
Abstract
Identification of individuals at highest risk of coronary artery disease (CAD)—ideally before onset—remains an important public health need. Prior studies have developed genome-wide polygenic scores to enable risk stratification, reflecting the substantial inherited component to CAD risk. Here we develop a new and significantly improved polygenic score for CAD, termed GPSMult, that incorporates genome-wide association data across five ancestries for CAD (>269,000 cases and >1,178,000 controls) and ten CAD risk factors. GPSMult strongly associated with prevalent CAD (odds ratio per standard deviation 2.14, 95% confidence interval 2.10–2.19, P < 0.001) in UK Biobank participants of European ancestry, identifying 20.0% of the population with 3-fold increased risk and conversely 13.9% with 3-fold decreased risk as compared with those in the middle quintile. GPSMult was also associated with incident CAD events (hazard ratio per standard deviation 1.73, 95% confidence interval 1.70–1.76, P < 0.001), identifying 3% of healthy individuals with risk of future CAD events equivalent to those with existing disease and significantly improving risk discrimination and reclassification. Across multiethnic, external validation datasets inclusive of 33,096, 124,467, 16,433 and 16,874 participants of African, European, Hispanic and South Asian ancestry, respectively, GPSMult demonstrated increased strength of associations across all ancestries and outperformed all available previously published CAD polygenic scores. These data contribute a new GPSMult for CAD to the field and provide a generalizable framework for how large-scale integration of genetic association data for CAD and related traits from diverse populations can meaningfully improve polygenic risk prediction.
Similar content being viewed by others

Main
Coronary artery disease (CAD) is the leading cause of death worldwide, and identification of at-risk individuals remains a critical public health need1. Especially if identified early, at-risk individuals can benefit from more efficiently targeted lifestyle interventions and cholesterol-lowering medications toward lifelong risk mitigation2. However, commonly used clinical risk estimators for CAD were optimized for use in middle-aged adult populations in historical cohort studies and consequently underperform in younger populations or individuals of non-European ancestries3,4,5,6. As CAD is a heritable disease, the increasing amount of widely available genetic data offers additional opportunities to substantially enhance CAD risk prediction early in life, which is likely to prove to be particularly valuable for those in the extremes of the inherited risk distribution7.
Polygenic scores integrate data derived from genome-wide association studies (GWASs)—which quantify the relationship between each of many common DNA variants and risk of disease—into a single quantitative and predictive metric of inherited risk. Several studies so far observed substantial gradients in CAD risk, even among participants with similar clinical risk factor profiles, according to a polygenic score8,9,10,11. Given this potential, polygenic scores are now being deployed clinically across some biobanks and returned through direct-to-consumer testing platforms12,13. Although the past decade has seen numerous advances in the predictive capacity of polygenic scores, score performance remains considerably lower than the theoretical maximum, the proportion of trait liability explained by common DNA variants, particularly among individuals of non-European ancestry14. Simulation studies suggest that even larger sample sizes of GWASs have the potential to more accurately estimate the effect size associated with each single nucleotide polymorphism to improve scores for CAD15. Polygenic scores integrating GWAS data from individuals of diverse ancestries in addition to that of the target population show relative improvement in predictive accuracy compared with methods only utilizing GWAS data from a single ancestry source16,17. Furthermore, the principles of genetic correlation suggest benefit in incorporating information from GWAS of related traits to refine polygenic prediction in the trait of interest18,19.
Alongside considerable—and warranted—enthusiasm for polygenic scores to enable a new era of preventive clinical medicine is recognition of several key limitations. First, polygenic scores have reduced predictive performance in individuals of non-European ancestry20. This largely stems from relative underrepresentation of other ancestries in prior GWAS discovery cohorts. Recent efforts have focused on conducting GWAS in larger and more ancestrally diverse populations and designing methods leveraging ancestry-specific linkage disequilibrium patterns to help improve score performance16,17,21,22. Second, although available scores associate strongly with prevalent disease, they perform less well in predicting incident disease, which would offer more clinical utility in enabling targeted interventions23. Finally, most risk prediction models so far are based on either genetic or clinical risk factors, but better integration of these modalities and estimation of a clinically actionable risk estimate is needed24,25.
In this Article, to address these needs, we used information from ancestrally diverse 269,000 CAD cases, over 1,178,000 controls and data from related traits in over two million individuals along with methods leveraging commonalities in mechanistic pathways to develop a new polygenic risk score for CAD.
Results
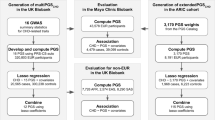
Summary statistics from GWAS for CAD, other atherosclerotic diseases (for example, ischemic stroke), and their risk factors (for example, diabetes, blood pressure and lipid concentrations) across over 1.4 million individuals from multi-ancestry cohorts were aggregated to design polygenic risk scores for CAD (Fig. 1 and Supplementary Table 1). These scores were trained within the UK Biobank cohort in 116,649 individuals of European ancestry and then validated in the remaining independent study population of 325,991 individuals (54.3% female, 7281 African, 1,464 East Asian, 308,264 European and 8,982 South Asian ancestry) (Supplementary Table 2)26. The participants in the training and validation cohorts are independent from the individuals analyzed in the previously conducted GWAS from which summary statistics were obtained27. A total of 51 candidate ancestry- and trait-specific scores were included in the genome-wide polygenic score (GPS) training analysis, with 32 scores carried forward on the basis of a stepwise process to identify those that significantly contributed to overall prediction and included in the weighting of GPSMult (Fig. 2a,b).

Polygenic scores were constructed using cohort-specific, ancestry-stratified summary statistics for CAD and CAD-related traits, resulting in 51 GPS across all traits and ancestries. For each trait (for example, CAD) the best-performing combination of cohort-specific, ancestry-stratified GPSs was determined using stepAIC, and their optimal mixing weights (β) were determined using logistic regression in 116,649 individuals of European ancestry in the UK Biobank training dataset. The selected GPSs were linearly combined using these mixing weights to yield multi-ancestry scores predicting CAD for each trait (layer 1). The best-performing combination of multi-ancestry, trait-specific GPSs was determined using stepAIC, and their optimal mixing weights (β) were determined using logistic regression in 116,649 individuals of European ancestry in the UK Biobank training dataset. The selected GPSs were linearly combined using these mixing weights to yield GPSMult (layer 2). Ancestries: AFR, African; EA, East Asian; EUR, European; HISP, Hispanic; SA, South Asian. Source GWAS traits: CAD27,33,34,38,56, body mass index (BMI)38,57, ischemic stroke38,58,59, diabetes mellitus (DM)59,60,61, peripheral artery disease (PAD)38,56,62, glomerular filtration rate (GFR)38,63, systolic blood pressure (SBP)38,64, diastolic blood pressure (DBP)38,64, LDL cholesterol38,65,66, HDL cholesterol38,65,66, triglycerides (TG)38,65,66.

a, The OR/SD with 95% CI for prevalent CAD risk of the multi-ancestry, trait-specific layer 1 GPSs was assessed in logistic regression models adjusted for age, sex, genotyping array and the first ten principal components of ancestry in the same training group of n = 116,649 independent UK Biobank European ancestry individuals. b, The contributing weights of each of the ancestry-stratified, cohort-specific GWAS-based GPS to each of the trait-based layer 1 polygenic scores are proportional to stacked bar size, which are colored according to ancestry of source GWAS, and normalized to 100% to reflect composition in the overall GPSMult. Of 51 ancestry- and trait-specific scores that were included in the GPS training analysis, 32 scores significantly contributed to overall prediction in GPSMult after optimization of score selection with stepAIC and weighting through logistic regression in the two layers.
Association of GPSMult with prevalent disease in UK Biobank
The resulting score, GPSMult demonstrated a strong association with prevalent CAD, with significant improvement from previously published scores. Among 308,264 European ancestry individuals in the hold-out validation dataset, GPSMult was associated with an odds ratio per standard deviation increase (OR/SD) of 2.14 (95% confidence interval (CI) 2.10–2.19) in a model adjusted for age, sex, genotyping array and the first ten principal components of genetic ancestry, with significant improvement from prior published scores from the Polygenic Score Catalog without UK Biobank participants in discovery data, where OR/SD ranged from 1.14 to 1.77 (Supplementary Table 3)28. This corresponded to a Nagelkerke R2 of 0.074 and a logit liability R2 of 0.187 (Extended Data Fig. 1). After adjusting for measured clinical risk factors including systolic and diastolic blood pressure, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, triglycerides, diabetes, body mass index and glomerular filtration rate, this risk estimate was only modestly attenuated to an OR/SD 2.07 (95% CI 2.02–2.13) (Supplementary Table 4). The associations between GPSMult and CAD were largely consistent across studied subgroups, but some evidence of heterogeneity was found when restricting to male participants (OR/SD 2.20, 95% CI 2.15–2.26, P < 0.001) when compared with female participants (OR/SD 1.94, 95% CI 1.86–2.03, P < 0.001), with P-heterogeneity <0.001 (Extended Data Fig. 2). Additionally, the association between GPSMult and CAD was stronger in younger individuals aged 45–54 years (OR/SD 2.17, 95% CI 2.04–2.31, P < 0.001) and 55–64 years (OR/SD 2.18, 95% CI 2.11–2.25, P < 0.001), when compared with older individuals aged 65–75 years (OR/SD 2.08, 95% CI 2.01–2.15, P < 0.001), consistent with recent studies (Extended Data Fig. 2)29,30.
GPSMult showed stronger association with CAD risk when compared with the previously published GPS2018 (ref. 9) in direct comparison using the same group of individuals for validation. Among individuals of European ancestry, individuals in the bottom and top centile of the polygenic score had a 0.8% and 12.3% prevalence of CAD, respectively, with GPS2018, compared with 0.6% and 16.3% prevalence of CAD with GPSMult (Fig. 3 a,b). GPSMult also outperformed GPS2018 in predicting prevalent CAD across ancestry groups in the UK Biobank, with OR/SD of 1.39 (95% CI 1.17–1.67) in African ancestry, 2.14 (95% CI 1.34–3.49) in East Asian ancestry and OR 2.02 (95% CI 1.83–2.23) in South Asian ancestry (Fig. 3c). Among individuals with CAD, the median percentile of GPSMult is significantly higher than that of the GPS2018, 75 (interquartile range 50–91) versus 69 (interquartile range 43–88) (Fig. 3d). Given improved stratification with this newly developed polygenic score, both tails of the score distribution were associated with a greater magnitude of risk when compared with GPS2018. With the GPS2018, the top 8.3%, 3.0% and 1.3% of the population had 3-fold, 4-fold and 5-fold greater odds for CAD relative to the middle quintile of the population, respectively, whereas with the GPSMult, the top 20.0%, 9.6% and 4.9% of the population had 3-fold, 4-fold and 5-fold greater odds for CAD relative to the middle quintile of the population, respectively (Fig. 3e, Extended Data Fig. 3a,b and Supplementary Table 5). Conversely, with the GPS2018, the bottom 1.7%, 0.5% and 0.1% of the population had 1/3, 1/4 and 1/5 the odds for CAD relative to the middle quintile of the population, respectively, whereas with the GPSMult, the bottom 13.9%, 1.7% and 0.2% of the population had 3-fold, 4-fold and 5-fold lower odds of CAD relative to the middle quintile of the population, respectively (Fig. 3f and Extended Data Fig. 3c,d).

a,b, The mean prevalence of CAD with 95% CI according to 100 groups of the UK Biobank European ancestry validation dataset consisting of n = 308,264 independent participants, binned according to the percentile of the GPS2018 (a) and GPSMult (b). c, The OR/SD with 95% CI for prevalent CAD of GPSMult was assessed in a logistic regression model adjusted for age, sex and the first ten principal components of ancestry in n = 7,281 independent individuals of African ancestry, n = 1,464 independent individuals of East Asian ancestry, n = 308,264 independent individuals of European ancestry, and n = 8,982 independent individuals of South Asian ancestry. d, Distributions of GPS2018 and GPSMult percentiles across the UK Biobank European ancestry validation dataset consisting of n = 308,264 independent participants. For all box plots: central line of each box, median; top and bottom edges of each box, first and third quartiles; whiskers extend 1.5× the interquartile range beyond box edges. e, Proportion of UK Biobank validation population with 3-, 4- and 5-fold increased risk for CAD versus the middle quintile of the population, stratified by GPS. The odds ratio assessed in a logistic regression model adjusted for age, sex, genotyping array and the first ten principal components of ancestry. f, Proportion of UK Biobank testing population with 1/3, 1/4, and 1/5 risk for CAD versus the middle quintile of the population, stratified by GPS. Odds ratio assessed in a logistic regression model adjusted for age, sex, genotyping array and the first ten principal components of ancestry.
Validation of GPSMult in external cohorts
GPSMult was also strongly associated with prevalent CAD in external cohorts, with significant improvement from prior published scores. Twenty-seven polygenic scores for CAD from the Polygenic Score Catalog and GPSMult were calculated in identical groups of individuals to facilitate direct comparison within individuals of African, European and Hispanic Ancestry in Million Veteran Program31 and South Asian ancestry in Genes & Health32 (Fig. 4 and Supplementary Tables 6 and 7). For each group, individuals were selected for inclusion that were not included in any of the published GWAS summary statistics33,34 used for GPSMult derivation. Among 33,096 individuals of African ancestry in the Million Veteran Program, GPSMult was associated with an OR/SD of 1.25 (95% CI 1.21–1.29, P < 0.001) for CAD in a model adjusted for age, sex, genotyping array and the first ten principal components of genetic ancestry, corresponding in a 73% (P < 0.001) relative improvement in effect size compared with GPS2018 and 39% (P = 0.008) improvement when compared with the recently published PRS2022 (ref. 27), respectively. Similarly, among 124,467 individuals of European ancestry in the Million Veteran Program, GPSMult was associated with an OR/SD of 1.72 (95% CI 1.69–1.75, P < 0.001), corresponding in a 46% (P < 0.001) and 13.6% (P < 0.001) relative improvement in effect size compared with GPS2018 and PRS2022 (ref. 27), respectively. Among 16,433 individuals of Hispanic ancestry in the Million Veteran Program, GPSMult was associated with an OR/SD of 1.61 (95% CI 1.53–1.70, P < 0.001), corresponding in a 66.8% (P < 0.001) and 13.9% (P = 0.11) relative improvement in effect size compared with GPS2018 and PRS2022, respectively. Lastly, among 16,874 individuals of South Asian ancestry in Genes & Health, GPSMult was associated with an OR/SD of 1.83 (95% CI 1.69–1.99, P < 0.02), corresponding to a 113% (P < 0.001) and 29% (P = 0.02) relative improvement in effect size compared with GPS2018 and PRS2022, respectively (Fig. 4).

The OR/SD with 95% CI for prevalent CAD risk was assessed for each polygenic score in a logistic regression model adjusted for age, sex, genotyping array and the first ten principal components of ancestry in the same group of individuals per cohort: n = 33,096 independent African ancestry individuals in the Million Veteran Program; n = 124,467 independent European ancestry individuals in the Million Veteran Program; n = 16,433 independent Hispanic ancestry individuals in the Million Veteran Program; n = 16,874 independent South Asian ancestry individuals in the Genes & Health Study, using high-performing published scores from the Polygenic Score Catalog (GPS2018 (ref. 9), metaGRS8, metaPRSCAD67, AnnoPredCAD68, PRSCSCHD69 and PRS2022 (ref. 27), as well as GPSMult28. Results for these and additional CAD polygenic scores published in the Polygenic Score Catalog are available in Supplementary Tables 6 and 7.
Association of GPSMult with incident disease in UK Biobank
The GPSMult was predictive of incident CAD events over median (interquartile range) 12.0 (11.2–12.7) years of follow-up across all four ancestral groups in the UK Biobank. Across the entire UK Biobank validation study population without prior CAD, an incident CAD event was observed in 1.1% of those in the lowest percentile of the GPSMult distribution versus 11.7% of those in the top percentile. Overall, GPSMult was associated with a hazard ratio per standard deviation (HR/SD) of 1.73 (95% CI 1.70–1.76, P < 0.001), compared with hazard ratio (HR) 1.49 (95% CI 1.46–1.52, P < 0.001) found with GPS2018. When stratified by ancestry, risk estimates were comparable across individuals of East Asian (HR/SD 1.72, 95% CI 1.13–2.60, P = 0.011), European (HR/SD 1.74, 95% CI 1.71–1.78, P < 0.001), and South Asian (HR/SD 1.62, 95% CI 1.49–1.77, P < 0.001) ancestry, but effect size was reduced among individuals of African ancestry (HR/SD 1.25, 95% CI 1.07–1.46, P = 0.004) (Fig. 5a). Across all individuals in the UK Biobank validation dataset, GPSMult demonstrated 38% relative improvement in effect size compared with GPS2018. Of this, 26% improvement resulted from larger sample size of the primary Coronary ARtery DIsease Genome wide Replication and Meta-analysis plus The Coronary Artery Disease Genetics consortium (CARDIOGRAMplusC4D) GWAS (excluding UK Biobank participants), 9% improvement from incorporation of multi-ancestry CAD summary statistics, and 3% improvement from leveraging genetic commonalities with CAD risk factors to refine score weighting (Fig. 5b). Incorporation of multi-ancestry and multi-trait genetic data resulted in greater relative gains in incident disease prediction for individuals in each ancestry, with improved relative effect sizes of 143%, 71%, 38% and 23% for individuals of African, East Asian, European and South Asian ancestry, respectively, compared with GPS2018 performance in those groups. Enhanced performance, indexed to the effect size observed in European ancestry with the GPS2018, was also observed across ancestries, with improved prediction in African ancestry (relative effect size 0.55, increased from 0.23) (Fig. 5b) and performance surpassing the reference score in East Asian ancestry (relative effect size 1.37, increased from 0.80) and South Asian ancestry (relative effect size 1.19, increased from 0.97).

a, Adjusted HR/SD of the polygenic score with corresponding 95% CIs and P values for incident CAD by ancestry, stratified by the version of the polygenic score, calculated from Cox proportional-hazards regression models adjusted for age, sex, genotyping array and the first ten principal components of ancestry in the UK Biobank validation dataset, consisting of n = 7,157 independent individuals of African ancestry, n = 1,442 independent individuals of East Asian ancestry, n = 297,772 independent individuals of European ancestry, and n = 8,440 independent individuals of South Asian ancestry. GPS2018 corresponds to a previously published polygenic score for CAD9. P values are derived from a Wald test implemented in the coxph function in R and are two-sided. b, The score effect sizes relative to the effect size of GPS2018 in European ancestry individuals. ‘>3-fold larger CAD GWAS’ designates a polygenic score generated using summary statistics of largely European ancestry from the most recent CARDIOGRAMplusC4D excluding the UK Biobank (GPSCADEUR). ‘Multi-ancestry CAD GWAS’ refers to the polygenic score generated by combining ancestry-specific polygenic scores generated using GWAS summary statistics from CARDIOGRAMplusC4D, Genes & Health, Biobank Japan, Million Veteran Program and FinnGEN biobanks in layer 1 (GPSCADANC). GPSMult designates polygenic score for CAD designed with summary statistics from multiple ancestries and multiple CAD-related traits in layer 2. Asterisk designates the reference group for calculating relative gain.
Disease risk in the extremes of the GPSMult distribution
We additionally hypothesized that the GPSMult could identify individuals in the extreme tails of its distribution with clinically important increase, or decrease, in risk. Current cardiovascular disease prevention guidelines recommend statin therapy for individuals with prior CAD, peripheral artery disease (PAD), ischemic stroke, diabetes mellitus or severe hypercholesterolemia (LDL ≥190 mg/dL to help mitigate their high risk of cardiovascular disease and mortality2. In the high end of GPSMult, we sought to identify individuals with genetic risk of equivalent magnitude to that of individuals with these guideline-endorsed indications for statin therapy. In prospective analyses of individuals without prior CAD, when compared with individuals in the middle quintile, those within the top 3 percentiles of GPSMult had equivalent disease risk of incident CAD as the recurrent event risk for an individual who had a CAD event before enrollment (Extended Data Fig. 4a). Furthermore, individuals without PAD in the top 8% of polygenic score distribution had incident CAD risk equivalent to individuals with prior PAD; individuals without diabetes in the top 21% of polygenic score distribution had incident CAD risk equivalent to individuals with prior diabetes; and individuals without severe hypercholesterolemia (estimated untreated LDL cholesterol ≥190 mg/dL) in the top 28% of polygenic score distribution had incident CAD risk equivalent to individuals with prior hypercholesterolemia (Extended Data Fig. 5a–c). Conversely, in the low end of the GPSMult distribution, individuals in the bottom 5 percentiles were associated with a significant reduction in incident CAD risk (HR 0.27, 95% CI 0.21–0.35, P < 0.001) when compared with the middle quintile (40–59%). When comparing individuals who smoke and are in the bottom 5 percentiles of GPSMult with nonsmokers in the middle quintile, the reduction in the absolute incidence of CAD associated with low GPSMult offsets approximately 60 pack-years of smoking. Furthermore, individuals in the 5th to 9th percentiles of GPSMult also had a significant reduction in CAD risk (HR 0.55, 95% CI 0.49–0.62, P < 0.001) when compared with the middle quintile. These individuals experienced comparable risk reduction as those individuals carrying variants in PCSK9-associated lifelong low levels of LDL cholesterol (Extended Data Fig. 4b)35,36.
Modeling of GPSMult with clinical risk predictors
A risk prediction approach integrating clinical and genetic risk using the American College of Cardiology/American Heart Association Pooled Cohort Equations (PCE)5, GPSMult and their interaction in a single model was used to predict 10-year risk of CAD in the UK Biobank validation population. Accounting for the interaction between the polygenic score and clinical risk estimate improves performance beyond the simple addition of the two, with lower GPSMult weighting with higher PCE estimates (interaction effect size −0.60, Pinteraction < 0.001). This combined model effectively improved risk prediction when compared with PCE alone. When binned into strata corresponding to clinical guideline recommendations5, this model suggested striking gradients in predicted CAD incidence across the GPSMult distribution, with significant differences observed in ancestry-based subgroups (Fig. 6a). The absolute gradient in risk predicted by this model from bottom to top centile was largest in South Asian ancestry individuals with high PCE risk (5.1% to 29.1%), compared with European ancestry individuals (2.6% to 20.6%).

a, The cumulative incidence of CAD over 10 years predicted by modeling GPSMult, AHA/ACC PCE 10-year risk estimate, and their interaction in the UK Biobank validation dataset binned according to the percentile of the GPSMult. Individuals were grouped by risk categories of the PCE (predicted 10-year risk of atherosclerotic cardiovascular disease as ‘low’ (<5%), ‘borderline’ (5% to <7.5%), ‘intermediate’ (≥7.5% to <20%) and ‘high’ (≥20%)), and stratified by ancestry. b, C-statistics are based on 10-year follow-up events from Cox regression models of listed variables. PCE includes age and sex variables in its risk estimation. c, The improvement in the predictive performance of the addition of the GPSMult to the PCE was evaluated using continuous and categorized NRI, with a risk probability threshold of 7.5% and CIs (95%) obtained from 100-fold bootstrapping.
When compared with the PCE risk estimate incorporating clinical risk factors alone, integration of the PCE with GPSMult contributed to significantly higher discrimination and predictive performance across the entire tested population. First, discrimination was assessed in Cox regression models including various covariables using Harrell’s C-statistic. A gradient in improvement was seen using baseline models with age and sex alone (C-statistic 0.710, 95% CI 0.706–0.715), PCE, which is inclusive of age and sex (C-statistic 0.739, 95% CI 0.735–0.744), and the model integrating PCE, GPSMult and their interaction term (C-statistic 0.763, 95% CI 0.759–0.768) (Fig. 6b). Similar improvements in C-statistic were observed for models tested in subgroups stratified by ancestry (Supplementary Table 8). Second, categorized net reclassification improvement (NRI) was calculated across the entire study population using a threshold of 7.5% (NRI 0.075) of the predicted 10-year risk of CAD, which is the clinically accepted estimated risk threshold for recommending initiation of statin therapy for prevention of CAD. The risk model combining PCE and GPSMult resulted in significant improvements in the categorical net reclassification index (NRI 7.0%, +8.1% for incident cases and −1.1% for noncases), with GPSMult resulting in greater up classification of risk largely in individuals who go on to develop disease (Fig. 6c). Third, when compared with established risk-enhancing factors for CAD, categorization within the top 10 percentiles of the GPSMult distribution corresponded to a significantly higher net reclassification over the use of PCE estimate alone (3.7%) as compared with other risk enhancers such as elevated lipoprotein (a) (with NRI 1.3%) (Extended Data Fig. 6). Similar results in NRI were observed across other ancestries (Supplementary Table 9). Additionally, similar trends in predictive performance, discrimination and reclassification were observed in a model that included integration of the QRISK clinical risk estimator, instead of the PCE, with GPSMult (Supplementary Tables 8 and 9).
Association of GPSMult with recurrent disease in UK Biobank
In addition to first events, the GPSMult predicted recurrent CAD events in individuals with prior CAD. GPSMult was associated with an HR/SD of 1.13 (95% CI 1.08–1.18, P < 0.001), comparable to prior studies37. Although a significantly less pronounced effect estimate as compared with the prediction of a first CAD event, the predictive performance of GPSMult in this context was comparable to that of diastolic blood pressure (HR 1.11, 95% CI 1.06–1.16, P < 0.001) and glycated hemoglobin (HR 1.07, 95% CI 1.02–1.12, P < 0.001) (Extended Data Fig. 7).
Discussion
A new polygenic score for CAD incorporating multi-ancestry summary statistics from GWAS for CAD and related risk factor traits on a large scale demonstrated significantly improved performance when compared to prior published scores. External validation in fully independent datasets derived from the Million Veteran Program and the Genes & Health studies confirmed enhanced prediction compared withpreviously published and available polygenic scores across multiple ancestries. The enhanced predictive capacity of this score was particularly pronounced in the extremes of the score distribution, enabling—in some cases—identification of healthy individuals with risk of CAD equivalent to those with pre-existing disease. When added to risk scores used in current clinical practice, GPSMult significantly improved discrimination and reclassification relevant to clinically important decision thresholds, such as the decision to initiate statin therapy.
This work builds on prior studies in providing a framework for optimizing a polygenic score for any trait, within the limitations of available GWAS with finite sample sizes and underrepresentation of diverse populations. The GPSMult incorporates CAD summary statistics from large non-European ancestry biobanks encompassing over 269,000 cases and over 1,178,000 controls, including many-fold larger representation of individuals of non-European ancestries than previously published efforts32,33,38,39. This results in substantial improvements in prediction for individuals of East and South Asian ancestry, reflecting greater representation of summary statistics from Biobank Japan and Genes & Health. However, the majority of improvement in effect size is attributable to use of summary statistics from the largest CAD GWAS so far (CARDIOGRAMplusC4D consortium, excluding UK Biobank participants), particularly in European ancestry individuals27. The additional incorporation of genetic associations with CAD-related risk factors across ancestries into calculating GPSMult significantly improves prediction beyond using summary statistics from CAD GWAS alone, with impact most notable in individuals of non-European ancestry. This may potentially be due to greater representation of these ancestries in the discovery GWAS for CAD risk factor traits. With these additions, the phenotypic variance explained by GPSMult for CAD calculated as R2 on the logit-liability scale was 0.187. Although this estimate remains below the estimated single nucleotide polymorphism heritability for CAD of 0.4–0.6, it surpasses the phenotypic variance explained of 0.155 by the largest component GWAS from the CARDIOGRAMplusC4D consortium27,40.
Overall, modest improvements in prediction were observed among individuals of African ancestry, in part due to underrepresentation of this group in GWASs so far, and these discrepancies warrant careful consideration as polygenic scores start to enter into clinical practice20. Due to smaller haplotype blocks observed in individuals of African ancestry, a 4- to 7-fold larger GWAS is needed to yield comparable prediction gains41. In the near term, the decreased effect size observed in individuals of African ancestry is likely to persist, and this has also been observed for other biomarkers and predictors in clinical practice42. Nevertheless, genetic ancestry has a considerable impact on certain aspects of polygenic risk prediction, such as the allele frequency of a given variant. In order to best to move polygenic scores into widespread practice, research efforts would benefit from transparent and systematic reporting of score performance across ancestries43, recruitment of more diverse study participants in cohorts such as the US AllofUs Research Program21, new statistical methods to enhance cross-ancestry portability16,17, more sophisticated quantitative metrics and ongoing dialog with a range of stakeholders, including patients44. Furthermore, as the population of admixed individuals that do not discretely mapping onto a single continental ancestry continues to increase, recently developed methods that account for more continuous representations of ancestry in polygenic scores may prove useful45.
Polygenic scores have the potential to enhance clinical decision making, although this warrants confirmation in prospective studies. Some such studies are already underway returning polygenic risk information to patients12,46, and medical societies have begun to provide provisional guidance on their use47. Furthering these goals, GPSMult is able to better identify individuals at the highest risk for developing incident CAD to potentially guide early preventive interventions48,49. Building on prior work advocating for use of polygenic scores as a risk-enhancing factor to guide decision making regarding statin therapy in individuals at borderline or intermediate CAD risk, the current work more strongly supports use in primary screening across the population to target interventions50. Current cardiovascular prevention guidelines recommend statin initiation for individuals solely on the basis of having any of the following conditions as they portend high risk of a new atherosclerotic cardiovascular disease event: prior CAD, ischemic stroke, PAD, diabetes or severe hypercholesterolemia2. Here we demonstrate that GPSMult identified 3% of the population with equivalent risk for a future CAD event as that in individuals who have had prior disease. Similarly, the top 8%, 21% and 28% of the GPSMult distribution—despite having no known CAD—had equivalent risk of incident CAD as individuals with prior PAD, diabetes mellitus and severe hypercholesterolemia, respectively. Because all three of these designations are currently clinical indications for statin therapy, a high GPSMult could be employed to identify additional individuals for cholesterol-lowering therapies as an adjunct to current guidelines. Furthermore, given the GPSMult’s ability to identify these individuals with the highest propensity for developing CAD, these scores could be employed to enrich for high-genetic-risk individuals in CAD prevention trials to maximize event rates and minimize drug trial costs51. The GPSMult could also be employed to identify the individuals with the highest risk of recurrent events for targeted, otherwise costly therapies that have been shown to be beneficial in this population52,53. Additionally, GPSMult also identifies individuals in the lower end of genetic risk who are seemingly protected from CAD with similar risk reduction as that of carriers of variants in the PCSK9 gene leading to lifelong reductions on LDL cholesterol35,36.
Furthermore, a risk model incorporating polygenic risk with the PCE estimated risk is applied to individuals across different ancestries to demonstrate improved predictive performance. This improved performance illustrates the potential for an integrated absolute risk prediction model24,25. For example, this model is particularly useful in differentiating risk in the high-risk South Asian ancestry population, where traditional clinical risk estimators often fail to capture the increased risk associated with this ancestry4. The integration of the GPSMult with PCE builds on prior efforts that demonstrated improvement in model discrimination by now showing nearly identical improvement in C-statistic (0.03) in between models incorporating (1) age and sex, (2) PCE alone and (3) combined genetic and clinical risk across the population23. However measures of C-statistic alone are not optimal or fully comprehensive in evaluating models that predict future risk54. GPSMult demonstrates nearly three-fold greater net reclassification of CAD cases/noncases when added to the PCE 10-year risk assessment to guide statin initiation as compared with established ‘risk-enhancing factors’. Further work is needed to incorporate additional risk factors. To aid in future model calibration efforts, there is a need for population-level disease incidence and mortality data disaggregated by ancestral subgroups12.
These results should be interpreted within the context of limitations. Polygenic scores were developed and validated in individuals of European ancestry and then externally validated in non-European ancestry populations, and this may have contributed to decreased predictive performance in these groups. These results underscore the need for larger and more representative GWAS studies. UK Biobank participants were recruited at age 40–69 years, raising the possibility of survivorship or selection bias that limits generalizability to younger patients; however, recent studies have demonstrated reliable performance of GPS in younger age groups7. All UK Biobank disease endpoints were similarly ascertained through participant self-report, diagnosis codes from inpatient admissions, national procedure, and death registries. Relatively few incident events were observed in individuals of non-European ancestry in the UK Biobank, and additional work is needed to evaluate this in larger populations and further validate optimal approaches to integrate GPS with clinical risk scores. Participants in research studies tend to be healthier than the general population—recalibration of disease risk models for a given target population may be needed before clinical deployment55.
In conclusion, incorporating GWAS data for CAD and related traits from multiple ancestries on a large scale leads to significantly improved performance of GPSMult in external validation among diverse ancestry populations when compared with previously published scores. This approach is readily generalizable to common complex diseases and traits, results in a polygenic score that is able to better identify individuals at the highest and lowest ends of risk, significantly reclassifies risk beyond clinical risk estimators, and has the potential to advance clinical decision making.
Methods
Study populations
The UK Biobank is a prospective cohort study that enrolled over 500,000 individuals between the ages of 40 and 69 years between 2006 and 2010 (refs. 26,70). A detailed questionnaire completed by UK Biobank participants at enrollment assessed self-report of sex, ancestry and lifestyle factors, including smoking. Anthropometric measurements including body mass index were measured at the initial enrollment visit. Biomarkers including serum lipid concentrations and renal function markers were assessed at time of enrollment as part of the study protocol. Diagnoses of PAD, diabetes and hypertension were determined on the basis of self-report, hospitalization records, procedure codes and death registry codes confirming a clinical diagnosis4,71.
Participants within the Million Veteran Program were recruited from more than 75 Veteran Affairs Medical Centers nationwide since 2011, with >885,000 individuals currently enrolled31. Each participant has consented to linkage to their electronic medical record, wherein self-reports of ancestry and sex, ICD9/10 diagnosis codes, Current Procedural Terminology codes, clinical laboratory measurements and reports of diagnostic imaging modalities are available. Participants were also asked to complete baseline and lifestyle questionnaires to further augment data contained in the electronic health record.
Genes & Health is a UK-based cohort of over 48,000 British Pakistani and Bangladeshi individuals recruited and consented for lifelong electronic health record access and genetic analysis32. Medical records are linked to ICD10, OPCS and SNOMED diagnosis and procedural codes across inpatient and hospital settings as well as clinical laboratory measurements, and a baseline questionnaire containing demographic information including self-report of sex and ancestry.
Clinical endpoints
Ascertainment of CAD at enrollment in the UK Biobank was based on self-report, hospitalization records, procedural codes or death registry confirming diagnosis of myocardial infarction or its acute complications, or a coronary revascularization procedure (coronary artery bypass graft surgery or percutaneous angioplasty/stent placement)71,72. The earliest date at which the diagnosis was ascertained was considered as the diagnosis date. For individuals with CAD before enrollment, recurrence of CAD was determined on the basis of diagnosis of a myocardial infarction or revascularization in the follow-up period after study enrollment73.
Within the Million Veteran Program, ICD9, ICD10 and Current Procedural Terminology codes from both inpatient and outpatient encounters were used to curate and classify CAD cases based on having a myocardial infarction or undergoing revascularization, identified as subjects with at least two codes (of any category) that occurred on distinct dates within a 12 month window33. Incident cases were identified as those with the first of the two qualifying codes occurring after enrollment. The remaining CAD cases, including through self-report, were considered prevalent.
In the Genes & Health study, ICD10 and SNOMED codes from the linked electronic health record were used to classify CAD cases defined as myocardial infarction or revascularization on the basis of first diagnosis date34. Prevalent cases were defined as events before enrollment while events occurring after enrollment were designated as incident disease.
GPS construction
Summary statistics from recent CAD GWAS studies (Genes & Health, FinnGen, Million Veteran Program, Biobank Japan and CARDIOGRAMplusC4D excluding UK Biobank samples) conducted in individuals of diverse ancestries were used to determine primary CAD score weights (Supplementary Table 1)27,32,33,38,39. UK Biobank participants were not included among these discovery cohorts to preserve them as an independent hold-out dataset for training and validation of the GPSMult (Supplementary Table 2). Ancestry-specific linkage disequilibrium reference panels were extracted from the 1000 Genomes Project phase 3 data to match with the ancestry for the discovery GWAS, and only unrelated samples were used74. GPSMult construction comprised a two-layer process, with layer 1 consisting of combining multiple polygenic scores derived from different ancestry-specific GWAS data for each trait, and layer 2 consisting of combining this multi-ancestry CAD polygenic score with similarly constructed multi-ancestry CAD-related trait scores predicting CAD (Fig. 1) to generate GPSMult.
Separate GPS were constructed for each ancestry-stratified CAD GWAS using the LDpred2 method, which is a Bayesian approach to calculate a posterior mean effect for all variants based on an effect size in the prior GWAS and subsequent shrinkage based on linkage disequilibrium75. Only HapMap3 variants—a set of 1,296,172 variants compiled by the International HapMap Project which capture common patterns of variation in a variety of human populations—were included for score calculation76. The default parameters used in the LDpred2 method included the proportion of variants assumed to be causal (cut-offs of P = 1.0 × 10−4, 1.8 × 10−4, 3.2 × 10−4, 5.6 × 10−4, 1.0 × 10−3, 1.8 × 10−3, 3.2 × 10−3, 5.6 × 10−3, 1.0 × 10−2, 1.8 × 10−2, 3.2 × 10−2, 5.6 × 10−2, 1.0 × 10−1, 1.8 × 10−1, 3.2 × 10−1, 5.6 × 10−1 and 1), the scale of heritability (s = 0.7, 1 and 1.4) and whether or not a sparse LD matrix was applied9,75,77. Combinations of these parameters resulted in 102 candidate GPSs for each set of ancestry-stratified GWAS summary statistics. We extracted the genotypes from centrally imputed data repository, manipulated and transformed the data by bgenix and BCFtools78,79, computed the polygenic scores by the Plink software parallelly for each chromosome, and combined the chromosome scores for each individual by the Datamash software80,81. The best GPS was selected among these candidates by assessing their performance in predicting prevalent CAD in an independent 116,649 individuals of White British ancestry from UK Biobank (this dataset was used in all the score selection procedures thereafter, and same group of individuals used to train previously published score GPS2018 who had not withdrawn consent in the interim)9. For example, using the GWAS data from CARDIOGRAMplusC4D excluding UK Biobank samples, the best-performing score predicting CAD (GPSCADEUR) was generated using LDpred2 parameters of P = 0.018, h2 scale = 1, and without sparse LD. For selecting the best combination of CAD GPS scores from each ancestry-specific CAD GWAS for mixing, the discriminative capacities (Akaike information criterion, AIC) of these GPS combinations for predicting CAD were assessed using the stepAIC function from R MASS package82. A logistic regression model was used to estimate the mixing weights for each individual ancestry-specific GPS. These GPSs were then linearly combined together into a single GPSCADANC score (layer 1, Fig. 1). Similar procedures were followed for other atherosclerotic diseases (ischemic stroke and PAD)58,62 and risk factor traits—LDL cholesterol, HDL cholesterol, triglycerides65,66, diabetes60, systolic blood pressure83, diastolic blood pressure, glomerular filtration rate63 and body mass index (Supplementary Table 1 and Fig. 1).
These multi-ancestry trait-specific GPSs were then linearly combined with the multi-ancestry GPSCADANC (from layer 1) to generate the final GPSMult (layer 2). Just as for layer 1, the discriminative capacities (AIC) of these GPS combinations for predicting CAD were assessed to identify the best combination of trait-level scores for mixing82. A logistic regression model was used to estimate the mixing weights for each individual trait-specific GPS as described above. These GPSs were then linearly combined together into a single GPSMult score (layer 2, Fig. 1). Of 51 GWAS- and ancestry-specific GPS that went through layers 1 and 2 of selection and mixing, 32 contributed to the final GPSMult, incorporating GWAS summary statistics from multiple ancestries and multiple CAD-related traits (Fig. 2). LDpred2 parameters selected for each score, whether the score survived after feature selection, and mixing weights from layers 1 and 2 are listed in Supplementary Table 1.
GPS validation
The GPSMult was compared with previously published polygenic scores with respect to effect size for CAD association. The variant effect sizes were downloaded from PGS Catalog and calculated in the same UK Biobank validation dataset of 308,264 European ancestry individuals for direct comparison8,9,23,28,49,68,69,84,85,86,87,88,89,90,91,92,93,94,95,96. For score accession numbers and performance metrics, see Supplementary Table 3. The validation datasets were composed of UK Biobank participants separate from those used to train the GPSMult. These individuals underwent genotyping using the UK BiLEVE Axiom Array or UK Biobank Axiom Array, containing over 800,000 variants spanning the genome26. Imputation was performed using the Haplotype Reference Consortium resource, the UK10K panel, and the 1000 Genomes panel74,97,98. We identified a subset of 488,243 participants with genotyping array data. After additional exclusion of 45,602 individuals for high heterozygosity or genotype missing rates, discordant reported versus genotypic sex, putative sex chromosome aneuploidy, excess relatedness (second-degree relative or closer), withdrawal of informed consent, or unreported ancestry and 116,649 individuals used for score training, 325,991 individuals (54.3% female, 2.2% African, 0.4% East Asian, 92.0% European and 2.7% South Asian) were included in the multi-ancestry internal validation cohort for subsequent analyses.
External validation was performed in the Million Veteran Program and Genes & Health studies. Among Million Veteran Program participants, 173,996 individuals not included in the previously published CAD GWAS33 were included and comprised 33,096 (21%) individuals of African ancestry and 124,467 (79%) individuals of European ancestry (Supplementary Table 2). Individuals were genotyped using the Affymetrix Axiom array and imputed to the TOPMed reference panel. Variants and sample quality control were previously described99. Within the Genes & Health study, individuals not included in the previously published CAD GWAS34 were included and comprised 16,874 participants of South Asian ancestry (Supplementary Table 2). These individuals underwent genotyping using the Illumina Infinium Global Screening Array v3 and imputed using the GenomeAsia pilot reference panel. Variants with low call rate (<0.99), rare variants with minor allele frequency <1%, and variants that failed the Hardy–Weinberg test (P < 1 × 10−6) in a subset of samples with low level of autozygosity were removed.
Across all cohorts, individuals were analyzed in distinct self-identified groups of African, East Asian, European, Hispanic and South Asian ancestries. The generated polygenic scores were residualized for the first ten principal components of genetic ancestry and then scaled to a mean of 0 and standard deviation of 1 for each ancestral group.
Statistical analysis
Comparison of baseline characteristics between individuals with high or average genetic risk based on polygenic score was performed with the chi-squared test for categorical variables, analysis of variance (ANOVA) for a subset of continuous variables with normal distributions, and Mann–Whitney U test for continuous variables with nonparametric distributions. Individuals with a given magnitude of increased risk were identified by comparing progressively higher percentile cut-offs to the middle quintile population in a logistic regression model predicting disease status and adjusted for baseline model covariates. Individuals were next binned into 100 groupings according to percentile of the GPSMult, and the unadjusted prevalence of CAD within each bin was determined.
Risk for prevalent disease was calculated using logistic regression models, including baseline model covariates defined as enrollment age, sex, genotyping array and the first ten principal components of genetic ancestry. Risk for incident CAD was calculated using Cox proportional-hazards regression models, including baseline model covariates. The proportion of phenotypic variance explained by the polygenic score or risk factor of interest on the observed scale was calculated using the Nagelkerke’s pseudo-R2 metric using the rcompanion R package—where R2 was calculated for the full model inclusive of the variable of interest plus the baseline model covariates minus R2 for the baseline model covariates alone. The proportion of phenotypic variance explained on the liability scale was similarly calculated using the logit liability R2 metric100.
To determine the polygenic risk equivalent of a CAD event comparable to risk experienced by those with prior CAD, a model was constructed comparing three groups and monitored for a CAD event in the follow-up period: individuals with prior CAD, individuals without prior CAD in different groupings of the top distribution of GPSMult (high GPSMult) and individuals in the middle quintile of GPSMult without prior CAD using the survminer R package. Sequentially lower percentile cut-offs for this high GPSMult group were tested to find the grouping with equivalent risk increase for CAD as those with prior CAD. This analysis was repeated for diabetes mellitus, PAD and severe hypercholesterolemia (LDL cholesterol ≥190 mg/dL). In the lower tail of GPSMult, the risk for incident CAD was calculated in individuals in the bottom 5 percentiles or 5th to 9th percentiles of GPSMult relative to those in the middle quintile, using Cox proportional-hazards regression models including baseline model covariates. The prevalence of CAD among individuals in the bottom 5 percentiles of GPSMult was calculated, stratified by 20 pack-years smoking increments and compared with the prevalence of CAD in nonsmokers in the middle 40th to 59th percentiles to estimate equivalent offset risk.
Cox proportional-hazards models were used to estimate HRs for incident CAD in the UK Biobank, with covariates of the first ten principal components. In model 1, only age and sex were modeled with the covariates. In model 2, only the clinical risk estimator—ACC/AHA PCE5 or QRISK3 (ref. 6)—was modeled with the covariates. In model 3, GPSMult, clinical risk estimator, and the interaction term of GPSMult with the clinical risk estimator and the first ten principal components of genetic ancestry are modeled. The 10-year incidence of CAD for individuals grouped by GPSMult percentile and stratified by ancestry group was quantified using model 3 standardized to four PCE risk levels (mean 10-year risk of atherosclerotic cardiovascular disease as low (<5%), borderline (5% to <7.5%), intermediate (≥7.5% to <20%), and high (≥20%)) and the means of each of the covariates. The discrimination of each of these predictive models was assessed using Harrell’s C-statistic. The improvement in predictive performance of the addition of the GPSMult to the PCE or QRISK3 was evaluated using continuous and categorized NRI, with a risk probability threshold of 7.5% and 95% CIs obtained from 100-fold bootstrapping with the nricens R package. All analyses were two-sided. In all analyses, a 95% CI that excluded unity was considered evidence of statistical significance. All statistical analyses were performed with the use of R software, versions 3.5 and 3.6 (R Project for Statistical Computing) and figures were generated using the ggplot2 R package.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are made available from the UK Biobank (https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access) to researchers from universities and other institutions with genuine research inquiries following institutional review board and UK Biobank approval. This research was conducted using the UK Biobank resource under application number 7089 and approved by the Mass General Brigham institutional review board. The genome-wide association data supporting the findings of this study are publicly available in Biobank Japan (http://jenger.riken.jp/en/result), FinnGen (https://www.finngen.fi/en/access_results), AGEN T2D (https://kp4cd.org/index.php/node/309), GIANT (https://portals.broadinstitute.org/collaboration/giant/), Global Lipids Genetics Consortium (http://csg.sph.umich.edu/willer/public/glgc-lipids2021) and Million Veteran Program (via dbGaP at https://ftp.ncbi.nlm.nih.gov/dbgap/studies/, under accession number phs001672), and upon request from CARDloGRAMplusC4D (http://www.cardiogramplusc4d.org/data-downloads/), MEGASTROKE (http://megastroke.org/download.html) and Genes & Health (https://www.genesandhealth.org/research/scientific-data-downloads). The full GPSMult weights are available in the Polygenic Score Catalog through accession ID PGS003725.
References
Roth, G. A. et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1736–1788 (2018).
Arnett, D. K. et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 140, e596–e646 (2019).
DeFilippis, A. P. et al. An analysis of calibration and discrimination among multiple cardiovascular risk scores in a modern multiethnic cohort. Ann. Intern. Med. 162, 266–275 (2015).
Patel, A. P., Wang, M., Kartoun, U., Ng, K. & Khera, A. V. Quantifying and understanding the higher risk of atherosclerotic cardiovascular disease among South Asian individuals. Circulation 144, 410–422 (2021).
Goff David, C. et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk. Circulation 129, S49–S73 (2014).
Hippisley-Cox, J., Coupland, C. & Brindle, P. Development and validation of QRISK3 risk prediction algorithms to estimate future risk of cardiovascular disease: prospective cohort study. Br. Med. J. 357, j2099 (2017).
Emdin, C. A. et al. Polygenic score assessed in young adulthood and onset of subclinical atherosclerosis and coronary heart disease. J. Am. Coll. Cardiol. 80, 280–282 (2022).
Inouye, M. et al. Genomic risk prediction of coronary artery disease in 480,000 adults. J. Am. Coll. Cardiol. 72, 1883–1893 (2018).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Hindy, G. et al. Genome-wide polygenic score, clinical risk factors, and long-term trajectories of coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 40, 2738–2746 (2020).
Sun, L. et al. Polygenic risk scores in cardiovascular risk prediction: a cohort study and modelling analyses. PLoS Med. 18, e1003498 (2021).
Hao, L. et al. Development of a clinical polygenic risk score assay and reporting workflow. Nat. Med. 28, 1006–1013 (2022).
Maamari, D. J. et al. Clinical implementation of combined monogenic and polygenic risk disclosure for coronary artery disease. JACC Adv. 1, 1–11 (2022).
Patel, A. P. & Khera, A. V. Advances and applications of polygenic scores for coronary artery disease. Annu. Rev. Med. 74, 141–154 (2023).
Zhang, Y., Qi, G., Park, J.-H. & Chatterjee, N. Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat. Genet. 50, 1318–1326 (2018).
Ruan, Y. et al. Improving polygenic prediction in ancestrally diverse populations. Nat. Genet. 54, 573–580 (2022).
Weissbrod, O. et al. Leveraging fine-mapping and multipopulation training data to improve cross-population polygenic risk scores. Nat. Genet. 54, 450–458 (2022).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Ning, Z., Pawitan, Y. & Shen, X. High-definition likelihood inference of genetic correlations across human complex traits. Nat. Genet. 52, 859–864 (2020).
Martin, A. R. et al. Current clinical use of polygenic scores will risk exacerbating health disparities. Nat. Genet. 51, 584–591 (2019).
All of Us Research Program Investigators et al. The ‘All of Us’ research program. N. Engl. J. Med. 381, 668–676 (2019).
Fatumo, S. et al. Promoting the genomic revolution in Africa through the Nigerian 100K Genome Project. Nat. Genet. 54, 531–536 (2022).
Elliott, J. et al. Predictive accuracy of a polygenic risk score-enhanced prediction model vs a clinical risk score for coronary artery disease. JAMA 323, 636–645 (2020).
Riveros-Mckay, F. et al. Integrated polygenic tool substantially enhances coronary artery disease prediction. Circ. Genom. Precis. Med. 14, e003304 (2021).
Weale, M. E. et al. Validation of an integrated risk tool, including polygenic risk score, for atherosclerotic cardiovascular disease in multiple ethnicities and ancestries. Am. J. Cardiol. 148, 157–164 (2021).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Aragam, K. G. et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat. Genet. 54, 1803–1815 (2022).
Lambert, S. A. et al. The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nat. Genet. 53, 420–425 (2021).
Manikpurage, H. D. et al. Polygenic risk score for coronary artery disease improves the prediction of early-onset myocardial infarction and mortality in men. Circ. Genom. Precis. Med. 14, e003452 (2021).
Neumann, J. T. et al. Prognostic value of a polygenic risk score for coronary heart disease in individuals aged 70 years and older. Circ. Genom. Precis. Med. 15, e003429 (2022).
Gaziano, J. M. et al. Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol. 70, 214–223 (2016).
Finer, S. et al. Cohort Profile: East London Genes & Health (ELGH), a community-based population genomics and health study in British Bangladeshi and British Pakistani people. Int. J. Epidemiol. 49, 20–21i (2020).
Tcheandjieu, C. et al. Large-scale genome-wide association study of coronary artery disease in genetically diverse populations. Nat. Med. 28, 1679–1692 (2022).
Huang, Q. Q. et al. Transferability of genetic loci and polygenic scores for cardiometabolic traits in British Pakistani and Bangladeshi individuals. Nat. Commun. 13, 4664 (2022).
Dron, J. S. et al. Association of rare protein-truncating DNA variants in APOB or PCSK9 with low-density lipoprotein cholesterol level and risk of coronary heart disease. JAMA Cardiol. 8, 258–267 (2023).
Cohen, J. C., Boerwinkle, E., Mosley, T. H. & Hobbs, H. H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 354, 1264–1272 (2006).
Howe, L. J. et al. Polygenic risk scores for coronary artery disease and subsequent event risk amongst established cases. Hum. Mol. Genet 29, 1388–1395 (2020).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424 (2021).
Locke, A. E. et al. Exome sequencing of Finnish isolates enhances rare-variant association power. Nature 572, 323–328 (2019).
Zdravkovic, S. et al. Heritability of death from coronary heart disease: a 36-year follow-up of 20 966 Swedish twins. J. Intern Med 252, 247–254 (2002).
Zhang, H. et al. Novel methods for multi-ancestry polygenic prediction and their evaluations in 5.1 million individuals of diverse ancestry. Preprint at bioRxiv https://doi.org/10.1101/2022.03.24.485519 (2023).
Myers, P. D. et al. Identifying unreliable predictions in clinical risk models. NPJ Digit. Med. 3, 1–8 (2020).
Wand, H. et al. Improving reporting standards for polygenic scores in risk prediction studies. Nature 591, 211–219 (2021).
Brockman, D. G. et al. Design and user experience testing of a polygenic score report: a qualitative study of prospective users. BMC Med. Genomics 14, 238 (2021).
Ding, Y. et al. Polygenic scoring accuracy varies across the genetic ancestry continuum. Nature https://doi.org/10.1038/s41586-023-06079-4 (2023).
Linder, J. E. et al. Returning integrated genomic risk and clinical recommendations: the eMERGE study. Genet. Med. 25, 100006 (2023).
O’Sullivan, J. W. et al. Polygenic risk scores for cardiovascular disease: a scientific statement from the American Heart Association. Circulation 146, e93–e118 (2022).
Khera, A. V. et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N. Engl. J. Med. 375, 2349–2358 (2016).
Natarajan, P. et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation 135, 2091–2101 (2017).
Aragam, K. G. et al. Limitations of contemporary guidelines for managing patients at high genetic risk of coronary artery disease. J. Am. Coll. Cardiol. 75, 2769–2780 (2020).
Fahed, A. C., Philippakis, A. A. & Khera, A. V. The potential of polygenic scores to improve cost and efficiency of clinical trials. Nat. Commun. 13, 2922 (2022).
Marston, N. A. et al. Predicting benefit from evolocumab therapy in patients with atherosclerotic disease using a genetic risk score: results from the FOURIER trial. Circulation 141, 616–623 (2020).
Damask, A. et al. Patients with high genome-wide polygenic risk scores for coronary artery disease may receive greater clinical benefit from alirocumab treatment in the ODYSSEY OUTCOMES trial. Circulation 141, 624–636 (2020).
Cook, N. R. Use and misuse of the receiver operating characteristic curve in risk prediction. Circulation 115, 928–935 (2007).
Fry, A. et al. Comparison of sociodemographic and health-related characteristics of UK Biobank participants with those of the general population. Am. J. Epidemiol. 186, 1026–1034 (2017).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023).
Locke, A. E. et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015).
Malik, R. et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 50, 524–537 (2018).
Zhou, W. et al. Global Biobank Meta-analysis Initiative: Powering genetic discovery across human disease. Cell Genomics 2, 100192 (2022).
Vujkovic, M. et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 52, 680–691 (2020).
Spracklen, C. N. et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature 582, 240–245 (2020).
Klarin, D. et al. Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nat. Med. 25, 1274–1279 (2019).
Hellwege, J. N. et al. Mapping eGFR loci to the renal transcriptome and phenome in the VA Million Veteran Program. Nat. Commun. 10, 3842 (2019).
Evangelou, E. et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 50, 1412–1425 (2018).
Graham, S. E. et al. The power of genetic diversity in genome-wide association studies of lipids. Nature 600, 675–679 (2021).
Klarin, D. et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet. 50, 1514–1523 (2018).
Koyama, S. et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet. 52, 1169–1177 (2020).
Ye, Y. et al. Interactions between enhanced polygenic risk scores and lifestyle for cardiovascular disease, diabetes, and lipid levels. Circ. Genom. Precis. Med. 14, e003128 (2021).
Tamlander, M. et al. Integration of questionnaire-based risk factors improves polygenic risk scores for human coronary heart disease and type 2 diabetes. Commun. Biol. 5, 158 (2022).
Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Patel, A. P. et al. Association of rare pathogenic DNA variants for familial hypercholesterolemia, hereditary breast and ovarian cancer syndrome, and lynch syndrome with disease risk in adults according to family history. JAMA Netw. Open 3, e203959 (2020).
Fahed, A. C. et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat. Commun. 11, 3635 (2020).
Patel, A. P. et al. Lp(a) (lipoprotein[a]) concentrations and incident atherosclerotic cardiovascular disease: new insights from a large national biobank. Arterioscler. Thromb. Vasc. Biol. 41, 465–474 (2021).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Privé, F., Arbel, J. & Vilhjálmsson, B. J. LDpred2: better, faster, stronger. Bioinformatics https://doi.org/10.1093/bioinformatics/btaa1029 (2020).
International HapMap Consortium. The International HapMap Project. Nature 426, 789–796 (2003).
Vilhjálmsson, B. J. et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet 97, 576–592 (2015).
Band, G. & Marchini, J. BGEN: a binary file format for imputed genotype and haplotype data. Preprint at bioRxiv https://doi.org/10.1101/308296 (2018).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575 (2007).
Zhang, Z. Variable selection with stepwise and best subset approaches. Ann. Transl. Med 4, 136 (2016).
Giri, A. et al. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat. Genet. 51, 51–62 (2019).
Wang, M. et al. Validation of a genome-wide polygenic score for coronary artery disease in South Asians. J. Am. Coll. Cardiol. 76, 703–714 (2020).
Mega, J. et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy. Lancet 385, 2264–2271 (2015).
Abraham, G. et al. Genomic risk score offers predictive performance comparable to clinical risk factors for ischaemic stroke. Nat. Commun. 10, 5819 (2019).
Ripatti, S. et al. A multilocus genetic risk score for coronary heart disease: case–control and prospective cohort analyses. Lancet 376, 1393–1400 (2010).
Tikkanen, E., Havulinna, A. S., Palotie, A., Salomaa, V. & Ripatti, S. Genetic risk prediction and a 2-stage risk screening strategy for coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 33, 2261–2266 (2013).
Tada, H. et al. Risk prediction by genetic risk scores for coronary heart disease is independent of self-reported family history. Eur. Heart J. 37, 561–567 (2016).
Paquette, M. et al. Polygenic risk score predicts prevalence of cardiovascular disease in patients with familial hypercholesterolemia. J. Clin. Lipidol. 11, 725–732.e5 (2017).
Hajek, C. et al. Coronary heart disease genetic risk score predicts cardiovascular disease risk in men, not women. Circ. Genom. Precis. Med. 11, e002324 (2018).
Pechlivanis, S. et al. Risk prediction for coronary heart disease by a genetic risk score—results from the Heinz Nixdorf Recall study. BMC Med. Genet. 21, 178 (2020).
Gola, D. et al. Population bias in polygenic risk prediction models for coronary artery disease. Circ. Genom. Precis. Med. 13, e002932 (2020).
Bauer, A. et al. Comparison of genetic risk prediction models to improve prediction of coronary heart disease in two large cohorts of the MONICA/KORA study. Genet. Epidemiol. 45, 633–650 (2021).
Mars, N. et al. Genome-wide risk prediction of common diseases across ancestries in one million people. Cell Genom. 2, 100118 (2022).
Lu, X. et al. A polygenic risk score improves risk stratification of coronary artery disease: a large-scale prospective Chinese cohort study. Eur. Heart J. 43, 1702–1711 (2022).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Walter, K. et al. The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 (2015).
Hunter-Zinck, H. et al. Genotyping array design and data quality control in the Million Veteran Program. Am. J. Hum. Genet. 106, 535–548 (2020).
Lee, S. H., Goddard, M. E., Wray, N. R. & Visscher, P. M. A better coefficient of determination for genetic profile analysis. Genet. Epidemiol. 36, 214–224 (2012).
Acknowledgements
This work was supported by the KL2/Catalyst Medical Research Investigator Training award from Harvard Catalyst (to A.P.P. and K.G.A.); the Sarnoff Cardiovascular Research Foundation Fellowship (to S.A.); grants 1K08HL153937 (to K.G.A.), 1K08HL161448 (to A.C.F.), R01HL1427 (to P.N.), R01HL148565 (to P.N.), R01HL148050 (to P.N.), 1RO1HL092577 (to P.T.E.), 1R01HL157635 (to P.T.E.) and 1R01HL157635 (to P.T.E.) from the National Heart, Lung, and Blood Institute; grants RG/18/13/33946 and CH/12/2/29428 from the British Heart Foundation (to A.S.B.) grants BRC-1215-20014 and NIHR203312 from the NIHR Cambridge Biomedical Research Centre (to A.S.B.) grant RE/18/1/34212 from the Cambridge British Heart Foundation Centre of Research Excellence (to A.S.B.); grants 862032 (to K.G.A.) 18SFRN34110082 (to P.T.E.), 17IFUNP3384001 (to K.G.A.) from the American Heart Association; grant MAESTRIA 965286 from the European Union (to P.T.E.); grants 1K08HG010155 (to A.V.K.) and 1U01HG011719 from the National Human Genome Research Institute (to A.P.P., P.N. and A.V.K.); a Hassenfeld Scholar Award from Massachusetts General Hospital (to P.N. and A.V.K.); a Merkin Institute Fellowship from the Broad Institute of MIT and Harvard (to A.V.K.). This research has been conducted using the UK Biobank Resource, and we thank the volunteers participating. This research is based on data from the Million Veteran Program, Office of Research and Development, Veterans Health Administration, and was supported by Veterans Administration awards I01–01BX003362 (P.S.T.), I01-BX004821 (P.W.F.W. and K.C.) and VA HSR RES 13–457 (VA Informatics and Computing Infrastructure). The content of this manuscript does not represent the views of the Department of Veterans Affairs or the US Government. Genes & Health is/has recently been core-funded by Wellcome (WT102627 and WT210561), the Medical Research Council (UK) (M009017, MR/X009777/1 and MR/X009920/1), Higher Education Funding Council for England Catalyst, Barts Charity (845/1796), Health Data Research UK (for London substantive site), and research delivery support from the NHS National Institute for Health Research Clinical Research Network (North Thames). Genes & Health is/has recently been funded by Alnylam Pharmaceuticals, Genomics PLC; and a Life Sciences Industry Consortium of AstraZeneca PLC, Bristol-Myers Squibb Company, GlaxoSmithKline Research and Development Limited, Maze Therapeutics Inc, Merck Sharp & Dohme LLC, Novo Nordisk A/S, Pfizer Inc, Takeda Development Centre Americas Inc. We thank Social Action for Health, Centre of The Cell, members of our Community Advisory Group, and staff who have recruited and collected data from volunteers. We thank the NIHR National Biosample Centre (UK Biocentre), the Social Genetic & Developmental Psychiatry Centre (King’s College London), Wellcome Sanger Institute, and Broad Institute for sample processing, genotyping, sequencing and variant annotation. We thank: Barts Health NHS Trust, NHS Clinical Commissioning Groups (City and Hackney, Waltham Forest, Tower Hamlets, Newham, Redbridge, Havering, Barking and Dagenham), East London NHS Foundation Trust, Bradford Teaching Hospitals NHS Foundation Trust, Public Health England (especially David Wyllie), Discovery Data Service/Endeavour Health Charitable Trust (especially David Stables) and NHS Digital, for GDPR-compliant data sharing backed by individual written informed consent. We thank all of the volunteers participating in Genes & Health.
Author information
Authors and Affiliations
Consortia
Contributions
Concept and design: A.P.P., M.W., Y.R., P.N. and A.V.K. Acquisition, analysis or interpretation of data: A.P.P., M.W., Y.R., S.K., S.L.C., X.Y., C.T., S.A., A.C.F., D.A.v.H., A.S.B., K.G.A., P.N. and A.V.K. Drafting of the manuscript: A.P.P., M.W. and A.V.K. Critical revision of the manuscript for important intellectual content: P.T.E., P.S.T., Y.V.S., K.C., P.W.F.W. and T.L.A.
Corresponding authors
Ethics declarations
Competing interests
S.A. has served as a scientific advisor to Third Rock Ventures. A.C.F. is a co-founder of Goodpath and reports a grant from Abbott Vascular. P.T.E. receives sponsored research support from Bayer AG and IBM Research; he has also served on advisory boards or consulted for Bayer AG, MyoKardia and Novartis. A.S.B. reports institutional grants from AstraZeneca, Bayer, Biogen, BioMarin, Bioverativ, Novartis, Regeneron and Sanofi. P.N. reports research grants from Allelica, Apple, Amgen, Boston Scientific, Genentech/Roche and Novartis, personal fees from Allelica, Apple, AstraZeneca, Blackstone Life Sciences, Foresite Labs, Genentech/Roche, GV, HeartFlow, Magnet Biomedicine and Novartis, scientific advisory board membership of Esperion Therapeutics, Preciseli and TenSixteen Bio, scientific co-founder of TenSixteen Bio, equity in Preciseli and TenSixteen Bio, and spousal employment at Vertex Pharmaceuticals, all unrelated to the present work. A.V.K. is an employee of Verve Therapeutics; has served as a scientific advisor to Amgen, Novartis, Silence Therapeutics, Korro Bio, Veritas International, Color Health, Third Rock Ventures, Illumina, Ambry and Foresite Labs; holds equity in Verve Therapeutics, Color Health and Foresite Labs; and is listed as a co-inventor on patent applications related to assessment and mitigation of risk associated with perturbations in body fat distribution. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling editor: Michael Basson, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Sequential improvements in R2 with GPSMult in the UK Biobank Study.
The proportion of phenotypic variance explained by the polygenic score predicting coronary artery disease (CAD) was calculated in the UK Biobank European ancestry validation cohort for each GPS score using the A: Nagelkerke’s pseudo-R2 metric, as the difference of the full model inclusive of the polygenic score plus age, sex, genotyping array, and the first ten principal components of ancestry minus R2 for the covariates alone; and B: logit-liability R2 metric. GPS2018 denotes previously published polygenic score for CAD9. > 3-fold larger CAD GWAS designates metrics for polygenic score generated using summary statistics from the most recent Coronary ARtery DIsease Genome wide Replication and Meta-analysis plus The Coronary Artery Disease Genetics consortium analysis (CARDIOGRAMplusC4D) excluding the UK Biobank. Multi-ancestry CAD GWAS refers to the polygenic score generated by combining ancestry-specific polygenic scores generated using discovery data from Genes & Health, Biobank Japan, Million Veteran Program, FinnGen, and CARDIOGRAMplusC4D (excluding UK Biobank). GPSMult designates polygenic score for CAD designed with summary statistics from multiple ancestries and multiple CAD-related traits.
Extended Data Fig. 2 GPSMult performance by sex and age subgroups.
The odds ratio per standard deviation (OR/SD) with 95% confidence intervals for prevalent coronary artery disease (CAD) risk of the GPSMult was assessed in a logistic regression model adjusted for age, sex, genotyping array, and the first ten principal components of genetic ancestry in the European ancestry validation dataset of the UK biobank (N = 308,264 independent participants) stratified by sex and age subgroups. P values are derived from a t-test implemented in the GLM function in R and are two-sided.
Extended Data Fig. 3 Coronary artery disease risk in the extreme ends of the polygenic score distribution.
Proportion of UK Biobank validation population with 3, 4, and 5-fold increased risk for CAD versus the middle quintile of the population identified by GPS2018 (A) and GPSMult (B). The odds ratio assessed in a logistic regression model adjusted for age, sex, genotyping array, and the first ten principal components of ancestry. Proportion of UK Biobank testing population with 1/3, 1/4, and 1/5 risk for CAD versus the middle quintile of the population, population identified by GPS2018 (C) and GPSMult (D). Odds ratio assessed in a logistic regression model adjusted for age, sex, genotyping array, and the first ten principal components of ancestry. GPS: Genome-wide polygenic score; CAD: coronary artery disease.
Extended Data Fig. 4 Extremes of risk for incident coronary artery disease identified by tail distributions of GPSMult.
A: Cumulative incidence of coronary artery disease (CAD) events (%) over length of the follow-up period stratified by presence of prior CAD or with no prior CAD and for the middle quintile or top 3% of the population for GPSMult risk, estimated using Cox proportional-hazards regression model adjusted for age, sex, genotyping array, and the first ten principal components of ancestry in the European ancestry UK Biobank validation dataset. The estimated 10-year CAD event risk was predicted using same model standardized to the mean of each of the covariates. B: Cumulative CAD risk (%) stratified by the bottom 5%, the 5–9% segment, and the 40–59% segment of the population for GPSMult risk, estimated using Cox proportional-hazards regression model adjusted for age, sex, genotyping array, and the first ten principal components of ancestry in the European ancestry validation dataset of the UK Biobank Study. The estimated 10-year CAD risk was predicted using same model standardized to the mean of each of the covariates. GPS: Genome-wide polygenic score.
Extended Data Fig. 5 Equivalents of increased risk for incident coronary artery disease event identified by high GPSMult in the UK Biobank Study.
A: Cumulative incidence of coronary artery disease (CAD) events (%) over length of follow-up stratified by presence of prior peripheral artery disease (PAD) or no prior PAD with GPSMult in the middle quintile or top 8% of the population, estimated using Cox proportional-hazards regression model adjusted for age, sex, genotyping array, and the first ten principal components of ancestry in the European ancestry UK Biobank validation dataset. B: Cumulative incidence of CAD events (%) over length of follow-up stratified by presence of prior diabetes mellitus (DM) or no prior DM with GPSMult in the middle quintile or top 21% of the population, estimated using Cox proportional-hazards regression model adjusted for age, sex, genotyping array, and the first ten principal components of ancestry in the European ancestry UK Biobank validation dataset. C: Cumulative incidence of CAD events (%) over length of follow-up stratified by presence of prior severe hypercholesterolemia (estimated untreated low-density lipoprotein cholesterol, LDL-C 190 mg/dL or higher), or no prior hypercholesterolemia with GPSMult in the middle quintile or top 28% of the population, estimated using Cox proportional-hazards regression model adjusted for age, sex, genotyping array, and the first ten principal components of ancestry in the European ancestry UK Biobank validation dataset.
Extended Data Fig. 6 Net reclassification with presence of high GPSMult or CAD risk enhancing factors over PCE 10-year risk estimates.
Net reclassification of coronary artery disease (CAD) cases and non-cases at the 7.5% threshold achieved by presence of established CAD risk enhancing factors or high GPSMult when added to a baseline model of just the American Heart Association/American College of Cardiology Pooled Cohort Equations5 in the European ancestry validation dataset of the UK Biobank.
Extended Data Fig. 7 Association of GPSMult and risk factors with incident and recurrent coronary artery disease events.
Hazards ratio per standard deviation (HR/SD) with 95% confidence intervals of variable of interest for incident disease assessed in individuals without prior coronary artery disease (CAD) followed for development of first CAD event with Cox proportional hazards regression model adjusted for age, sex, genotyping array, and the first ten principal components of ancestry in the European ancestry validation dataset of the UK Biobank Study (N = 308,264 independent participants). HR/SD of variable of interest for recurrent disease assessed in individuals with prior CAD followed for development of recurrent CAD event with Cox proportional hazards regression model adjusted for age, sex, genotyping array, and the first ten principal components of ancestry. P values are derived from a Wald test implemented in the coxph function in R and are two-sided. *LDL-C, HDL-C, and triglyceride values were adjusted for cholesterol-lowering medication status, as previously described57. BP: Blood pressure. BMI: Body-mass index. HgbA1c: Glycated hemoglobin. LDL-C: Low-density lipoprotein cholesterol. HDL-C: High-density lipoprotein cholesterol.
Supplementary information
Supplementary Information
Supplementary Tables 1–9.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Patel, A.P., Wang, M., Ruan, Y. et al. A multi-ancestry polygenic risk score improves risk prediction for coronary artery disease. Nat Med 29, 1793–1803 (2023). https://doi.org/10.1038/s41591-023-02429-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-023-02429-x
This article is cited by
-
Clinical data mining: challenges, opportunities, and recommendations for translational applications
Journal of Translational Medicine (2024)
-
Predicting the presence of coronary plaques featuring high-risk characteristics using polygenic risk scores and targeted proteomics in patients with suspected coronary artery disease
Genome Medicine (2024)
-
Genetic variation across and within individuals
Nature Reviews Genetics (2024)
-
Integration of polygenic and gut metagenomic risk prediction for common diseases
Nature Aging (2024)
-
What Causes Premature Coronary Artery Disease?
Current Atherosclerosis Reports (2024)
