
Abstract
Intelligent image-activated cell sorting (iIACS) is a machine-intelligence technology that performs real-time intelligent image-based sorting of single cells with high throughput. iIACS extends beyond the capabilities of fluorescence-activated cell sorting (FACS) from fluorescence intensity profiles of cells to multidimensional images, thereby enabling high-content sorting of cells or cell clusters with unique spatial chemical and morphological traits. Therefore, iIACS serves as an integral part of holistic single-cell analysis by enabling direct links between population-level analysis (flow cytometry), cell-level analysis (microscopy), and gene-level analysis (sequencing). Specifically, iIACS is based on a seamless integration of high-throughput cell microscopy (e.g., multicolor fluorescence imaging, bright-field imaging), cell focusing, cell sorting, and deep learning on a hybrid software–hardware data management infrastructure, enabling real-time automated operation for data acquisition, data processing, intelligent decision making, and actuation. Here, we provide a practical guide to iIACS that describes how to design, build, characterize, and use an iIACS machine. The guide includes the consideration of several important design parameters, such as throughput, sensitivity, dynamic range, image quality, sort purity, and sort yield; the development and integration of optical, microfluidic, electrical, computational, and mechanical components; and the characterization and practical usage of the integrated system. Assuming that all components are readily available, a team of several researchers experienced in optics, electronics, digital signal processing, microfluidics, mechatronics, and flow cytometry can complete this protocol in ~3 months.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



















Similar content being viewed by others
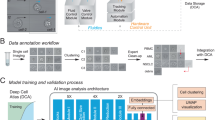
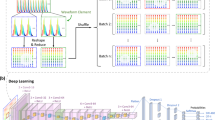
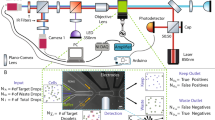
Data and code availability
The data and code are available as Supplementary Data and upon reasonable request.
Change history
17 October 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Nitta, N. et al. Intelligent image-activated cell sorting. Cell 175, 266–276 (2018).
Mikami, H. et al. Ultrafast confocal fluorescence microscopy beyond the fluorescence lifetime limit. Optica 5, 117–126 (2018).
Kanno, H., Mikami, H., Kaya, Y., Ozeki, Y. & Goda, K. Simple, stable, compact implementation of frequency-division-multiplexed microscopy by inline interferometry. Opt. Lett. 44, 467–470 (2019).
Shivhare, P. K., Bhadra, A., Sajeesh, P., Prabhakar, A. & Sen, A. K. Hydrodynamic focusing and interdistance control of particle-laden flow for microflow cytometry. Microfluid. Nanofluidics 20, 86 (2016).
Park, J. W. et al. Acoustofluidic harvesting of microalgae on a single chip. Biomicrofluidics 10, 034119 (2016).
Grenvall, C., Antfolk, C., Bisgaard, C. Z. & Laurell, T. Two-dimensional acoustic particle focusing enables sheathless chip Coulter counter with planar electrode configuration. Lab Chip 14, 4629–4637 (2014).
Sakuma, S., Kasai, Y., Hayakawa, T. & Arai, F. On-chip cell sorting by high-speed local-flow control using dual membrane pumps. Lab Chip 17, 2760–2767 (2017).
Lecun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521, 436–444 (2015).
Goodfellow, I., Bengio, Y., Courville, A. & Bengio, Y. Deep Learning (MIT Press, Cambridge, 2016).
LeCun, Y. et al. Backpropagation applied to handwritten zip code recognition. Neural Comput. 1, 541–551 (1989).
Krizhevesky, A., Sutskever, I. & Hinton, G. E. ImageNet classification with deep convolutional neural networks. In Proc. 25th International Conference on Neural Information Processing Systems (NIPS 2012) (eds Pereira, F., Burges, C. J. C., Bottou, L. & Weinberger, K. Q.) 1097–1105 (Curran Associates, 2012).
Herzenberg, L. A. et al. The history and future of the fluorescence activated cell sorter and flow cytometry: a view from Stanford. Clin. Chem. 48, 1819–1827 (2002).
Tung, J. W. et al. Modern flow cytometry: a practical approach. Clin. Lab. Med. 27, 453–468 (2007).
Liu, L., Cheung, T. H., Charville, G. W. & Rando, T. A. Isolation of skeletal muscle stem cells by fluorescence-activated cell sorting. Nat. Protoc. 10, 1612–1624 (2015).
Hayatsu, N. et al. Analyses of a mutant Foxp3 allele reveal BATF as a critical transcription factor in the differentiation and accumulation of tissue regulatory T cells. Immunity 47, 268–283 (2017).
de St Groth, B. F., Zhu, E. rhu., Asad, S. & Lee, L. Flow cytometric detection of human regulatory T cells. Methods Mol. Biol. 707, 263–279 (2011)
Shapiro, H. M. Practical Flow Cytometry (John Wiley & Sons, 2005).
Herzenberg, L. A., Gottlinger, C., Muller, W., Radbruch, A. & Recktenwald, D. Flow Cytometry and Cell Sorting (Springer, 1992).
Lindmo, T., Peters, D. C. & Sweet, R. G. Flow Cytometry and Sorting (Wiley-Liss, 1990).
Kawata, S., Hori, M., Kado, H. & Tamiya, E. Biological Imaging and Sensing (Springer, 2004).
Wang, P. & Wu, C. Micro/Nano Cell and Molecular Sensors (Springer, 2016).
Boutros, M., Heigwer, F. & Laufer, C. Microscopy-based high-content screening. Cell 163, 1314–1325 (2015).
Caicedo, J. C. et al. Data-analysis strategies for image-based cell profiling. Nat. Methods 14, 849–863 (2017).
Boutros, M. & Ahringer, J. The art and design of genetic screens: RNA interference. Nat. Rev. Genet. 9, 554–566 (2008).
Carpenter, A. E. Image-based chemical screening. Nat. Chem. Biol. 3, 461–465 (2007).
Boutros, M. et al. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 303, 832–835 (2004).
Lum, L. et al. Identification of Hedgehog pathway components by RNAi in Drosophila cultured cells. Science 299, 2039–2045 (2003).
Kiger, A. et al. A functional genomic analysis of cell morphology using RNA interference. J. Biol. 2, 27 (2003).
Liu, T., Sims, D. & Baum, B. Parallel RNAi screens across different cell lines identify generic and cell type-specific regulators of actin organization and cell morphology. Genome Biol. 10, R26 (2009).
Arpali, S. A., Arpali, C., Coskun, A. F., Chiang, H. H. & Ozcan, A. High-throughput screening of large volumes of whole blood using structured illumination and fluorescent on-chip imaging. Lab Chip 12, 4968–4971 (2012).
Zhang, Y. et al. High-throughput screening of encapsulated islets using wide-field lens-free on-chip imaging. ACS Photonics 5, 2081–2086 (2018).
Lei, C., Guo, B., Cheng, Z. & Goda, K. Optical time-stretch imaging: principles and applications. Appl. Phys. Rev. 3, 011102 (2016).
Mikami, H. et al. High-speed imaging meets single-cell analysis. Chem 4, 2278–2300 (2018).
Mikami, H., Gao, L. & Goda, K. Ultrafast optical imaging technology: principles and applications of emerging methods. Nanophotonics 5, 497–509 (2016).
Porichis, F. et al. High-throughput detection of miRNAs and gene-specific mRNA at the single-cell level by flow cytometry. Nat. Commun. 5, 5641 (2014).
Wu, J. L. et al. Ultrafast laser-scanning time-stretch imaging at visible wavelengths. Light Sci. Appl. 6, e16196 (2017).
Mahjoubfar, A. et al. Time stretch and its applications. Nat. Photonics 11, 341–351 (2017).
Lai, Q. T. K. et al. High-throughput time-stretch imaging flow cytometry for multi-class classification of phytoplankton. Opt. Express 24, 28170–28184 (2016).
Han, Y. & Lo, Y. Imaging cells in flow cytometer using spatial-temporal transformation. Sci. Rep. 5, 13267 (2015).
Han, Y., Gu, Y., Zhang, A. C. & Lo, Y. H. Review: imaging technologies for flow cytometry. Lab Chip 16, 4639–4647 (2016).
Rane, A. S., Rutkauskaite, J., DeMello, A. & Stavrakis, S. High-throughput multi-parametric imaging flow cytometry. Chem 3, 588–602 (2017).
Miura, T. et al. On-chip light-sheet fluorescence imaging flow cytometry at a high flow speed of 1 m/s. Biomed. Opt. Express 9, 3424–3433 (2018).
Jiang, Y. et al. Label-free detection of aggregated platelets in blood by machine-learning-aided optofluidic time-stretch microscopy. Lab Chip 17, 2426–2434 (2017).
George, T. C. et al. Distinguishing modes of cell death using the ImageStream® multispectcal imaging flow cytometer. Cytometry A 59A, 237–245 (2004).
Kobayashi, H. et al. Label-free detection of cellular drug responses by high-throughput bright-field imaging and machine learning. Sci. Rep. 7, 12454 (2017).
Muñoz, H. E. et al. Single-cell analysis of morphological and metabolic heterogeneity in Euglena gracilis by fluorescence-imaging flow cytometry. Anal. Chem. 90, 11280–11289 (2018).
Guo, B. et al. High-throughput, label-free, single-cell, microalgal lipid screening by machine-learning-equipped optofluidic time-stretch quantitative phase microscopy. Cytometry A 91A, 494–502 (2017).
George, T. C. et al. Quantitative measurement of nuclear translocation events using similarity analysis of multispectral cellular images obtained in flow. J. Immunol. Methods 311, 117–129 (2006).
Basiji, D. A., Ortyn, W. E., Liang, L., Venkatachalam, V. & Morrissey, P. Cellular image analysis and imaging by flow cytometry. Clin. Lab. Med. 27, 653–670 (2007).
Lee, D., Mehta, N., Shearer, A. & Kastner, R. A hardware accelerated system for high throughput cellular image analysis. J. Parallel Distrib. Comput. 113, 167–178 (2018).
Goda, K. & Jalali, B. Dispersive Fourier transformation for fast continuous single-shot measurements. Nat. Photonics 7, 102–112 (2013).
Wong, T. T. W. et al. Asymmetric-detection time-stretch optical microscopy (ATOM) for ultrafast high-contrast cellular imaging in flow. Sci. Rep. 4, 3656 (2014).
Lau, A. K. S., Shum, H. C., Wong, K. K. Y. & Tsia, K. K. Optofluidic time-stretch imaging-an emerging tool for high-throughput imaging flow cytometry. Lab Chip 16, 1743–1756 (2016).
Lei, C. et al. High-throughput imaging flow cytometry by optofluidic time-stretch microscopy. Nat. Protoc. 13, 1603–1631 (2018).
Guo, B. et al. Optofluidic time-stretch quantitative phase microscopy. Methods 136, 116–125 (2018).
Goda, K. et al. High-throughput single-microparticle imaging flow analyzer. Proc. Natl. Acad. Sci. USA 109, 11630–11635 (2012).
Lei, C., Nitta, N., Ozeki, Y. & Goda, K. Optofluidic time-stretch microscopy: recent advances. Opt. Rev. 25, 464–472 (2018).
Lei, C. et al. GHz optical time-stretch microscopy by compressive sensing. IEEE Photonics J. 9, 1–8 (2017).
Hiraki, K. et al. All-IP-Ethernet architecture for real-time sensor-fusion processing. In Proc. SPIE BiOS 9720 97200D (2016). https://doi.org/10.1117/12.2212016
Inaba, M. & Hiraki, K. Network processing hardware. In Proc. Second Asian International Conference on Technologies for Advanced Heterogeneous Network (eds Cho, K. & Jacquet, P.) 103–112 (Springer, 2006).
Okada, K. et al. Protocol design for all-IP computer architecture. In Proc. International Conference on Information Networking 2008 (ICOIN2008) (eds Kaiser, B., Madden, S. & Suri, S.) 1–5 (IEEE, 2008).
Hao, N., Budnik, Ba & Gunawardena, J. Tunable signal processing through modular control of transcription factor translocation. Science 339, 460–464 (2013).
Von Erlach, T. C. et al. Cell-geometry-dependent changes in plasma membrane order direct stem cell signalling and fate. Nat. Mater. 17, 237–242 (2018).
Sarioglu, A. F. et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat. Methods 12, 685–691 (2015).
Moor, A. E. et al. Global mRNA polarization regulates translation efficiency in the intestinal epithelium. Science 357, 1299–1303 (2017).
Zenker, J. et al. A microtubule-organizing center directing intracellular transport in the early mouse embryo. Science 357, 925–928 (2017).
Pernas, L., Bean, C., Boothroyd, J. C. & Scorrano, L. Mitochondria restrict growth of the intracellular parasite Toxoplasma gondii by limiting its uptake of fatty acids. Cell Metab. 27, 886–897 (2018).
Cho, E. H. et al. Characterization of circulating tumor cell aggregates identified in patients with epithelial tumors. Phys. Biol. 9, 016001 (2012).
Aceto, N. et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122 (2014).
Molnar, B., Ladanyi, A., Tanko, L., Sréter, L. & Tulassay, Z. Circulating tumor cell clusters in the peripheral blood of colorectal cancer patients. Clin. Cancer Res. 7, 4080–4085 (2001).
Wang, L. et al. Chloroplast-mediated regulation of CO2-concentrating mechanism by Ca2+-binding protein CAS in the green alga Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 113, 12586–12591 (2016).
Mackinder, L. C. M. et al. A spatial interactome reveals the protein organization of the algal CO2-concentrating mechanism. Cell 171, 133–147 (2017).
Ohnuki, S. & Ohya, Y. High-dimensional single-cell phenotyping reveals extensive haploinsufficiency. PLoS Biol. 16, 1–23 (2018).
Suzuki, G. et al. Global study of holistic morphological effectors in the budding yeast Saccharomyces cerevisiae. BMC Genomics 19, 149 (2018).
Iwaki, A., Ohnuki, S., Suga, Y., Izawa, S. & Ohya, Y. Vanillin inhibits translation and induces messenger ribonucleoprotein (mRNP) granule formation in Saccharomyces cerevisiae: application and validation of high-content, image-based profiling. PLoS ONE 8, e61748 (2013).
Treiser, M. D. et al. Cytoskeleton-based forecasting of stem cell lineage fates. Proc. Natl. Acad. Sci. USA 107, 610–615 (2010).
Thery, M. et al. Anisotropy of cell adhesive microenvironment governs cell internal organization and orientation of polarity. Proc. Natl. Acad. Sci. USA 103, 19771–19776 (2006).
Wu, C. Y. et al. Shaped 3D microcarriers for adherent cell culture and analysis. Microsyst. Nanoeng. 4, 21 (2018).
Lancaster, M. A. & Knoblich, J. A. Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345, 1247125 (2014).
Orange, J. S. Formation and function of the lytic NK-cell immunological synapse. Nat. Rev. Immunol. 8, 713–725 (2008).
Dustin, M. L., Chakraborty, A. K. & Shaw, A. S. Understanding the structure and function of the immunological synapse. Cold Spring Harb. Perspect. Biol. 2, a002311 (2010).
Ingham, P. W. The molecular genetics of embryonic pattern formation in Drosophila. Nature 335, 25–34 (1988).
Mullins, M. C., Hammerschmidt, M., Haffter, P. & Nüsslein-Volhard, C. Large-scale mutagenesis in the zebrafish: in search of genes controlling development in a vertebrate. Curr. Biol. 4, 189–202 (1994).
Fabritius, A. et al. Imaging-based screening platform assists protein engineering. Cell Chem. Biol. 25, 1554–1561 (2018).
Környei, Z. et al. Cell sorting in a Petri dish controlled by computer vision. Sci. Rep. 3, 1–10 (2013).
Das, A. et al. Adaptive from innate: human IFN-γ+ CD4+ T cells can arise directly from CXCL8-producing recent thymic emigrants in babies and adults. J. Immunol. 199, 1696–1705 (2017).
Jin, A. et al. A rapid and efficient single-cell manipulation method for screening antigen-specific antibody-secreting cells from human peripheral blood. Nat. Med. 15, 1088–1092 (2009).
Yoshimoto, N. et al. An automated system for high-throughput single cell-based breeding. Sci. Rep. 3, 1191 (2013).
Dura, B. et al. Longitudinal multiparameter assay of lymphocyte interactions from onset by microfluidic cell pairing and culture. Proc. Natl. Acad. Sci. USA 113, E3599–E3608 (2016).
Ogunniyi, A. O., Story, C. M., Papa, E., Guillen, E. & Love, J. C. Screening individual hybridomas by microengraving to discover monoclonal antibodies. Nat. Protoc. 4, 767–782 (2009).
Yao, X. et al. Tumor cells are dislodged into the pulmonary vein during lobectomy. J. Thorac. Cardiovasc. Surg. 148, 3224–3231 (2014).
Piatkevich, K. D. et al. A robotic multidimensional directed evolution approach applied to fluorescent voltage reporters. Nat. Chem. Biol. 14, 352–360 (2018).
Brasko, C. et al. Intelligent image-based in situ single-cell isolation. Nat. Commun. 9, 1–7 (2018).
Grys, B. T. et al. Machine learning and computer vision approaches for phenotypic profiling. J. Cell Biol. 216, 65–71 (2017).
Hennig, H. et al. An open-source solution for advanced imaging flow cytometry data analysis using machine learning. Methods 112, 201–210 (2017).
Abadi, M. et al. Tensorflow: a system for large-scale machine learning. In Proc. 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI ’16) 265–283 (USENIX, 2016).
Chollet, F. Keras: the Python deep learning library. https://keras.io (2015).
Kasai, Y., Sakuma, S. & Arai, F. On-chip multi-sorting using high-speed and high-accuracy flow control. In Proc. 22nd International Conference on Miniaturized Systems for Chemistry and Life Sciences (MicroTAS2018) (eds Tseng, F.-G. & Lee, G.-B.) 1237–1238 (Chemical and Biological Microsystems Society, 2018).
Paszke, A. et al. Automatic differentiation in PyTorch. In Proc. 31st Conference on Neural Information Processing Systems (NIPS 2017) (eds Guyon, I. et al.)1–4 (Curran Associates, 2017).
Tokui, S., Oono, K., Hido, S. & Clayton, J. Chainer: a next-generation open source framework for deep learning. In Proc. Conference on Neural Information Processing Systems (NIPS 2015) (eds Cortes, C., Lawrence, N. D., Lee, D. D., Sugiyama, M. & Garnett, R.) 1–4 (Curran Associates, 2015).
Carpenter, A. E. et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100.1–R100.11 (2006).
Abrams, C. S. et al. Direct detection of activated platelets and platelet-derived microparticles in humans. Blood 75, 128–138 (1990).
Shalek, A. K. et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510, 363–369 (2014).
Kalisky, T. & Quake, S. R. Single-cell genomics. Nat. Methods 8, 311–314 (2011).
Rotem, A. et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 33, 1165–1172 (2015).
Yamano, T. et al. Light and low-CO2-dependent LCIB-LCIC complex localization in the chloroplast supports the carbon-concentrating mechanism in Chlamydomonas reinhardtii. Plant Cell Physiol. 51, 1453–1468 (2010).
Acknowledgements
This work was supported primarily by the ImPACT program of the Council for Science, Technology, and Innovation (Cabinet Office, Government of Japan) and partly by the JSPS Core-to-Core Program and White Rock Foundation. We thank M. Kanematsu, M. Urakawa, A. Komiya, and S. Aihara for assistance. N.N. is an ISAC Marylou Ingram Scholar.
Author information
Authors and Affiliations
Contributions
K.G. conceived iIACS. A.I., N.N., T. Iino, and K.G. designed the protocol. A.I., H.M., K. Hiramatsu, S.S., Y.K., T. Iino, T.Y., A.Y., Y. Oguchi, N.S., Y.S., T. Ito, K. Hiraki, S.M., T.H., F.A., T.S., Y. Ozeki, and N.N. performed the experiments. H.F. and Y.Y. helped prepare the blood and microalgal samples. T.E., M.Y., and T.S. developed the digital image-processing algorithms. K. Hiraki developed the all-IP network. A.I., H.M., K. Hiramatsu, S.S., Y.K., T. Iino, T.Y., A.Y., Y. Ozeki, F.A., T.S., Y. Oguchi, N.N., and K.G. prepared the figures and tables. K.G. supervised the work with the help of T. Ito, Y. Hoshino, Y. Hosokawa, A.N., S.U., T.S., Y. Ozeki, and N.N. A.I., H.M., K. Hiramatsu, S.S., Y.S., M.Y., D.D., T.S., N.N., and K.G. mainly wrote the manuscript. All authors contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
H.M. and K.G. are inventors on a patent covering the FDM microscope. S.S., F.A., and T.H. are inventors on a patent application covering the dual-membrane push–pull cell sorter. N.N., T.S., and K.G. are inventors on a patent covering the data analysis and display method. N.N. is the president of CYBO, Inc. N.N., T.S., and K.G. are shareholders of CYBO, Inc.
Additional information
Peer review information: Nature Protocols thanks Kenneth K. Y. Wong and other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Nitta, N. et al. Cell 175, 266–276.e13 (2018): https://www.cell.com/cell/fulltext/S0092-8674(18)31044-4
Mikami, H. et al. Optica 5, 117–126 (2018): https://doi.org/10.1364/OPTICA.5.000117
Sakuma, S., Kasai, Y., Hayakawa, T. & Arai, F. Lab Chip 17, 2760–2767 (2017): https://pubs.rsc.org/en/content/articlelanding/2017/lc/c7lc00536a
Supplementary information
Supplementary Video 1
Operation of the iIACS machine. The iIACS machine is composed of optical, microfluidic, electrical, computational, and mechanical parts. An interdisciplinary team of trained operators is needed to run the iIACS machine. First, a sample of suspended cells is prepared before a sorting run. Second, a tube containing the sample is placed at the injection port for the sorting run. Third, the process of each subsystem is monitored on multiple computer panels during the sorting run. Fourth, when the sorting run is finished, collection and waste tubes containing sorted and unsorted cells, respectively, are removed from the iIACS machine. Fifth, cells in the tubes are inspected under an optical microscope to evaluate the results of the sorting run. Sixth, the microscope images are automatically analyzed and then manually verified. Finally, the operators discuss the outcomes and reach a conclusion.
Supplementary Data 1
AutoCAD design file for the microfluidic chip.
Supplementary Data 2
SolidWorks design file for the optics–microfluidic integration unit.
Supplementary Data 3
Source codes for the IA node.
Supplementary Data 4
Source codes for the TM node.
Rights and permissions
About this article

Cite this article
Isozaki, A., Mikami, H., Hiramatsu, K. et al. A practical guide to intelligent image-activated cell sorting. Nat Protoc 14, 2370–2415 (2019). https://doi.org/10.1038/s41596-019-0183-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-019-0183-1
This article is cited by
-
High-throughput microbial culturomics using automation and machine learning
Nature Biotechnology (2023)
-
Label-free liquid biopsy through the identification of tumor cells by machine learning-powered tomographic phase imaging flow cytometry
Scientific Reports (2023)
-
Computer vision meets microfluidics: a label-free method for high-throughput cell analysis
Microsystems & Nanoengineering (2023)
-
A guide to molecular and functional investigations of platelets to bridge basic and clinical sciences
Nature Cardiovascular Research (2022)
-
Deep learning-based image processing in optical microscopy
Biophysical Reviews (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
