dipole moments of the binding pocket
Homology modeling
When no experimental structure has been determined for the target protein, structural models can be generated if a template structure with high sequence identity is known. Common programs used for homology modeling include Modeller52, Rosetta53, ICM27 and I-Tasser54. These two principles can improve the chances of success:
-
Typically, the higher the sequence identity between the target and the template, the better the accuracy of the model55. Particular focus should be given to identity within the target binding pocket; if there is a choice, choose the template that has the highest identity in the binding pocket
-
Incorporation of a ligand during the modeling process or ligand-steered homology modeling approaches56,57 will help prevent the pocket from collapsing inward, and will better orient the side chains of binding residues32,58,59
When it is unknown how a ligand binds within the pocket, orthogonal experimental data can guide the modeling such as iterating between docking and modeling60,61. In the case of MRGPRX262 and GPR6863, for instance, the authors predicted multiple binding poses of a known active ligand and used mutagenesis and binding assays to test these predictions. A binding mode and receptor structure were identified that was consistent with the mutagenesis data and used for subsequent preparation in the prospective screen. Despite many difficulties, homology models have been successful in identifying novel ligands from prospective docking campaigns41,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76; though it is also true that, given the choice, most investigators will prefer to use a well-determined experimental structure.
Control calculations
Docking undersamples ligand–protein configurations and conformations, and its scoring of these configurations for fit remains highly approximate. Unlike methods like quantum mechanics, or certain lattice calculations in statistical mechanics, docking has surrendered ‘ground truth’ to be able to pragmatically search among a large and growing chemical space. Accordingly, control calculations are critical to the success of a docking campaign. As with experimental controls, they do not ensure prospective success, but they do guard against obvious sources of failure, and can help one understand where things have gone wrong if they do. Through a key control, we assess whether the prepared binding pocket and docking parameters can prioritize known ligands over presumed inactive molecules. In an optimized binding pocket, these known actives should rank higher against a background of decoy molecules in a retrospective screen, and reasonable poses should be predicted.
As it is more likely to know true actives than true inactives, it is common practice to use property-matched decoys77, which are compounds that have similar physical properties as the actives, but unrelated topologies, and so are presumed inactive. The DUDE-Z server (tldr.docking.org) was built specifically to generate decoys for a given list of ligands by matching the following physical properties: molecular weight, hydrophobicity (LogP), charge, the number of rotatable bonds, and the number of hydrogen bond donors and acceptors78.
The performance of the parameterized binding pocket to enrich known ligands over decoys can be evaluated by receiver-operator characteristic (ROC) curves, quantifying the true positive rate as a function of false positive rate (Fig. 2)17,77,79,80,81. The area under the curve (AUC) is a well-regarded metric to monitor the performance of a virtual screen by a single number17 (Fig. 2a). The log transformation of the false positive rate enhances the effect of early enrichment for true positives17 (Fig. 2b). This is important because, in a docking campaign with hundreds of millions of molecules, only compounds ranked within the top 0.1% are often closely evaluated (see ranks in Table 1). For example, if a retrospective docking challenge shows that known actives are only identified starting around the tenth percentile, novel actives may be missed in a prospective screen. In this setting, higher LogAUC values correspond to better discrimination between actives and inactives and provide a sanity check on the ability of the docking parameters to identify actives.

a, ROC curves for two models used for retrospective docking screens plotting the rate of true positives found against decoys found. The AUC can be used to describe the ability of the models to identify true positive, known ligands against a background of decoys. In this format, the two models have similar AUCs, suggesting similar performance. b, Semilogarithmic ROC curves focus on the early enrichment, i.e., determine if true positives are identified within the e.g. top 10% (gray area) of docked decoys. The LogAUC is calculated as the difference between the semilogarithmic AUC of the model and the random semilogarithmic AUC (dashed line). In this format, it is clear that model 2 outperforms model 1 in early enrichment with a LogAUC value more than double of model 1.
A second criterion is the pose fidelity of the docked ligands to their experimental structures. The validity of predicted binding poses can be assessed qualitatively by visual inspection of reported key interactions between protein and ligand or, in the best case, quantitatively by calculating root mean square deviations between predicted and experimentally determined poses82. During pocket modeling and docking parameter optimization, one will often insist that the retrospective controls lead to both high LogAUC values and good pose fidelity; often there will be some trade-off between the two. While calibrating the scoring functions, it is also important to monitor the contributions of each energy term to the total score and ensure they match the properties of the binding pocket. If one term dominates, the scoring may have been overoptimized to that term, while other protein–ligand interactions are underweighted, leading to dominance by a certain type of molecule. For instance, if the docking score of a polar solvent-exposed pocket is inappropriately dominated by VDW energies, large molecules may score high due to nonspecific surface contacts with the target protein.
In addition to property-matched decoys, other chemical matter can be used to evaluate different aspects of the docking model (Fig. 3). A test set including molecules with extreme physical properties (Extrema set), such as a wide variety of different net charges, can be screened to measure whether the docking model succeeds in prioritizing molecules with net charges corresponding to known actives78. If there is a difference in the charge of top-ranked compounds from the extrema set and the charges of the known ligands, the scoring may be biased. In another useful control experiment, a small fraction of the purchasable chemical space (e.g., readily available ‘in-stock’ compounds from multiple vendors) representing the characteristics of the ultra-large make-on-demand screening library can be docked against the protein model. This control serves two purposes: to test whether the positive control molecules remain among the highest scored compounds and to examine if intriguing novel compounds rise to the top of the rank-ordered docking list. It may even be fruitful to purchase and experimentally test a few promising and structurally diverse in-stock compounds as it could inform the docker if the binding pocket model is likely to find hits in the ultra-large library screen. If top ranked compounds do not form expected interactions, further optimization may be beneficial.

For DUDE-Z decoys, properties of the decoys are either forced to match (green) or be different (red). Properties that are neither selected for or against are highlighted in yellow. In the Extrema set, the charge state is explicitly sampled.
Another control, if available, are true inactives from a previous discovery campaign. These can be used as a background against known actives to provide a ‘real-world’ benchmark of the performance of the system. Lastly, for a protein that has little or no known chemical matter (i.e., reported ligands), enrichment calculations with known actives against a background of property-matched decoys may be impossible. Here, the docking parameters can be calibrated by docking ‘Extrema’, which challenges the docking with extremes of physical properties, and ‘in-stock’ compound sets, which probe how the docking will perform on a representative subset of the library78,83. It remains true that, without known ligands as positive controls, one is at a substantial disadvantage at setting up docking campaigns, increasing the risk of failure.
Prospective screen
Once the docking model is calibrated, large libraries of molecules can be virtually screened against the target protein. For this virtual screen, it makes sense to focus on compounds that are readily available for testing. The ZINC20 database (http://zinc20.docking.org/) enumerates over 14 billion commercially available chemical products, of which ~700 million are available with calculated 3D conformer libraries ready for docking. Most of the enumerated compounds belong to the make-on-demand libraries of Enamine and WuXi. Further, ZINC20 allows one to preselect subsets of molecules for docking, reducing computation time. For instance, ZINC20 allows users to download ready-to-dock subsets of molecules within user-defined ranges of molecular weight (MWT), LogP and net charge, as well as predefined sets such as fragments (MWT ≤ 250 amu) or lead-like molecules (250 ≤ MWT < 350 amu; LogP ≤ 3.5). The result of a prospective screen is a list of molecules rank-ordered by docking score.
Hit-picking
A well-controlled docking calculation can concentrate likely ligands among the top-ranked molecules. But even if it was able to do so among the top 0.1% of the ranked library, in a screen of 1 billion molecules this would still leave 1 million molecules to consider, and with the errors inherent in docking, many of these will be false positives. Accordingly, we rarely pick the top N-ranked compounds by docking to test experimentally, but rather will use additional filters to identify promising hits within the top scoring 300,000–1,000,000 molecules. These filters can catch problematic features missed by the primary docking function, ensure dissimilarity to known ligands and promote diversity among the prioritized compounds.
Compounds may be filtered for both positive and negative features84 (Table 2). For instance, one may insist that a docked orientation has favored interactions with key residues. Conversely, molecules with strained conformations should be discarded. Molecules with unsatisfied, buried hydrogen-bond donors and acceptors may also be deprioritized. Compounds with metabolic liabilities85,86 or that closely resemble colloidal aggregators87 can also be filtered out, despite otherwise favorable scores. Further, as closely related compounds will likely dock in similar poses with similar score, we typically cluster compounds by 2D structure similarity after all other filters have been used and only select the best scoring cluster representative for testing. In such a large dataset, clustering can be computationally expensive, so reduced scaffold clustering such as Bemis–Murcko88 analysis is useful to efficiently parse the compounds. In principle, many of these filters could be included directly in the scoring functions, but balancing them against other scoring terms can demand extensive optimization and will likely increase the compute time, which will become a hindrance for ultra-large library docking. Lastly, visual examination of the docked poses has been useful for selection of compounds to purchase. Following the criteria in Table 2, we typically inspect up to 5,000 compounds visually after applying automated filtering and clustering steps.
Experiments to test docking hits
The success of a docking campaign is ultimately measured by its ability to reveal novel chemotypes that can be shown to experimentally bind to the target, typically in binding or functional assays (Fig. 4). However, common artifacts should be controlled for: chemotypes likely to interfere with specific assays89 (e.g., the controversial90 pan assay interference chemotypes), covalent adducts, redox cyclers91 and aggregators87,92.

In general, a primary screen will use a limited number of compound concentrations to test for activity at a target. Compounds that pass a set threshold of activity in the primary screen will be moved forward to secondary confirmation of activity that is not attributed to colloidal aggregation. Identity of the compound should be confirmed if it passes these stages and before proceeding to optimization by stereoisomer purification, selection of analogs and/or experimental structure determination.
Among the most common of these mechanisms is colloidal aggregation, which can account for >90% of all primary hits89,93. This aggregates sequester proteins with little selectivity, partially denaturing and inhibiting them32,92,94,95,96, occasionally even activating them97,98, a common problem both in HTS and also in docking screens92. Aggregators tend to have high LogP values and limited aqueous solubility, so we prefer to dock and test molecules with LogP ≤ 3.5. Chemical stability or reactivity can also contribute to experimental artifacts, and reactive scaffolds should be avoided. The ZINC20 database allows users to select screening libraries in the lead-like chemical space (i.e., low LogP) and exclude reactive compounds9,99. Unless controlled for in the experimental setting (Box 1), these artifacts can lead to false positives.
Once convinced that one’s hits are not artifactual, more detailed testing on-target is warranted. For all targets, this involves determining concentration response curves, typically with 7–12 points at half-log intervals, with the transition being sampled over at least two log orders of concentration (Fig. 4). For enzymes, determining a true Ki will illuminate mechanism, though this can be laborious and it might only make sense to do this for characteristic lead molecules. For receptors such as GPCRs and ion channels, functional assays are typically performed to determine if the new ligand is acting as an agonist, an antagonist or an allosteric modulator. For GPCRs, initial screening assays may differ from secondary confirmatory assays, and showing that a molecule is active in more than one assay is often quite useful.
Before initial hits are advanced for optimization, it is important for make-on-demand libraries to confirm the identity of the hit compound. Limitations of virtual library enumeration, chemical synthesis and downstream purification can result in mismatches between the in silico predicted compound and the in vitro tested compound. For example, in the screen against the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) macrodomain protein, a number of purine derivatives with an N9 substitution were requested but N3-substituted compounds were synthesized instead83. Accordingly, it is worth fully characterizing promising compounds before costly lead optimization is undertaken; we ourselves have made the mistake of designing analogs based on an in silico starting scaffold that was different than the true hit delivered from the chemists. At a minimum, the identity and purity of the compounds should be determined. High-resolution mass spectrometry or quantitative 1D proton nuclear magnetic resonance spectroscopy can be used to detect gross variation between the tested sample and the expected molecule; these data can be obtained from most vendors, or the experiments could be performed independently.
The most direct experiment to test a docking pose prediction is the determination of an experimental protein structure in complex with the docking hit (Fig. 4). Such structures illuminate crucial details of ligand recognition, including adjustments of binding site residues or the docking hit in the binding pocket12,100,101. The key question to answer with high-resolution structures is whether docking worked for the right reasons, i.e., does the predicted binding mode agree with an experimental structure? Previous studies suggest that hits from virtual screening generally compare fairly well with experimental high-resolution structures; i.e., key anchor points are predicted correctly12,83,102,103. However, as docking is typically performed against rigid protein structures, conformational changes, especially in flexible loops, will complicate pose prediction104. Water-mediated protein–ligand interactions are generally difficult to explore de novo45,105, though experimentally determined and structurally conserved water molecules can be included in virtual screens100. Lastly, there are few cases where docking hits showing on-target activity revealed binding positions at unexpected and nontargeted subsites. For example, the β2 adrenergic receptor allosteric modulator AS408 was predicted to bind to an extracellular subpocket but crystallographically determined to bind to the membrane-facing surface of the receptor106. In contrast, the pose of an inverse agonist also identified at the β2 adrenergic receptor from in silico docking39 was confirmed by X-ray crystallography107, demonstrating the variability in outcomes even at the same target.
If the protein target can be readily expressed and purified in milligram quantities, crystallography and cryo electron microscopy can guide hit discovery early in the campaign. In campaigns against targets where high-resolution structures are more challenging to obtain (e.g., for transmembrane receptors), an experimental protein–ligand complex might only be achievable for the most potent hit compound. Nonetheless, it is usually worth the effort to confirm the predicted binding site and ligand pose, or to identify unexpected interactions between the discovered hit and the target106.
Next steps: selecting analogs for hit to lead optimization
In the fortunate event that the virtual screen was successful and docking hits were confirmed by different experiments, the newly obtained scaffolds are blueprints for exploring structure–activity relationships and lead optimization.
A concern about molecules from make-on-demand libraries is that they will initially be delivered as racemic or stereomeric mixtures since the production of pure stereoisomers requires more sophisticated synthesis routes. If confirmed hits were observed from stereomeric mixtures, the measured potency of the mixture may be artifactually lower if only one stereoisomer is active at the target (e.g., the concentration of the active stereoisomer is less than the total mixture concentration). Purified stereoisomers can typically be purchased from the make-on-demand compound supplier or separated in-house.
Hit optimization can be performed in several ways. Synthetic chemistry groups may obtain synthesis routes of the parent compound from the supplier allowing the generation of medicinal chemistry-inspired analog series108. Alternatively, chemoinformatic tools can be employed to virtually search the purchasable chemical space for structurally related scaffolds or identifying molecules with a common substructure to the parent hit compound (analog-by-catalog). Searching for similar molecules within the Enamine REAL space can be conducted using the SmallWorld (sw.docking.org) search engine99. Molecules with an identical substructure as the parent compound can be found using Arthor (arthor.docking.org)99. Additionally, the supplier of the parent compound might be able to provide a collection of molecules resembling the hit scaffold. While we cannot provide clear guidance or a best-practice protocol for hit-to-lead optimization, analog-by-catalog of docking hits obtained for the melatonin 1 (MT1) receptor and AmpC led to compounds with improved potencies compared with the initial parent compounds12,13. Currently, we recommend subtle changes to the starting compound or modifications to test particular interactions as suggested from the docked pose.
It is our hope that the above guidelines will be useful for outlining a docking campaign from start to finish. As shown, the prospective docking step is actually only one small component of the overall pipeline (Fig. 1). Control calculations using retrospective datasets and docking setup optimization make up the bulk of the process. Hit picking from the prospective screen requires careful perusal of the data, and experimental design of the confirmatory assays is critical for defining success of the campaign. We ourselves find that these guidelines insulate against the more common sources of failure in large library docking campaigns. Lastly, we want to mention that docking campaigns against protein targets without experimental structure, i.e., requiring homology modelling, or without known active chemical matter for retrospective control calculations are particularly risky and should not be initiated naively.
This concludes our general guidelines for large library docking. In the last section, we turn to a detailed protocol to set up, optimize and prospectively screen a target of interest using DOCK3.7, though any docking program can use the controls presented.
While the protocol should be general for most proteins, we provide example data from a recent campaign against the MT1 receptor13, a target particularly well suited for docking: a crystal structure had been determined; the orthosteric pocket is compact and almost completely encloses the ligands, simplifying the biophysics; many such ligands exist for retrospective calculations and for optimization; and in vitro assays to test docking hits were well established. We note that the MT1 receptor is ideal for large library docking, and so the achieved hit rate, and the hit potencies, were unusually high. Still, the docking optimization strategies below have been useful against a wide spectrum of targets, including the Nsp3 macrodomain of SARS-CoV-283 and highly solvated pockets as in β-lactamase12, and were developed against the spectrum of targets in the DUD-E benchmark77, ranging from ion channels to kinases to soluble enzymes. It should be clear that the optimization strategies sketched remain rooted in retrospective controls, and so will work best against targets with precedented ligands, and against targets with well-formed and readily liganded binding sites.
Docking campaigns with DOCK3.7 and ZINC20
In DOCK3.7, ligands are placed in the target pocket by mapping ligand atoms onto predefined hot-spots, so-called matching spheres. Matching spheres are generated from the coordinates of heavy atoms from an input bound ligand structure, if available, and supplemented with coordinates based on the negative image of the binding site generated from the program SphGen34. Ligand rigid fragment (e.g., rings) are mapped onto matching spheres using a bipartite graph algorithm79,109. Different ligand conformations and orientations are sampled in the binding pocket using precalculated 3D conformer libraries (flexibases, db2 files)9,99. During conformer library building, each molecule is divided into its rigid fragments, and different conformations of rigid fragment substituents are generated.
Ligand poses are evaluated using a physics-based scoring function (Escore) combining VDW, electrostatic (ES) and ligand desolvation (lig_desol) energy terms (Equation 1):
In order to allow rapid scoring of new poses (typically 1,000 poses per molecule per second), contributions of the target protein pocket are mapped onto pregenerated scoring grids. The VDW surface of the binding pocket is converted into a scoring grid by ChemGrid15. Electrostatic potentials within the binding pocket are estimated by numerically solving the Poisson–Boltzmann equation using QNIFFT16,110. Context-dependent desolvation energy scoring grids, both polar and apolar, are generated by the Solvmap program17. Thereby, ligand desolvation energies are computed as the sum of the atomic desolvation of each ligand atom scaled by the extent to which it is buried within the binding site. Atomic desolvation energies for ligand atoms are calculated during ligand building using AMSOL111 and are included in the ligand conformer library. Both, the electrostatic and ligand desolvation scoring grids depend on the dielectric boundary between the low dielectric protein (εr = 2) and high dielectric solvent (εr = 80) environments. Consequently, these scoring grids can be fine-tuned by modulating the protein–solvent dielectric interface (see below Steps 41–44).
The following protocol (Fig. 5) prepares a starting structure (Steps 1–4), generates the scoring grids and matching spheres (Steps 5–10), and collects a set of control ligands for model evaluation (Steps 11–33). Control ligands are docked retrospectively (Steps 34–58) to determine the model’s ability to identify actives from a pool of inactives. Optimization of sampling (Steps 59–64) and scoring (Steps 65–77) is evaluated with the same controls. Model biases are evaluated using an Extrema control set (Steps 78–88) to examine for charge preferences in the scoring grids and with a small-scale in-stock screen (Steps 89–97) to ensure a computationally expensive large-scale prospective screen (Steps 98–103) is likely to yield interesting chemical matter. Hit picking (Steps 104–108), though critical to success of these prospective experiments, is beyond the scope of the protocol as it is highly user- and target-specific, and we suggest referring to Table 2 for insight. Additionally, we caution against regarding hits from a screen as true positives until common artifacts are ruled out (Box 1). All controls in silico and in vitro are applicable to any docking program or computer-aided drug discovery campaign. The details of grid preparation and modification are DOCK3.7-specific, but the principles are transferrable to other software.

Collecting and preparing materials (blue) requires obtaining a structure or model and ligand control sets and setting them up for retrospective control calculations (yellow). In each control calculation, modifications may demand returning to a previous step and reoptimizing. In the absence of known actives for robust retrospective analysis, one may jump to testing the prospective performance with a small library. With a final setup, large-scale prospective screening (orange) can proceed, followed by in vitro testing of docking hits (green). The numbers refer to steps described in the Procedure.
Materials
Software
-
DOCK3.7.5: apply for a license from http://dock.docking.org/Online_Licensing/dock_license_application.html. Licenses are free for nonprofit academic research. Once your application is approved, you will be directed to a download for the source code. The code should run without issue on most Linux environments, but can be optimized by recompiling with gfortran if needed. Questions related to installation can be addressed to dock_fans@googlegroups.com
-
Python: the current code uses python2.7, which will also need to be installed. Additional python dependencies that are required for running scripts can be found in the file $DOCKBASE/install/environ/python/requirements.txt
-
AMSOL: free academic licenses can be obtained from https://comp.chem.umn.edu/amsol/
-
(Optional) 3D ligand building software: if interested in 3D ligand building in-house (not necessary for this protocol), licenses will also need to be obtained for ChemAxon (https://chemaxon.com/), OpenEye Omega (https://www.eyesopen.com/omega) and Corina (https://www.mn-am.com/products/corina). We note that, for many campaigns, 3D molecular structures with all necessary physical properties may be downloaded directly from ZINC20. This tutorial makes use of a webserver for 3D ligand building that is suitable for small control sets. Please apply for an account at https://tldr.docking.org/, which is free of charge
-
Chimera: this application is recommended for grid visualization with the VolumeViewer feature (see Troubleshooting). The ViewDock feature, also within Chimera, is useful to examine the docking results
Equipment
-
Hardware: initial tests of this tutorial (grid preparation and small retrospective screens) can be performed on a single workstation, but more intensive docking will need access to a high-performance computing cluster (we regularly use 1,000 cores)
-
Queuing system: DOCK3.7 comes with submission scripts for SGE, Slurm and PBS job schedulers. If using a different scheduler, scripts may need to be adapted for your specific computing cluster
-
Protein structure: structures can be downloaded from www.rcsb.org or generated by the user in the case of homology models
-
Example data: an example set of files used in this protocol including ligand and decoy sets, default docking grids and optimized docking grids can be downloaded from http://files.docking.org/dock/mt1_protocol.tar.gz. The example dataset uses the MT1 structure (PDB: 6ME3) co-crystallized with 2-phenylmelatonin
Equipment setup
Define the pathway to critical software
The environment will need to be defined in order to run the following commands correctly. The two most important paths that need to be defined are to DOCKBASE and python. We provide an environment script in the example dataset that will need to be modified for a user’s settings. Always run the following command with updated paths specific to your file directory before working with DOCK3.7:
source env.csh
Procedure
Section 1: set up binding pocket for docking calculations
Timing 30 min to 1 h
-
1
Download the structure from the PDB or use in-house structures. Preference should be given to structures of high resolution and with a ligand bound in the site that will be docked into. Apo structures are useable, but tend to yield poorer results due to unconstrained binding site geometries32.
-
2
Visualize the binding pocket in PyMOL or Chimera to decide on the relevance of protein cofactors.
-
Delete components from the structure that do not contribute to ligand binding such as lipids, water molecules and buffer components
-
Fusion proteins engineered for protein stability that are near the binding pocket should be removed, and the resulting chain break should be either capped or the loop remodeled
-
Protein cofactors such as heme or ions should also be kept or discarded depending on their relevance to ligand binding in the pocket
-
Additionally, examine the residues in the binding pocket for incomplete sidechain, multiple rotamers or engineered mutations, and revert or rebuild as necessary
-
It is helpful to examine the pocket with the electron density if available for making these decisions. For the example MT1, we deleted the fusion protein, capped non-native termini and reverted two mutations in the binding pocket (6me3_cleaned.pdb)
-
-
3
Save the rec.pdb file. This file will contain any component that will remain static during the docking such as structural waters and cofactors.
-
4
Save the xtal-lig.pdb file. This file will contain the atoms of the bound ligand, which will be used to generate the matching spheres that guide ligand sampling in the pocket.
-
5
Generate the scoring grids and matching spheres with blastermaster from these two inputs (rec.pdb and xtal-lig.pdb)
$DOCKBASE/proteins/blastermaster/blastermaster.py --addhOptions=" -HIS -FLIPs " –v
-
6
Inspect the output, in particular the rec.crg.pdb file located in the new working directory to ensure protonation of polar residues and side chain flips of glutamine and asparagine side chains look accurate. Examine histidine tautomers (HID, HIE and HIP) as well. If everything looks as it should, proceed to Step 10; otherwise, proceed to Step 7.
Generating a rec.crg.pdb file manually
-
7
If the automatically protonated rec.crg.pdb file does not fit to the expected protonation state of key residues, the rec.crg.pdb file can be generated manually using various protein modeling software packages including Rosetta112, Chimera50 or Maestro113. This may include manually flipping side chain rotamers and setting the pH value for pKa calculation of charged residues. After the modeling step is completed, new rec.pdb, rec.crg.pdb and xtal-lig.pdb files need to be provided to blastermaster. To be compatible with the united atom AMBER force field, rec.crg.pdb should only contain polar hydrogen atoms, and explicit histidine tautomer names. rec.pdb and xtal-lig.pdb only need to contain heavy atom coordinates. In Step 8, we provide a script that produces the required files.
-
8
Assuming the protein modeling software resulted in a fully protonated protein–ligand complex, protein atoms should be listed as ATOM records, while ligand atoms should be listed as HETATM records. Further, ensure that cofactors are given the correct heading in the PDB file (i.e., static metal ions should be given ATOM records if they are to be included in the scoring grids).
bash $DOCKBASE/proteins/protein_prep/prep.sh protonated_input.pdb $PWD
The outputs are new rec.pdb and xtal-lig.pdb files, as well as a rec.crg.pdb file inside a working directory. HID, HIE and HIP naming is generated automatically from the protonation state of the HIS residues and again should be checked for accuracy.
Note: this script requires a path to the Chimera binary file and may need to be modified by the user.
-
9
Generate scoring grids and matching spheres from the new files (rec.pdb, xtal-lig.pdb, and working/rec.crg.pdb), and examine the outputs as in Step 6:
$DOCKBASE/proteins/blastermaster/blastermaster.py --addNOhydrogensflag –v
Checking the files
-
10
Check that all files are generated. At the end of a successful blastermaster run, the directory should contain an INDOCK file and two directories: working and dockfiles. The working directory contains all the intermediate files used for grid generation. The dockfiles directory contains the scoring grid files and matching spheres file. The INDOCK file contains all parameters to control the docking program, such as sampling options and location of input files, i.e., docking grids and 3D ligand conformer libraries (see the INDOCK Guide in Supplementary Information). In our experience, the automated grid generation with blastermaster.py successfully produces reliable docking parameters; however, nonstandard amino acids or particular atom types require additional information and adjustments of force field parameters. Instructions on how to check and adopt the grid generation are provided in the Troubleshooting section below and in the Blastermaster Guide (Supplementary Information). For the provided example files generated for MT1, the protein–ligand complex was modeled using the automated preparation pipeline in Maestro, Schrodinger48.
Section 2: collect and build the control ligand set
Timing Minutes to hours for ligand building, depending on the number of molecules
-
11
Collect the known actives (positive controls) for retrospective analysis. For a given target, these can be found in the scientific literature, patent literature or public databases such as IUPHAR/BPS114, ChEMBL115 or ZINC9,99,116, or available in-house.
Curate the actives list
Critical
While it may be possible to find dozens of actives, it is likely that many come from the same chemical series. For a rigorous control analysis, redundant (i.e., highly similar) compounds should be clustered and the most potent compound selected.
-
12
Sort all knowns by potency.
-
13
Cluster these based on 2D similarity using any preferred method. We suggest calculating clusters based on ECFP4 Tanimoto similarities with a cutoff of 0.35.
-
14
Use the most potent compound from each cluster as the representative of that scaffold. It is best to refine these actives to a set that is representative of the chemical space intended for prospective screening (i.e., with limits on molecular weight) so that the retrospective analysis will match the prospective aims.
-
15
Find out whether the known ligands of the target are neutral or charged as one criteria of the docking parameter calibration is the ability to score ligands with corresponding charges well.
-
16
Save a final list of actives’ SMILES as ligands.smi. Typically, 10–30 diverse actives represent a good control set. For targets with less than this value, the controls are still useful but may not be as informative. For the example of MT1 we extracted a set of 28 agonist and antagonists from the IUPHAR/BPS database13.
Build the 3D conformer library of the actives
-
17
Go to the ‘build3d’ application on tldr.docking.org79.
-
18
Add the ligands.smi file to the ‘Input’ section.
-
19
Select ‘db2’ for the file type.
-
20
Click ‘Go’ to submit.
-
21
Download the build3d_results.tar.gz file, and move it to your work directory.
-
22
Decompress the tar file:
tar xvfz build3d_results.tar.gz
-
23
The results are in a build3d_results/ directory with two subdirectories failed/ and finished/ indicating the build status of the compounds. In example data provided, all ligands were built to completion.
-
24
To build a split database index file, or path file to the ligands, use the following command:
ls -d $PWD/build3d_results/finished/*.db2.gz > actives.sdi
-
25
To generate a list of actives’ IDs:
ls build3d_results/finished/*0.db2.gz | awk -F"/" '{print $NF}' | awk -F"_" '{print $1}' > actives.id
Curate and build the property-matched decoy set
-
26
Go to the ‘dudez’ application on tldr.docking.org78.
-
27
Submit the ligands.smi file to the input section.
-
28
Click ‘Go’ to start the calculation. The ZINC database will be scanned for compounds that match the following six properties of the input ligands: molecular weight, LogP, charge, number of rotatable bonds, number of hydrogen bond donors, and number of hydrogen bond acceptors. Each compound will then be compared with the actives for 2D similarity, with similar compounds being discarded. A final set of 50 decoys per input active will be calculated.
-
29
Download the decoys_dudez.tar.gz file, and move it to your work directory.
-
30
Extract the files:
tar xvfz decoys_dudez.tar.gz
The files will live in a new directory new_decoys.
-
31
To obtain the split database index file of the decoys:
ls -d $PWD/newdecoys/decoys/*.db2.gz > decoys.sdi
-
32
To obtain a list of all the decoys’ IDs:
awk ‘{print $2}’ newdecoys/decoys.smi > decoys.id
-
33
Collect output files. At this point, four files should be generated: actives.sdi, actives.id, decoys.sdi and decoys.id.
Section 3: run retrospective docking calculations to test the binding pocket parameters
Timing Minutes to hours depending on number of molecules and compute cores
-
34
In the directory where blastermaster was run (i.e., contains the INDOCK file and dockfiles directory), copy over the ligand and decoys .sdi files and .id files.
-
35
Combine the two .sdi files:
cat actives.sdi ligands.sdi » controls.sdi
-
36
Check values in the INDOCK file
-
Set the atom_maximum to 100. This value is a hard cutoff such that if a ligand has more than this number of heavy atoms it will not be docked. For retrospective calculations, we want all ligands to be docked and scored regardless of size, so we use a large value here
-
Set the mol2_maximum_cutoff to 100. This value is a cutoff for saving 3D coordinates of docked poses. If a ligand scores worse (i.e., more positive) than this cutoff, the pose will not be saved. Setting a low value is useful in large-scale prospective screens to save on disk space and computation time, but for retrospective screens we want all information saved for analysis
-
-
37
Set up the docking directory
$DOCKBASE/docking/setup/setup_db2_zinc15_file_number.py ./ controls controls.sdi 1 count
This script will separate the controls.sdi file into one directory called controls0000. For this first calculation, the size of the sdi file is manageable on a single core, though splitting it will be faster (i.e., 10). For control calculations with 10,000–100,000 ligands and for large-scale prospective screens, the sdi file should be split into multiple directories and the jobs distributed over multiple cores. A dirlist file is generated that lists all of the split directories prepared for docking.
-
38
Dock the control set. At this point, it is possible to submit the data to a computer cluster or do the calculation on a single computer.
-
(A)
Docking without submitting to a cluster
-
(i)
Move into the controls0000 directory. You will find INDOCK (pointing to the dockfiles/ directory) and a split_database_index file. These are all the inputs that the DOCK program needs to run the calculation.
-
(ii)
Run the docking calculation:
$DOCKBASE/docking/DOCK/bin/dock.csh
-
(iii)
The output files from the docking program are OUTDOCK, listing docking scores of successfully docked molecules, computational performance or potential error messages, and a zipped mol2 file (test.mol2.gz) containing the 3D poses of docked molecules.
-
(iv)
Move back into the directory that contains dirlist when done.
-
(i)
-
(B)
Docking with submitting to a cluster
-
(i)
In the directory that contains the dirlist (created in the previous step), run one of the following commands depending on cluster architecture:
-
SGE: $DOCKBASE/docking/submit/submit.csh
-
Slurm: $DOCKBASE/docking/submit/submit_slurm.csh
-
PBS: $DOCKBASE/docking/submit/submit_pbs.csh
Critical step
If you use another cluster scheduler, adopt these scripts to your needs.
-
-
(i)
-
(A)
-
39
When the job has completed, the last line of the OUTDOCK file should contain an elapsed time. If this is not the last line of the OUTDOCK file, an error has occurred (see Troubleshooting) and the following script will not run.
-
40
Extract the scores of all docked compounds
python $DOCKBASE/analysis/extract_all_blazing_fast.py ./ dirlist extract_all.txt 100
The arguments are the dirlist, the name of the file to be written (extract_all.txt) and the max energy to be kept (this value should match the mol2_maximum_cutoff value in the INDOCK file). The important output file for future analysis is the extract_all.sort.uniq.txt file, containing the rank-ordered list of docked compounds with only the highest score for each compound.
-
41
Get the poses of the docked compounds
python $DOCKBASE/analysis/getposes_blazing_faster.py ./ extract_all.sort.uniq.txt 10000 poses.mol2 test.mol2.gz
The arguments are the path where the docking is located (./), the name of the extract_all.sort.uniq.txt file, the number of poses to get (10000, i.e., set to larger than the number of compounds docked for retrospective calculations since we want to get all), the name of the output mol2 file (poses.mol2), and the name of the input mol2 files (test.mol2.gz), containing 3D coordinates of predicted poses from the docking calculation (located in the controls0000 directory).
-
42
Get the poses of just the actives
python $DOCKBASE/analysis/collect_mol2.py actives.id poses.mol2 actives.mol2
The arguments are the file containing active IDs, the pose file containing all of the compounds both actives and decoys, and the name of the output file to be written.
-
43
Calculate the LogAUC early enrichment
python $DOCKBASE/analysis/enrich.py -i . -l actives.id -d decoys.id
Inputs are the working directory (‘.’ if in the directory where the extract_all file is located) and the two files containing the IDs of actives and decoys. The output is a roc.txt and roc_own.txt file. Both files report an AUC and LogAUC and contain the grid points for plotting receiver operator curve (ROC) plots. The roc.txt file is calculated over all inputs in the ID files, and the roc_own.txt file is calculated only over the compounds that successfully docked.
-
44
To generate a plot (roc_own.png) of the roc_own.txt file:
python $DOCKBASE/analysis/plots.py -i . -l actives.id -d decoys.id
-
45
Calculate the charge distribution by DOCK score
python $DOCKBASE/analysis/get_charges_from_poses.py poses.mol2 charges
Inputs are the poses files and the name of the output file to be written. The output charges file is a list of DOCK scores and the charge of the ligand with that score.
-
46
To generate a plot of total DOCK score by ligand charge (charge_distributions_vs_energy.png) run:
python $DOCKBASE/analysis/plot_charge_distribution.py charges
-
47
Plot the contribution of each score term in the energy function outlined in Equation 1 (energy_distributions.png):
python $DOCKBASE/analysis/plot_energy_distributions.py extract_all.sort.uniq.txt
-
48
Collate the output files. The important outputs of these steps are: actives.mol2, roc_own.png, energy_distributions.png and charge_distributions_vs_energy.png.
Evaluate the docked poses of the actives
-
49
Open the rec.pdb and xtal-lig.pdb in Chimera, and prepare the visualization of the binding pocket to the user’s preference.
-
50
In the Tools tab, select Structure/Binding Analysis and click ViewDock.
-
51
In the menu, navigate to your directory and open actives.mol2.
-
52
For file type, select any of the DOCK options. This will open a window (ViewDock) to navigate through all of the molecules in this file such as in Fig. 6a.
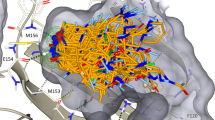
Fig. 6: Controls for docking optimization. 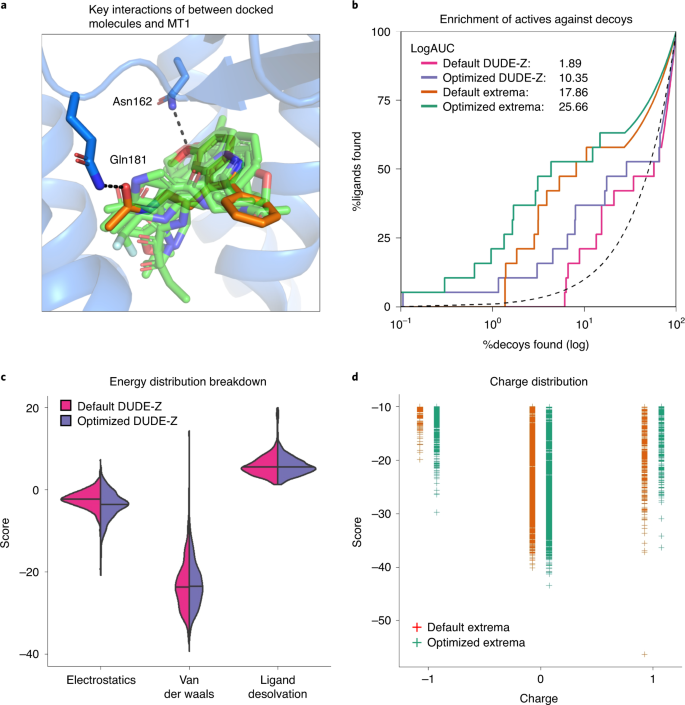
a, The receptor (blue) is shown with the crystallized ligand (orange). Docked control actives are shown in green and yield similar poses and interactions as the crystal ligand. The two residues, Asn162 and Gln181, that have their dipoles artificially increased (‘polarized’) to enhance the weight of polar interactions are shown hydrogen bonding to the crystal ligand. b, A log-transformed ROC plot is shown comparing the rate of identifying ligands versus decoys. A random selection would follow the dashed black line. The area under this dashed line is subtracted from the values reported for LogAUC such that a curve above the line would have a positive LogAUC, a curve below the line would have a negative LogAUC, and a curve following the dashed line would yield a LogAUC value of zero. Shown are the curves for the default settings and optimized settings for either the DUDE-Z control set and the Extrema control set. In both cases, the overall LogAUC value increases and the early enrichment improves. c, The energy distribution breakdown shows the individual score terms for each scored molecule in the docked setup. Based on this breakdown, it is clear that VDW interactions primarily drive ligand recognition. However, in the optimized setup in which electrostatic spheres with a radius of 1.9 that extend the dielectric boundary are used, the electrostatic score term shifts to more negative values. The desolvation spheres at the dielectric boundary in the optimized setup, with a radius of 0.1, have only minor effects on the ligand desolvation score term. d, In the Extrema challenge, the top-ranking ligands are plotted by their charge and DOCK score. In the Default settings, there is a preference for neutral ligands followed closely by monocations. The Optimized settings enhance the preference for neutrals.
-
53
Under the ‘Column’ tab, several details about the docked ligands can be listed, e.g., Total Energy, Electrostatic or Van der Waals terms.
-
54
In the Tools tab, under Structure/Binding Analysis, ‘Find HBonds’ can be used to identify hydrogen bonds between the protein and ligand molecules.
-
55
Evaluate the docked poses, asking the following questions:
-
Do the ligands occupy the part of the pocket as expected?
-
Do they make the types of interactions anticipated from what is known about the ligands and the pocket?
-
Are there aberrant or unsatisfied interactions? For example, in MT1, it is known from the crystal structure that ligands can form hydrogen bonds with Asn162 and Gln181. Issues with binding poses are usually a sampling problem, but scoring terms such as electrostatics can influence specific interactions
-
If many actives do not dock, is there a reasonable explanation (e.g., selected controls are larger than the pocket volume as has been observed when attempting to dock antagonists into agonist pockets)? Optimization of sampling is performed with a matching sphere scan as detailed in Steps 59–64
-

Evaluate the enrichment and scoring metrics
-
56
Evaluate the docking parameters’ ability to discriminate actives from decoys. The roc_own.png file shows the plot of the rate of finding actives as a function of decoys. The AUC is indicative of this discriminatory power (Fig. 6b). A positive LogAUC value is a sign that actives are enriched over decoys, a value near 0 represents random selection, and a negative value demonstrates that the model prefers decoys over actives. This may be a result of poor poses, improper scoring or both. It is best to optimize poses before pushing forward on scoring discrimination. Even for good LogAUC values, it is important to evaluate the poses as in Step 55 as it is possible to get good scoring actives that do not dock in the correct pose.
-
57
Examine the energy contributions of the docked poses. In the energy_distribution.png file (Fig. 6c), the total DOCK scores for the docked poses are broken down into the main components of the scoring function: electrostatics, VDW and polar ligand desolvation.
Do the contributions of the various score terms match the properties of the binding pocket? In the MT1 pocket, the VDW interactions dominate because of its largely hydrophobic nature. For targets forming salt bridges to ligands, electrostatic terms should at least be balanced with VDW scores. If the balance does not match with what is expected for the pocket, the strength of the scoring terms can be modified in Steps 65–68.
-
58
Evaluate the charge preference of the docking parameters. If the electrostatics term dominates in Step 57, the scoring function will likely prefer highly charged ligands. This can be measured in the charge_distributions_vs_energy.png plot (Fig. 6d), which shows the charge state of the top scoring molecules. DUDE-Z decoys should have charges matching the active ligands. However, in property-unmatched decoys such as the Extrema set78 (Steps 78–88) and in-stock set (Steps 89–97), it is important to ensure the docking is not biased toward charge extremes, which can suggest an overweighting of electrostatic terms; this can be addressed in Steps 65–68.
Section 4: optimize poses by modifying matching spheres
Timing Minutes to build; ≤1 h to dock
-
59
Create a new directory matching_sphere_scan/ that contains the INDOCK file and working/ and dockfiles/ directory from the previous directory.
-
60
Run the following script to create sets of matching spheres in which the matching spheres that do not map to atoms in the original xtal-lig.pdb file are perturbed as in Fig. 7a: (~1 min)
Fig. 7: Matching and dielectric boundary spheres drive changes to sampling and scoring in DOCK3.7. 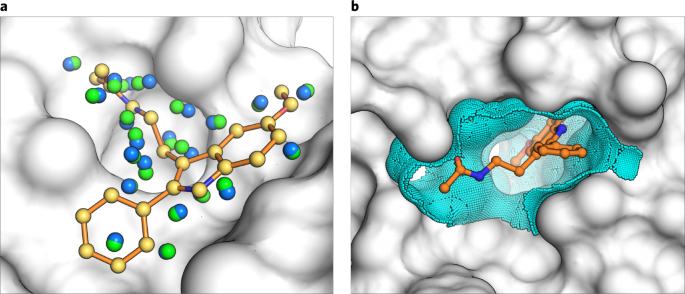
a, the crystal ligand is shown as orange sticks in the receptor pocket (gray). Matching spheres derived from the coordinates of the crystal ligand are shown in yellow and remain fixed during sphere perturbation. Random spheres (blue) are calculated with the program SphGen, and a set of spheres are selected that are near the crystal ligand. In a matching sphere scan, only the random spheres are perturbed and a new set is obtained (green). b, the crystal ligand (orange) is again shown in the context of the receptor binding pocket (gray). Dielectric boundary spheres (cyan) cover the binding surface around the crystal ligand to alter the electrostatic or desolvation potentials at the boundary between solvent and protein.
python $DOCKBASE/proteins/optimization/scramble-matching-spheres.py -i $PWD/dockfiles -o $PWD -s 0.5 -n 50
The arguments are: -i, the path to the dockfiles/ directory; -o, the path to where new docking directories should be created; -s, the maximum distance to move a matching sphere (0.5 recommended); -n, the number of perturbed sphere combinations to create. For a single receptor target, we recommend 50–100 sets of sphere sets.
-
61
Each new directory should contain an INDOCK file and dockfiles/ directory. Check that the matching_spheres.sph file in the dockfiles/ directory is unique to each new directory created.
-
62
For each directory, copy over the controls.sdi file.
-
63
Run the docking calculation and extract the poses as in Steps 37–48.
-
64
Examine the new actives.mol2 files and LogAUC values from the different matching sphere sets. Ideally, a set of spheres will increase the LogAUC and improve the binding poses of the actives. For MT1, the LogAUC increased from ~2 to 5. While not a large change, there was improved placement of the flexible components of the active ligands, which suggests better sampling and pose identification in a prospective screen.
If the poses did not improve during this matching sphere scan, it may indicate that there is a problem with the binding pocket model, the ligand set, the sampling of ligand conformations, or the placement of the crystallographic matching spheres. Examining each of these can help improve the binding poses in this first control calculation. Alternatively, the scoring function needs to be optimized to score the expected pose better (see Steps 65–68).

Section 5: optimize ligand scoring by modulating the protein–water dielectric interface (adding a layer of dielectric boundary spheres)
Timing 15 min on cluster; 20–30 min per radius on local machine
-
65
Create a new directory with the INDOCK file and working/ and dockfiles/ directories from the best matching sphere set (or the default settings if no matching sphere scan was run).
-
66
Generate the boundary modifying spheres (Fig. 7b)
$DOCKBASE/proteins/optimization/boundary-sphere-scan.sh -b $PWD -v
This version of the script will run on an SGE cluster. Modify the submission script and scheduler as needed for different cluster architecture. To run on a local machine, use the script:
$DOCKBASE/proteins/optimization/boundary-sphere-scan_no_cluster.sh -b $PWD -v
-
67
Combine the different radii boundary spheres (5 min):
python $DOCKBASE/proteins/optimization/combine-grids.py -b $PWD
This script generates a set of combinations of boundary modifying spheres across both ligand desolvation and low dielectric spheres. These directories have everything needed to run individual retrospective calculations except for the controls.sdi file that will need to be copied into each one. Repeat the retrospective calculations from Steps 37–48.
-
68
Following retrospective calculations across the combinations of dielectric boundary sphere radii, select a combination that ideally improves LogAUC (Fig. 6b) and helps to balance the score distributions in the energy_distribution.png plot (Fig. 6c). If there are a number of equally performing combinations, subsequent control calculations can help identify the best set for your system (see Steps 78–97). In the case of MT1, we chose electrostatic spheres of radius 1.9 and desolvation spheres of radius 0.1 as they increased LogAUC and enhanced the charge distribution of compounds to favor neutrals to match known MT1 ligands (Fig. 6d).
Section 6: (optional): polarize (or depolarize) residues to effect electrostatics
Timing 15 min
Critical
If after these scans a particular interaction is still missed or erroneously captured, it may be necessary to modify the partial charges of a particular residue. For example, in the case of MT1, the docking scores are largely dominated by VDW interactions (Fig. 6c). However, two residues Gln181 and Asn162 form hydrogen bonds with the known ligands. For these specific interactions, a global modification to the dielectric boundary (Steps 65–68) may not be sufficient and, instead, a local modification to partial charges as outlined in Steps 69–77 may enhance favorable scores for these interactions. For targets where this is not necessary, proceed to Step 78.
-
69
Make a new directory for polarizing residues. Copy over rec.pdb, xtal-lig.pdb, working/rec.crg.pdb, working/amb.crg.oxt and working/prot.table.ambcrg.ambH from the previous docking setup.
-
70
In the rec.crg.pdb file, rename the residues to be polarized with a unique three-letter amino acid code. For example, in the MT1 receptor, we chose to polarize GLN181 and ASN162 to enhance the polar interactions between actives and these hydrogen bonding residues. Accordingly, we renamed GLN181 as GLD and ASN162 as ASM (see Fig. 8).
Fig. 8: Polarizing effects specific atoms’ electrostatic potential. 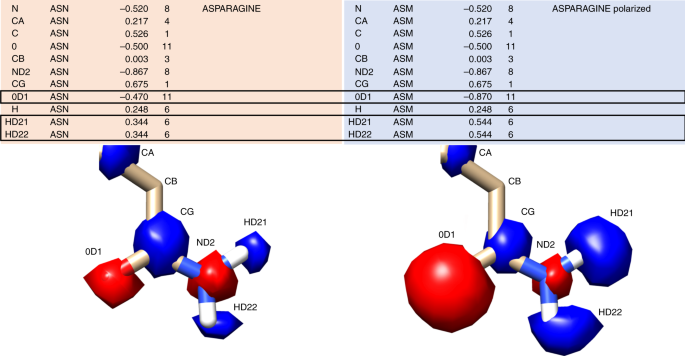
In contrast to global modifications to the electrostatic potential with the incorporation of thin spheres, polarizing allows for very specific modifications to a residue’s charge status. A canonical asparagine (ASN) from the prot.table.ambcrg.ambH file is shown with its polarized version ASM in which the carbonyl becomes more electronegative while the amide hydrogens become more electropositive to maintain the overall charge. The electrostatic potential corresponding to each atom is shown as spheres, with red corresponding to negative charge and blue corresponding to positive charge.
sed -i 's/GLN A 181/GLD A 181/g' rec.crg.pdb sed -i 's/ASN A 162/ASM A 162/g' rec.crg.pdb

Generate partial charges for the new residues
Critical
The two files that read in partial charges are the amb.crg.oxt and prot.table.ambcrg.ambH files located withing the working/ directory. These files contain default partial charges for all amino acids and some special residues such as ions. Make the following changes in both amb.crg.oxt and prot.table.ambcrg.ambH files.
-
71
Create new sections for the polarized residues in the two files by copying the standard residue partial charges.
-
72
Rename the polarized residues to match the three-letter codes used in the rec.crg.pdb file. The names are all capitalized in the prot.table.ambcrg.ambH file as shown in Fig. 8 and all lowercase in the amb.crg.oxt file.
-
73
Redistribute the charge around the atom of interest, making sure that the net charge remains the same. We suggest testing modifications of 0.2 or 0.4 charge units for a given atom. For example, for an ASN-to-ASM change, where we want to enhance the electronegativity of the sidechain carbonyl (OD1), increase the charge by 0.4 units, resulting in a change from −0.470 to −0.870. As −0.4 charge was added to the residue, +0.4 must be distributed to other atoms in the residue to maintain the net charge. One option is to add +0.2 to each of the sidechain amide hydrogens (HD21 and HD22) as in Fig. 8, though the charge could have been distributed to the backbone amide or another atom not involved in binding.
Running the grid generation
-
74
Before running the grid generation, check that the following files are present: rec.pdb, xtal-lig.pdb, the modified amb.crg.oxt and prot.table.ambcrg.ambH files, and a working/ directory with the modified rec.crg.pdb file.
-
75
Run blastermaster with the following command:
$DOCKBASE/proteins/blastermaster/blastermaster.py --addNOhydrogensflag --chargeFile=amb.crg.oxt --vdwprottable=prot.table.ambcrg.ambH –v
-
76
Rerun control calculations as in Step 37–48. When examining the new poses, consider the following questions:
-
Does the polarized residue promote or discourage the desired interaction?
-
Are the new interactions scored more favorably?
-
-
77
Rerun the dielectric boundary modifying sphere scan in Steps 65–68. We recommend doing this because the new polarized residue(s) will have altered the electrostatics grid. It may be necessary to test different combinations and strengths (0.2, 0.4) of the polarized residues to identify the best-performing set of parameters that enhances correct interactions, improves poses and ideally improves LogAUC. However, due to the modification to the electrostatic potential introduced by these changes, it is important to ensure these improvements do not come at the cost of biasing the screen towards overly charged molecules. This bias can be checked in the next section Steps 78–88.
Section 7: Extrema charge control calculations
Timing 2–3 h for compound building
Generate the Extrema set
Critical
Compounds in the extrema set come from the area of chemical space occupied by the known actives (molecular weight and LogP) but distinct charges (−2, −1, 0, +1, +2). These decoys will be used to measure biases introduced into the scoring function owing to modifications to the electrostatic potential.
-
78
On tldr.docking.org, navigate to the ‘extrema’ application.
-
79
Upload the SMILES of your active ligands used in section II to the webpage.
-
80
Select 500 or 1,000 molecules per charge, and click the ‘Go’ button.
-
81
After a few minutes, a results file will be available to download and extract. Within the file should be five SMILES files grouped by charge. Extract the SMILES file from the download:
unzip extrema_results.zip
-
82
Build the extrema set.
-
83
For each SMILES file, submit the file to the ‘build3d’ application on tldr.docking.org. Depending on the number of ligands in each file, and the activity on the website, each file should take 2–3 h to build.
-
84
When all sets are built and downloaded, combine the path to all compounds in a split database index file along with the known actives.
Run the extrema set
-
85
Navigate to a directory containing the INDOCK file and dockfiles/ directory that you want to test. The dockfiles/ directory should contain the set of matching spheres and the dielectric boundary sphere combination selected from the previous steps.
-
86
Prepare the docking run. In the previous steps, the control ligand set was small enough to run in a single run. This control set often has 10,000+ compounds, and it is best to split this into multiple jobs. To do this run:
$DOCKBASE/docking/setup/setup_zinc15_file_number.py ./ extrema extrema.sdi 100 count
-
87
Submit the docking calculations over a cluster as in Step 38B.
-
88
Repeat the analysis in Steps 39–48 with a focus on the charge_distributions_vs_energy.png plot (Fig. 6d). An optimal set of docking parameters will have good LogAUC with early enrichment, and the top-ranking compounds will contain charges that match the charges of the known actives. Sometimes multiple boundary-modifying sphere combinations will be tested in this Extrema calculation to find a set that enhances the scoring preference for compounds similar to knowns.
Section 8: run a ‘small’-scale in-stock screen
Timing 1–24 h depending on download speed
-
89
To collect an in-stock screening library, go to http://zinc20.docking.org/tranches/home/.
-
90
At the top of the tranche viewer (Fig. 9), select ‘3D’ representation, set Purchasability to ‘In-stock’, and select the charge states of interest to your target (i.e., only 0). Next, select the molecular weight range you want to screen. Finally, select a LogP range tailored to your target. If no specific LogP range is desired, we recommend LogP ≤ 3.5 to avoid insoluble compounds that might aggregate and yield false positives in the experimental assay.
Fig. 9: Navigating the ZINC20 Tranche Viewer. 
Several options are available at http://zinc20.docking.org/tranches/home/ for selecting different subsets of ligands for virtual screening. Important criteria such as selecting between 2D/3D, purchasability, charge, molecular weight and logP are highlighted. To download compounds, different methods such as downloading as an index file or directly downloading with cURL and WGET are shown.
-
91
When only the tranches of interest are selected, click the download button in the upper right corner of the screen. Select ‘DOCK37’ as the download format and ‘cURL’ or ‘WGET’ as the download method. When done, click ‘Download’ to obtain a text file with all the cURL or wget commands needed to download the set of selected tranches.
-
92
Prepare a split database index file with the path to all of the newly downloaded compounds.
-
93
Split the docking run over 1,000 jobs to efficiently run the screen as the number of compounds is usually ~1–4 million.
$DOCKBASE/docking/setup/setup_zinc15_file_number.py ./ instock instock.sdi 1000 count
-
94
Run the screen on a cluster as in Step 38B.
-
95
Run the analysis of the screen to generate a LogAUC. It should be on par or better than LogAUC values seen previously, as the set of decoy molecules grows in size and departs further from the set of known ligands.
-
96
Test out hit picking strategies. Use any metric for filtering hits (Table 2), and visually examine the hits that pass the selected filters.
-
Are the compounds forming the expected interactions with the binding pocket?
-
Are they sampling in the correct region of the pocket?
If there are any issues, determine if it is a sampling or scoring problem and alter the previous steps (matching spheres or dielectric boundary modifying spheres, respectively) to optimize. If the top poses capture expected interactions, then it is worth moving into a large-scale prospective screen.
-
-
97
Choose settings for the large-scale docking experiment. If multiple docking parameters were tested in this section (i.e., section 8), one method for selecting the best settings for large-scale docking is to choose the parameters that yield the greatest number of viable compounds after all of the post-docking filters have been applied. This will ensure a large number of hits in the large-scale campaign. Alternatively, one can purchase hits from the different screens and see which setup leads to more true actives in an experimental setting. Even if just a few low-potency hits are obtained at this point, it may suggest which interactions are critical for activity in the pocket.

Section 9: large-scale docking
Timing 1–5 d, depending on number of compounds and compute nodes
-
98
Create a new directory for the large-scale campaign with the final set of parameters, i.e., the INDOCK file and the dockfiles/ directory.
-
99
Change the mol2_maximum_cutoff in the INDOCK file to a value likely to eliminate ~90% of the docked molecules to save on disk storage as compounds with total scores worse than a certain value (more positive) are unlikely to be considered hits from the large screen and should not be saved (use the results of the in-stock screen to help inform on the best mol2_maximum_cutoff values to use).
-
100
Obtain the ZINC20 library. The ZINC20 library contains a prebuilt 3D conformer library of nearly 700 million fully protonated and tautomerized compounds (i.e., protomers). The library is largely built on Enamine’s readily accessible library, which consists of molecules that have a >80% likelihood of synthesis in one or two reaction steps11. This library can be downloaded from https://files.docking.org/3D/ or using the tranche browser as in Fig. 9 and consists of ~60 TB of data.
-
101
Select properties of the ZINC20 library for the screen such as molecular weight, charge and LogP. We recommend screening compounds separately by MWT range (i.e., fragments ≤ 250 amu) as larger compounds often score better due to their ability to form additional interactions within the binding pocket117. To obtain a split database index (sdi) file for a predownloaded ZINC20 database, use the ‘DOCK Database Index’ download format in the tranche viewer (Fig. 9) and provide a path to the downloaded database in the ‘ZINC DB Root’ field.
-
102
Split the docking campaign over 1,000 or more jobs. Typically, ~100 million compounds can be screened per day on an academic 1,000 core cluster of recent vintage (as of 2020).
$DOCKBASE/docking/setup/setup_zinc15_file_number.py ./ largescaledocking zinc20library.sdi 1000 count
-
103
Run the screen on a cluster as in Step 38B.
Section 10: hit picking
Timing Days to weeks
-
104
Extract the scores of the top compounds as in Step 40 with the maximum score cutoff set to the mol2_maximum_cutoff set in Step 99
python $DOCKBASE/analysis/extract_all_blazing_fast.py ./dirlist extract_all.txt -50
-
105
Collect between 300,000 and 1,000,000 poses from the best-scoring compounds.
python $DOCKBASE/analysis/getposes_blazing_faster.py ./ extract_all.sort.uniq.txt 1000000 poses.mol2 test.mol2.gz
-
106
Use any number of post-docking filters including but not limited to those described in Table 2.
-
107
From the compounds that remain after filtering, visually examine up to 5,000 molecules depending on how viable the docked compounds look. We recommend that visual examination is done by more than one person, because of the different knowledge base that each individual brings to the selection process (i.e., a medicinal chemist will prioritize different features over a molecular biologist and vice versa)19. This step should be done before prioritizing compounds for purchase, as we have found human-picked compounds have better efficacy than compounds obtained from fully automated hit-picking12. By using post-docking filters as in Step 106 prior to visual examination, one can search more deeply through the list of top ranked molecules to identify molecules for testing.
-
108
Choose 50–200 compounds for wet-lab testing depending on price, capability of testing and confidence of success as informed from retrospective calculations.
Troubleshooting
Structure preparation
Alternative conformations or missing side chains should be completely modeled before starting the grid generation (blastermaster.py). Otherwise, any superfluous or missing atoms will result in erroneous VDW surface or partial charge calculations (see below).
Blastermaster
Section 1, Step 5
All input and output files as well as options and flags of the blastermaster program are listed in the Blastermaster Guide (see Supplementary Information). Of important note is that blastermaster requires protein and ligand structures to be provided as PDB files called rec.pdb (and working/rec.crg.pdb in case a protonated structure is used) and xtal-lig.pdb. Alternative file names will not be recognized in the default settings.
Section 1, Step 10
If blastermaster does not successfully finish the grid generation, log files in the working directory will yield the required information to backtrace the error (see Blastermaster Guide in Supplementary Information). We suggest running blastermaster in the verbose mode (-v) as it allows to easily detect at which step the program failed.
Most common errors are related to unknown atom or residue types in rec.pdb (and/or rec.crg.pdb). Missing parameters for specific atom types can be added to the following parameter files:
$DOCKBASE/proteins/defaults/radii (required for surface calculation by the dms program) $DOCKBASE/proteins/defaults/amb.crg.oxt (partial charges for qnifft) $DOCKBASE/proteins/defaults/vdw.siz (Van der Waals radii for qnifft) $DOCKBASE/proteins/defaults/prot.table.ambcrg.ambH (atom typing for chemgrid) $DOCKBASE/proteins/defaults/vdw.parms.amb.mindock (Van der Waals parameters for minimizer)
In our experience, most protein input structures can be converted into complete docking grids with the default parameters provided in DOCKBASE. For some modifications such as capped termini or structural waters, parameters are included in the provided files (e.g., amb.crg.oxt) but may use slightly different atom names compared with default names from modeling programs such as Chimera, PyMol or Maestro. In these cases, we suggest adapting the corresponding atom names in the input protein structure files (rec.pdb, rec.crg.pdb) rather than adding more atom types to the default parameter files. Common naming errors occur for disulfide bonded cysteines (CYX in prot.table.ambcrg.ambH) and water atoms (HOH, TIP, WAT and SPC in prot.table.ambcrg.ambH).
The qnifft program may crash if too many atoms (>50,000) are present in rec.crg.pdb. While we do rarely encounter this problem, particularly large proteins or other molecular complexes may have to be reduced in atom number (e.g., removing fusion proteins or distal monomers). Typically, protein segments >20 Å away from the binding pocket are unlikely to influence the resulting docking grids.
In certain cases, blastermaster may be able to calculate all grids, even though some parameters might be missing. For example, if nonstandard amino acids are used, the protonation state as well as partial charges or VDW surfaces may not be computed correctly, but will not cause blastermaster to crash.
To check if all force field parameters were assigned to the protein structure correctly, we suggest visual inspection of docking grids (Chimera). The scoring grids (vdw.vdw, trim.electrostatics.phi, as well as ligand.desolv.heavy and ligand.desolv.hydrogen) are found in the dockfiles directory. The following scripts can be run in the dockfiles directory to convert the grids into dx files for visualization.
Convert VDW grid:
python $DOCKBASE/proteins/grid_visualization/create_VDW_DX.py
Input: vdw.vdw, vdw.bump (default)
Output: vdw.dx (default)
Convert electrostatics grid:
python $DOCKBASE/proteins/grid_visualization/create_ES_DX.py trim.electrostatics.phi trim.electrostatics.dx
Input: trim.electrostatics.phi
Output: trim.electrostatics.dx
Convert desolvation grid:
python $DOCKBASE/proteins/grid_visualization/create_LigDeSolv_DX.py
Input: ligand.desolv.heavy (default)
Input: ligdesolv.dx (default)
To visualize the grids, first open the rec.crg.pdb file in Chimera. The dx files can be opened with the Volume Viewer application. The vdw.dx file should resemble the surface of the receptor. The ligdesolv.dx file demonstrates various solvation levels of the pocket (the grid itself is a continuum). By changing the level of the representation, the volume should fill the pocket illustrating the ligand moving from a less solvated state to the most solvated state. For the trim.electrostatics.dx file, it is best to set the minimum and maximum levels to −100 and 100, respectively. Then, set the colors to red for −100 and blue for 100. This now shows regions of positive partial charges (blue) and negative partial charges (red). For all grids, ensure that the representations match what is expected for the binding pocket and that there are no regions of missing grid points. If there are obvious holes in any of the grids, in close proximity to the ligand, certain residue types were not identified correctly and corresponding parameters could not be assigned.
A list of all partial charges assigned to rec.crg.pdb is given in working/qnifft.atm (column 11). If the total charge of the system does not sum up to an integer value, certain partial charges may not have been found in $DOCKBASE/proteins/defaults/amb.crg.oxt. Furthermore, if many partial charges for a given residue (or other structural components such as metals or water molecules) are 0, proper parameters were likely not assigned. Specific errors in partial charge assignment can be found in working/OUTPARM.
Docking
Section 3, Step 38A(ii)
The DOCK executable ($DOCKBASE/docking/DOCK/bin/dock64) will only read a file called INDOCK.
Section 4, Step 64
If ligands dock in poses far from the binding pocket, visualize the matching spheres to ensure they occupy the same area as the xtal-lig. The matching_spheres.sph file in the dockfiles directory contains the set of matching spheres. To view this file in a protein visualization application, they first must be converted to a pdb file.
csh $DOCKBASE/proteins/showsphere/doshowsph.csh matching_spheres.sph 1 matching_spheres.pdb
Input: matching_spheres.sph
Output: matching_spheres.pdb
The output file, matching_spheres.pdb, can be visualized with the receptor and ligand in either Chimera or PyMOL. The matching spheres should align to the ligand’s heavy atoms, and additional random spheres should be scattered around the ligand as in Fig. 7a.
If many ligands fail to dock (>50%), a number of issues may be at fault. It is common for 10–20% of ligands to fail to dock in retrospective calculations because of mismatches between the pocket and ligand properties. In the OUTDOCK file, two common statements may indicate these issues: ‘Grids too small’ and ‘Skip size’. The ‘Grids too small’ statement indicates that the ligands do not fit in the binding pocket. The ‘Skip size’ statement is used when the number of atoms in the ligand exceeds the atom_maximum value in the INDOCK file. Adjusting this number to a higher value will ensure that larger ligands will be docked and scored. If the OUTDOCK file shows many compounds getting scored but the poses are not being saved, the mol2_maximum_cutoff value may be too stringent. Relaxing this term will save more poses, but be careful to not go too high as the size of the output files will correspondingly increase and may be an issue for disk storage capacity. Additional INDOCK parameters can be adjusted according to the INDOCK Guide available in the Supplementary Information.
Anticipated results
At the end of this protocol, a receptor will have been converted into a dock-readable binding pocket, the system optimized on retrospective control calculations, and the system screened prospectively against a large chemical library. While this protocol has been specific to a single target that yielded successful results13, we have equally applied this protocol to a number of targets with general success demonstrating its broad applicability12,83,118. We have attempted to curate all of the relevant steps, though expert intuition brought by each user for their target will result in a different selection of final hits for purchase. For these intuition-based steps, we recommend multiple people review the data before making the final decision regarding which compounds to buy.
If none of the purchased compounds demonstrates activity at the target of interest as determined experimentally, there remain several reasons for failure, including:
-
The calculations were performed using the wrong conformation of the binding pocket
-
The compound library did not contain enough examples of the class of compound most likely to dock
-
Retrospective optimization was not possible due to lack of known chemical matter
-
The experimental structure contained ambiguities inside chain or loop placement due to poor electron density or refinement errors
-
Incorrect or low-quality homology model
For example, targets that bind dianions or dications will suffer from much smaller library sizes, while targets that bind large protein ligands may be difficult to modulate with small molecules. As in experimental biology, no set of retrospective controls will ever guarantee prospective success, and the many approximations in docking ensure that we still measure success by hit rates.
Nevertheless, these controls and optimization steps can reduce obvious sources of failure, and allow one to better design a subsequent campaign. The experience of the field by now suggests that the approach is likely enough to succeed to be worth the investment, while the novel ligands that result can bring genuinely new biological insight to a field, a longstanding goal of the structure-based enterprise.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
An example set of files used in this protocol, including ligand and decoy sets, default docking grids and optimized docking grids, can be downloaded from http://files.docking.org/dock/mt1_protocol.tar.gz. The example dataset uses the MT1 structure (PDB: 6ME3) co-crystallized with 2-phenylmelatonin.
Software availability
DOCK3.7 can be downloaded after applying for a license from http://dock.docking.org/Online_Licensing/index.htm. Licenses are free for nonprofit research.
Change history
09 December 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41596-021-00650-x
References
Mayr, L. M. & Bojanic, D. Novel trends in high-throughput screening. Curr. Opin. Pharmacol. 9, 580–588 (2009).
Keserü, G. M. & Makara, G. M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discov. 8, 203–212 (2009).
Keiser, M. J., Irwin, J. J. & Shoichet, B. K. The chemical basis of pharmacology. Biochemistry 49, 10267–10276 (2010).
Bohacek, R. S., McMartin, C. & Guida, W. C. The art and practice of structure-based drug design: a molecular modeling perspective. Med. Res. Rev. 16, 3–50 (1996).
Brenner, S. & Lerner, R. A. Encoded combinatorial chemistry. Proc. Natl Acad. Sci. USA 89, 5381–5383 (1992).
Fitzgerald, P. R. & Paegel, B. M. DNA-encoded chemistry: drug discovery from a few good reactions. Chem. Rev. https://doi.org/10.1021/acs.chemrev.0c00789 (2020).
Grebner, C. et al. Virtual screening in the Cloud: how big is big enough? J. Chem. Inf. Model 60, 24 (2020).
Davies, E. K., Glick, M., Harrison, K. N. & Richards, W. G. Pattern recognition and massively distributed computing. J. Comput. Chem. 23, 1544–1550 (2002).
Sterling, T. & Irwin, J. J. ZINC 15—ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337 (2015).
Patel, H. et al. SAVI, in silico generation of billions of easily synthesizable compounds through expert-system type rules. Sci. Data 7, 384 (2020).
Grygorenko, O. O. et al. Generating multibillion chemical space of readily accessible screening compounds. iScience 23, 101681 (2020).
Lyu, J. et al. Ultra-large library docking for discovering new chemotypes. Nature 566, 224–229 (2019).
Stein, R. M. et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 579, 609–614 (2020).
Gorgulla, C. et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature 580, 663 (2020).
Meng, E. C., Shoichet, B. K. & Kuntz, I. D. Automated docking with grid‐based energy evaluation. J. Comput. Chem. 13, 505–524 (1992).
Sharp, K. A., Friedman, R. A., Misra, V., Hecht, J. & Honig, B. Salt effects on polyelectrolyte-ligand binding: comparison of Poisson–Boltzmann, and limiting law/counterion binding models. Biopolymers 36, 245–262 (1995).
Mysinger, M. M. & Shoichet, B. K. Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model. 50, 1561–1573 (2010).
Adeshina, Y. O., Deeds, E. J. & Karanicolas, J. Machine learning classification can reduce false positives in structure-based virtual screening. Proc. Natl Acad. Sci. USA 117, 18477–18488 (2020).
Irwin, J. J. & Shoichet, B. K. Docking screens for novel ligands conferring new biology. J. Med. Chem. 59, 4103–4120 (2016).
Mobley, D. L. & Dill, K. A. Binding of small-molecule ligands to proteins: “what you see” is not always “what you get. Structure 17, 489–498 (2009).
Bissantz, C., Folkers, G. & Rognan, D. Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J. Med. Chem. 43, 4759–4767 (2000).
Tirado-Rives, J. & Jorgensen, W. L. Contribution of conformer focusing to the uncertainty in predicting free energies for protein-ligand binding. J. Med. Chem. 49, 5880–5884 (2006).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2009).
Kramer, B., Rarey, M. & Lengauer, T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins 37, 228–241 (1999).
Halgren, T. A. et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759 (2004).
Morris, G. M. et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19, 1639–1662 (1998).
Abagyan, R., Totrov, M. & Kuznetsov, D. ICM—a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 15, 488–506 (1994).
Goodsell, D. S. & Olson, A. J. Automated docking of substrates to proteins by simulated annealing. Proteins 8, 195–202 (1990).
Mcgann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model 51, 578–596 (2011).
Jones, G., Willett, P., Glen, R. C., Leach, A. R. & Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267, 727–748 (1997).
Corbeil, C. R., Williams, C. I. & Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des. 26, 775–786 (2012).
McGovern, S. L. & Shoichet, B. K. Information decay in molecular docking screens against Holo, Apo, and modeled conformations of enzymes. J. Med. Chem. 46, 2895–2907 (2003).
Rueda, M., Bottegoni, G. & Abagyan, R. Recipes for the selection of experimental protein conformations for virtual screening. J. Chem. Inf. Model. 50, 186–193 (2010).
Kuntz, I. D., Blaney, J. M., Oatley, S. J., Langridge, R. & Ferrin, T. E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 161, 269–288 (1982).
Halgren, T. A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 49, 377–389 (2009).
Ngan, C. H. et al. FTMAP: extended protein mapping with user-selected probe molecules. Nucleic Acids Res. 40, W271–W275 (2012).
Wang, S. et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 358, 381–386 (2017).
Katritch, V. et al. Structure-based discovery of novel chemotypes for adenosine A(2A) receptor antagonists. J. Med. Chem. 53, 1799 (2010).
Kolb, P. et al. Structure-based discovery of beta2-adrenergic receptor ligands. Proc. Natl Acad. Sci. USA 106, 6843–6848 (2009).
De Graaf, C. et al. Crystal structure-based virtual screening for fragment-like ligands of the human histamine H 1 receptor. J. Med. Chem. 54, 8195–8206 (2011).
Mysinger, M. M. et al. Structure-based ligand discovery for the protein–protein interface of chemokine receptor CXCR4. Proc. Natl Acad. Sci. USA 109, 5517–5522 (2012).
Powers, R. A., Morandi, F. & Shoichet, B. K. Structure-based discovery of a novel, noncovalent inhibitor of AmpC β-lactamase. Structure 10, 1013–1023 (2002).
Zarzycka, B. et al. Discovery of small molecule CD40–TRAF6 inhibitors. J. Chem. Inf. Model. 55, 294–307 (2015).
Huang, N. & Shoichet, B. K. Exploiting ordered waters in molecular docking. J. Med. Chem. 51, 4862–4865 (2008).
Balius, T. E. et al. Testing inhomogeneous solvation theory in structure-based ligand discovery. Proc. Natl Acad. Sci. USA 114, E6839–E6846 (2017).
Weichenberger, C. X. & Sippl, M. J. NQ-Flipper: recognition and correction of erroneous asparagine and glutamine side-chain rotamers in protein structures. Nucleic Acids Res. 35, W403–W406 (2007).
Word, J. M., Lovell, S. C., Richardson, J. S. & Richardson, D. C. Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 285, 1735–1747 (1999).
Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234 (2013).
Bas, D. C., Rogers, D. M. & Jensen, J. H. Very fast prediction and rationalization of pKa values for protein–ligand complexes. Proteins 73, 765–783 (2008).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Bandyopadhyay, D., Bhatnagar, A., Jain, S. & Pratyaksh, P. Selective stabilization of aspartic acid protonation state within a given protein conformation occurs via specific “molecular association”. J. Phys. Chem. B 124, 5350–5361 (2020).
Webb, B. & Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 54, 5.6.1–5.6.37 (2016).
Bender, B. J. et al. Protocols for molecular modeling with Rosetta3 and RosettaScripts. Biochemistry 55, 4748–4763 (2016).
Yang, J. et al. Template-based protein structure prediction in CASP11 and retrospect of I-TASSER in the last decade. Proteins 84, 233–246 (2016).
Jaiteh, M., Rodríguez-Espigares, I., Selent, J. & Carlsson, J. Performance of virtual screening against GPCR homology models: impact of template selection and treatment of binding site plasticity. PLoS Comput. Biol. 16, e1007680 (2020).
Cavasotto, C. N. et al. Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J. Med. Chem. 51, 581–588 (2008).
Phatak, S. S., Gatica, E. A. & Cavasotto, C. N. Ligand-steered modeling and docking: a benchmarking study in class A G-protein-coupled receptors. J. Chem. Inf. Model. 50, 2119–2128 (2010).
Kaufmann, K. W. & Meiler, J. Using RosettaLigand for small molecule docking into comparative models. PLoS One 7, e50769 (2012).
Bordogna, A., Pandini, A. & Bonati, L. Predicting the accuracy of protein–ligand docking on homology models. J. Comput. Chem. 32, 81–98 (2011).
Katritch, V., Rueda, M., Lam, P. C.-H., Yeager, M. & Abagyan, R. GPCR 3D homology models for ligand screening: lessons learned from blind predictions of adenosine A2a receptor complex. Proteins 78, 197–211 (2010).
Schafferhans, A. & Klebe, G. Docking ligands onto binding site representations derived from proteins built by homology modelling. J. Mol. Biol. 307, 407–427 (2001).
Lansu, K. et al. In silico design of novel probes for the atypical opioid receptor MRGPRX2. Nat. Chem. Biol. 13, 529–536 (2017).
Huang, X.-P. et al. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 527, 477–483 (2015).
Trauelsen, M. et al. Receptor structure-based discovery of non-metabolite agonists for the succinate receptor GPR91. Mol. Metab. 6, 1585–1596 (2017).
Kolb, P. et al. Limits of ligand selectivity from docking to models: in silico screening for A1 adenosine receptor antagonists. PLoS One 7, e49910 (2012).
Daga, P. R., Polgar, W. E. & Zaveri, N. T. Structure-based virtual screening of the nociceptin receptor: hybrid docking and shape-based approaches for improved hit identification. J. Chem. Inf. Model. 54, 2732–2743 (2014).
Diaz, C. et al. A strategy combining differential low-throughput screening and virtual screening (DLS-VS) accelerating the discovery of new modulators for the Orphan GPR34 receptor. Mol. Inf. 32, 213–229 (2013).
Langmead, C. J. et al. Identification of novel adenosine A 2A receptor antagonists by virtual screening. J. Med. Chem. 55, 1904–1909 (2012).
Tikhonova, I. G. et al. Discovery of novel agonists and antagonists of the free fatty acid receptor 1 (FFAR1) using virtual screening. J. Med. Chem. 51, 625–633 (2008).
Martí-Solano, M., Schmidt, D., Kolb, P. & Selent, J. Drugging specific conformational states of GPCRs: challenges and opportunities for computational chemistry. Drug Discov. Today 21, 625–631 (2016).
Carlsson, J. et al. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat. Chem. Biol. 7, 769–778 (2011).
Männel, B. et al. Structure-guided screening for functionally selective D2 dopamine receptor ligands from a virtual chemical library. ACS Chem. Biol. 12, 2652–2661 (2017).
Khare, P. et al. Identification of novel S-adenosyl-l-homocysteine hydrolase inhibitors through homology-model-based virtual screening, synthesis, and biological evaluation. J. Chem. Inf. Model. 52, 777–791 (2012).
Li, S. et al. Identification of inhibitors against p90 ribosomal S6 kinase 2 (RSK2) through structure-based virtual screening with the inhibitor-constrained refined homology model. J. Chem. Inf. Model. 51, 2939–2947 (2011).
Eberini, I. et al. In silico identification of new ligands for GPR17: a promising therapeutic target for neurodegenerative diseases. J. Comput. Aided Mol. Des. 25, 743–752 (2011).
Frimurer, T. M. et al. Model-based discovery of synthetic agonists for the Zn2+-sensing G-protein-coupled receptor 39 (GPR39) reveals novel biological functions. J. Med. Chem. 60, 886–898 (2017).
Mysinger, M. M., Carchia, M., Irwin, J. J. & Shoichet, B. K. Directory of Useful Decoys, Enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 55, 6 (2012).
Stein, R. M. et al. Property-unmatched decoys in docking benchmarks. J. Chem. Inf. Model. 61, 699–714 (2020).
Coleman, R. G., Carchia, M., Sterling, T., Irwin, J. J. & Shoichet, B. K. Ligand pose and orientational sampling in molecular docking. PLoS One 8, e75992 (2013).
Huang, N., Shoichet, B. K. & Irwin, J. J. Benchmarking sets for molecular docking. J. Med. Chem. 49, 6789–6801 (2006).
Jain, A. N. & Nicholls, A. Recommendations for evaluation of computational methods. J. Comput. Aided Mol. Des. 22, 133–139 (2008).
Allen, W. J. & Rizzo, R. C. Implementation of the Hungarian algorithm to account for ligand symmetry and similarity in structure-based design. J. Chem. Inf. Model. 54, 518–529 (2014).
Schuller, M. et al. Fragment binding to the Nsp3 macrodomain of SARS-CoV-2 identified through crystallographic screening and computational docking. Sci. Adv. 7, eabf8711 (2021).
Fischer, A., Smieško, M., Sellner, M. & Lill, M. A. Decision making in structure-based drug discovery: visual inspection of docking results. J. Med. Chem. 64, 2489–2500 (2021).
Kirchmair, J. et al. Predicting drug metabolism: experiment and/or computation? Nat. Rev. Drug Discov. 14, 387–404 (2015).
Kirchmair, J. et al. Computational prediction of metabolism: sites, products, SAR, P450 enzyme dynamics, and mechanisms. J. Chem. Inf. Model. 52, 617–648 (2012).
Irwin, J. J. et al. An aggregation advisor for ligand discovery. J. Med. Chem. 58, 7076–7087 (2015).
Bemis, G. W. & Murcko, M. A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 39, 2887–2893 (1996).
Jadhav, A. et al. Quantitative analyses of aggregation, autofluorescence, and reactivity artifacts in a screen for inhibitors of a thiol protease. J. Med. Chem. 53, 37–51 (2010).
Capuzzi, S. J., Muratov, E. N. & Tropsha, A. Phantom PAINS: problems with the utility of alerts for Pan-Assay Interference Compound S. J. Chem. Inf. Model. 57, 417–427 (2017).
Baell, J. B. & Holloway, G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740 (2010).
McGovern, S. L., Caselli, E., Grigorieff, N. & Shoichet, B. K. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 45, 1712–1722 (2002).
Feng, B. Y. et al. A high-throughput screen for aggregation-based inhibition in a large compound library. J. Med. Chem. 50, 2385–2390 (2007).
Ganesh, A. N. et al. Colloidal drug aggregate stability in high serum conditions and pharmacokinetic consequence. ACS Chem. Biol. 14, 751–757 (2019).
Coan, K. E. D. & Shoichet, B. K. Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 130, 9606–9612 (2008).
Coan, K. E. D., Maltby, D. A., Burlingame, A. L. & Shoichet, B. K. Promiscuous aggregate-based inhibitors promote enzyme unfolding. J. Med. Chem. 52, 2067–2075 (2009).
Wolan, D. W., Zorn, J. A., Gray, D. C. & Wells, J. A. Small-molecule activators of a proenzyme. Science 326, 853–858 (2009).
Zorn, J. A., Wolan, D. W., Agard, N. J. & Wells, J. A. Fibrils colocalize caspase-3 with procaspase-3 to foster maturation. J. Biol. Chem. 287, 33781–33795 (2012).
Irwin, J. J. et al. ZINC20—a free ultralarge-scale chemical database for ligand discovery. J. Chem. Inf. Model. 60, 6065–6073 (2020).
Teotico, D. G. et al. Docking for fragment inhibitors of AmpC -lactamase. Proc. Natl Acad. Sci. USA 106, 7455–7460 (2009).
Chen, Y. & Shoichet, B. K. Molecular docking and ligand specificity in fragment-based inhibitor discovery. Nat. Chem. Biol. 5, 358–364 (2009).
Kolb, P. & Irwin, J. J. Docking screens: right for the right reasons? Curr. Top. Med. Chem. 9, 755–770 (2009).
Wu, Y., Lou, L. & Xie, Z.-R. A pilot study of all-computational drug design protocol–from structure prediction to interaction analysis. Front. Chem. 8, 81 (2020).
Greenidge, P. A., Kramer, C., Mozziconacci, J. C. & Sherman, W. Improving docking results via reranking of ensembles of ligand poses in multiple X-ray protein conformations with MM-GBSA. J. Chem. Inf. Model. 54, 2697–2717 (2014).
Mahmoud, A. H., Masters, M. R., Yang, Y. & Lill, M. A. Elucidating the multiple roles of hydration for accurate protein-ligand binding prediction via deep learning. Commun. Chem. 3, 19 (2020).
Liu, X. et al. An allosteric modulator binds to a conformational hub in the β2 adrenergic receptor. Nat. Chem. Biol. 16, 749–755 (2020).
Wacker, D. et al. Conserved binding mode of human β 2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J. Am. Chem. Soc. 132, 11443–11445 (2010).
Manglik, A. et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190 (2016).
Ewing, T. J. A. & Kuntz, I. D. Critical evaluation of search algorithms for automated molecular docking and database screening. J. Comput. Chem. 18, 1175–1189 (1997).
Gallagher, K. & Sharp, K. Electrostatic contributions to heat capacity changes of DNA-ligand binding. Biophys. J. 75, 769–776 (1998).
Wei, B. Q., Baase, W. A., Weaver, L. H., Matthews, B. W. & Shoichet, B. K. A model binding site for testing scoring functions in molecular docking. J. Mol. Biol. 322, 339–355 (2002).
Leaver-Fay, A. et al. Rosetta3. in Methods in Enzymology 545–574 (2011); https://doi.org/10.1016/B978-0-12-381270-4.00019-6
Madhavi Sastry, G., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234 (2013).
Armstrong, J. F. et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2020: extending immunopharmacology content and introducing the IUPHAR/MMV Guide to MALARIA PHARMACOLOGY. Nucleic Acids Res. 48, D1006–D1021 (2019).
Mendez, D. et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 47, D930–D940 (2019).
Irwin, J. J., Raushel, F. M. & Shoichet, B. K. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry 44, 12316–12328 (2005).
Verdonk, M. L. et al. Virtual screening using protein−ligand docking: avoiding artificial enrichment. J. Chem. Inf. Comput. Sci. 44, 793–806 (2004).
Alon, A. et al. Crystal structures of the σ 2 receptor template large-library docking for selective chemotypes active in vivo. Preprint at bioRxiv https://doi.org/10.1101/2021.04.29.441652 (2021).
Babaoglu, K. et al. Comprehensive mechanistic analysis of hits from high-throughput and docking screens against β-lactamase. J. Med. Chem. 51, 2502–2511 (2008).
Lorber, D. M. & Shoichet, B. K. Flexible ligand docking using conformational ensembles. Protein Sci. 7, 938–950 (1998).
Alhossary, A., Handoko, S. D., Mu, Y. & Kwoh, C.-K. Fast, accurate, and reliable molecular docking with QuickVina 2. Bioinformatics 31, 2214–2216 (2015).
Quiroga, R. & Villarreal, M. A. Vinardo: a scoring function based on Autodock Vina improves scoring, docking, and virtual screening. PLoS One 11, e0155183 (2016).
Bottegoni, G., Kufareva, I., Totrov, M. & Abagyan, R. Four-dimensional docking: a fast and accurate account of discrete receptor flexibility in ligand docking. J. Med. Chem. 52, 397–406 (2009).
Cho, Y., Ioerger, T. R. & Sacchettini, J. C. Discovery of novel nitrobenzothiazole inhibitors for Mycobacterium tuberculosis ATP phosphoribosyl transferase (HisG) through virtual screening. J. Med. Chem. 51, 5984–5992 (2008).
Verdonk, M. L., Cole, J. C., Hartshorn, M. J., Murray, C. W. & Taylor, R. D. Improved protein–ligand docking using GOLD. Proteins 52, 609–623 (2003).
Li, C. et al. Identification of diverse dipeptidyl peptidase IV inhibitors via structure-based virtual screening. J. Mol. Model. 18, 4033–4042 (2012).
Friesner, R. A. et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196 (2006).
Rai, B. K. et al. Comprehensive assessment of torsional strain in crystal structures of small molecules and protein–ligand complexes using ab initio calculations. J. Chem. Inf. Model. 59, 4195–4208 (2019).
Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. The Cambridge structural database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 72, 171–179 (2016).
Gu, S., Smith, M. S., Yang, Y., Irwin, J. J. & Shoichet, B. K. Ligand strain energy in large library docking. Preprint at bioRxiv https://doi.org/10.1101/2021.04.06.438722 (2021).
Xing, L., Klug-Mcleod, J., Rai, B. & Lunney, E. A. Kinase hinge binding scaffolds and their hydrogen bond patterns. Bioorg. Med. Chem. 23, 6520–6527 (2015).
Peng, Y. et al. 5-HT2C receptor structures reveal the structural basis of GPCR polypharmacology. Cell 172, 719–730.e14 (2018).
Bissantz, C., Kuhn, B. & Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 53, 5061–5084 (2010).
Rogers, D. & Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754 (2010).
Alexander, N., Woetzel, N. & Meiler, J. Bcl::Cluster: a method for clustering biological molecules coupled with visualization in the Pymol Molecular Graphics System. in 2011 IEEE 1st International Conference on Computational Advances in Bio and Medical Sciences (ICCABS) 2011, 13–18 (IEEE, 2011).
Bender, A. & Glen, R. C. A discussion of measures of enrichment in virtual screening: comparing the information content of descriptors with increasing levels of sophistication. J. Chem. Inf. Model. 45, 1369–1375 (2005).
Simeonov, A. et al. Fluorescence spectroscopic profiling of compound libraries. J. Med. Chem. 51, 2363–2371 (2008).
Lea, W. A. & Simeonov, A. Fluorescence polarization assays in small molecule screening. Expert Opin. Drug Disco. 6, 17–32 (2011).
Thorne, N., Auld, D. S. & Inglese, J. Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Curr. Opin. Chem. Biol. 14, 315–324 (2010).
Walters, W. P. & Namchuk, M. Designing screens: how to make your hits a hit. Nat. Rev. Drug Discov. 2, 259–266 (2003).
Thorne, N. et al. Firefly luciferase in chemical biology: a compendium of inhibitors, mechanistic evaluation of chemotypes, and suggested use as a reporter. Chem. Biol. 19, 1060–1072 (2012).
Sassano, M. F., Doak, A. K., Roth, B. L. & Shoichet, B. K. Colloidal aggregation causes inhibition of G protein-coupled receptors. J. Med. Chem. 56, 2406–2414 (2013).
Owen, S. C., Doak, A. K., Wassam, P., Shoichet, M. S. & Shoichet, B. K. Colloidal aggregation affects the efficacy of anticancer drugs in cell culture. ACS Chem. Biol. 7, 1429–1435 (2012).
McLaughlin, C. K. et al. Stable colloidal drug aggregates catch and release active enzymes. ACS Chem. Biol. 11, 992–1000 (2016).
McGovern, S. L. & Shoichet, B. K. Kinase inhibitors: not just for kinases anymore. J. Med. Chem. 46, 1478–1483 (2003).
Acknowledgements
This work was supported by NIH grants R35GM122481 (to B.K.S.) and GM133836 (to J.J.I.). J.C. was supported by grants from the Swedish Research Council (2017-04676) and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement: 715052). B.J.B. was partly supported by an NIH NRSA fellowship F32GM136062. C.M.W. was partly supported by the National Institutes of Health Training Grant T32 GM007175, NSF GRFP and UCSF Discovery Fellowship. We thank members of the Shoichet lab for feedback on the procedures described.
Author information
Authors and Affiliations
Contributions
B.J.B. and S.G. wrote the manuscript with additional input from all authors. B.J.B., S.G., A.L., J.L., C.W., R.M.S., E.F., T.E.B., J.C., J.J.I. and B.K.S. developed the protocol. B.J.B., S.G., A.L., J.L., C.W., R.M.S. and T.E.B. contributed scripts. E.F. tested the protocol. Research was supervised by J.C., J.J.I. and B.K.S.
Corresponding author
Ethics declarations
Competing interests
B.K.S. and J.J.I. are founders of Blue Dolphin Lead Discovery LLC, which undertakes fee-for-service ligand discovery.
Additional information
Peer review information Nature Protocols thanks Claudio Cavasotto, Vincent Zoete and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Stein, R. M. et al. Nature 579, 609–614 (2020): https://doi.org/10.1038/s41586-020-2027-0
Lyu, J. et al. Nature 566, 224–229 (2019): https://www.nature.com/articles/s41586-019-0917-9
Schuller, M. et al. Sci. Adv. 7, eabf8711 (2021): https://advances.sciencemag.org/content/7/16/eabf8711
Key data used in this protocol
Stein, R. M. et al. Nature 579, 609–614 (2020): https://doi.org/10.1038/s41586-020-2027-0
Supplementary information
Supplementary Information
INDOCK Guide and Blastermaster Guide.
Rights and permissions
About this article

Cite this article
Bender, B.J., Gahbauer, S., Luttens, A. et al. A practical guide to large-scale docking. Nat Protoc 16, 4799–4832 (2021). https://doi.org/10.1038/s41596-021-00597-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-021-00597-z
This article is cited by
-
One substrate many enzymes virtual screening uncovers missing genes of carnitine biosynthesis in human and mouse
Nature Communications (2024)
-
Machine learning accelerates pharmacophore-based virtual screening of MAO inhibitors
Scientific Reports (2024)
-
Exploring the combinatorial explosion of amine–acid reaction space via graph editing
Communications Chemistry (2024)
-
DynamicBind: predicting ligand-specific protein-ligand complex structure with a deep equivariant generative model
Nature Communications (2024)
-
EasyDock: customizable and scalable docking tool
Journal of Cheminformatics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
